

Abstract
Much is known about the neurobiology of memory storage for learned fear. In contrast, the molecular mechanisms underlying extinction of fear memory are just beginning to be delineated. Here, we investigate the role of protein kinase A (PKA) in extinction of memory for contextual fear by using conventional and temporally regulated transgenic approaches that allow us to inhibit PKA activity in neurons within brain regions thought to be involved in extinction. Strikingly, reduction of PKA activity facilitated the development of extinction, without interfering with storage of the original fear memory. Moreover, inhibition of PKA facilitated extinction of both recent and remote contextual fear memories. The finding that PKA, which is required for the acquisition of fear memory, is a constraint for extinction provides the first genetic support for the idea that fear extinction is itself a genuine learning process with its own specific molecular requirements, rather than simply the erasure of a previously learned process. Further, these experiments represent the first genetic evidence that protein kinases may be constraints for the extinction of fear.
Keywords: reconsolidation, extinction, fear, learning memory, pavlovian conditioning, protein kinase, protein synthesis, tetracycline, transgenic
Introduction
Many studies of the neural and genetic basis of memory have demonstrated that fearful memories can form quickly and are extremely difficult to eliminate (Maren and Quirk, 2004; Fanselow and Poulous, 2005; Phelps and LeDoux, 2005). In the laboratory, fear is created through pavlovian conditioning paradigms, in which a biologically neutral stimulus such as a tone or a context [the conditioned stimulus (CS)], is paired with an aversive event [the unconditioned stimulus (US)], such as a mild footshock in rodent studies. Memory for the CS–US association is robust and long lasting, but if the CS is presented repeatedly in the absence of the US, the fear response weakens through a process known as experimental extinction (for review, see Myers and Davis, 2002; Delamater, 2004). Numerous studies have suggested that extinction is an active learning process that results in new memories that compete with and suppress the expression of memories formed during conditioning (Pavlov, 1927; Bouton, 1993; Rescorla, 2001; Bouton et al., 2006). Indeed, the original memory is not eliminated during extinction and extinguished fear responses return with time (Pavlov, 1927; Lattal and Abel, 2004; Rescorla, 2004). Relatively little is known about the molecular mechanisms that mediate extinction and how these mechanisms compare with those that mediate initial memory storage.
Much is known about the molecular mechanisms and neural systems that underlie the formation of fear memories (Abel and Lattal, 2001; Tonegawa et al., 2003; Rodrigues et al., 2004; Fanselow and Poulos 2005). Extinction appears to rely on many of these same molecular and neural systems, but a number of studies suggest that extinction may also engage different processes (Lattal and Abel, 2001; Cain et al., 2002; Marsicano et al., 2002; Davis et al., 2006; Lattal et al., 2006). One molecular pathway that regulates memory formation across species is the cAMP/protein kinase A (PKA) signaling cascade. This pathway is critical for initial memory formation (Byrne and Kandel, 1996; Abel et al., 1997; Siwicki and Ladewski, 2003), but its role in extinction remains unclear (Koh and Bernstein, 2003; Szapiro et al., 2003; Cammarota et al., 2005; McNally et al., 2005; Tronson et al., 2006). In the following experiments, we take a transgenic approach to the study of PKA and extinction. This approach allows us to regulate PKA in a molecular and cell-type specific manner that is difficult with pharmacological approaches.
We assessed the role of neuronal PKA in the extinction of context-evoked fear by using transgenic mice that express a dominant-negative form of PKA from birth and transgenic mice in which the expression of this same dominant-negative form of PKA could be temporally regulated using the tetracycline system. Both approaches revealed that reduction of PKA activity in forebrain neurons facilitates extinction of contextual fear without affecting the original fear memory. Moreover, the use of the tetracycline regulated system enabled us to show that inhibition of neuronal PKA activity acutely in the adult forebrain was sufficient to cause impairments in fear memory formation. Together, these data define opposite roles for PKA in the formation of initial memories and extinction memories, and they provide strong genetic evidence for the idea that acquisition and extinction are distinct learning processes.
Materials and Methods
Mice.
For some of our experiments, we used previously characterized transgenic mice referred to as calmodulin-dependent protein kinase IIα (CaMKIIα)-R(AB) that expressed the dominant-negative form of PKA R(AB) in forebrain neurons from birth (Abel et al., 1997). Specifically, we used 8- to 12-week-old male and female transgenic R(AB) mice from line 2 and wild-type littermates as controls. For genotyping R(AB) transgenic mice, tail DNA was prepared and analyzed by Southern blotting using a transgene-specific probe as described previously (Abel et al., 1997). Bitransgenic mice in which the expression of R(AB) could be temporally limited by the tTA system were used for some experiments. To generate mouse lines carrying R(AB) under the control of the operator sequences to which the tetracycline-controlled transactivator binds, the cDNA encoding R(AB) was subcloned into the EcoRV site of pMM400 (Mayford et al., 1996b). The tetO-R(AB) transgene [R(AB) cDNA under the control of tetracycline operator sequences] was purified away from vector sequences and microinjected into pronuclei of B6/CBA F1 zygotes that were then implanted in pseudopregnant females. Founders were back-crossed to C57BL/6J mice. Fourteen tetO-R(AB) transgenic mouse lines were established. To generate bitransgenic animals, mice from these 14 lines were crossed to mice from a line that carried the tetracycline-controlled transcriptional transactivator tTA driven by the CaMKIIα promoter (line B) obtained from Mark Mayford (Scripps Institute, La Jolla, CA) (Mayford et al., 1996b). Levels of expression by in situ hybridization, PKA activity assays in extracts from different brain regions, and long term memory for contextual fear conditioning were assessed in bitransgenic mice from the different tetO-R(AB) lines (data not shown). The tetO-R(AB) mouse line showing the highest level of expression of R(AB) and the most profound deficits in long-term memory for contextual fear conditioning (line 7184) was selected for the experiments described in this paper. We used 8- to 12-week-old male and female double transgenic mice and wild-type littermates as controls (no sex differences were observed). For genotyping tTA line B and tetO-R(AB) transgenic mice, tail DNA was prepared and analyzed by Southern blotting using transgene-specific probes. Mice had access to food and water ad libitum and were maintained on a 12 h light/dark cycle with behavioral testing occurring during the light phase. All mice were bred onto a C57B/6J background for more than 10 generations. TetO-linked R(AB) expression was suppressed using mouse chow containing 40 mg/kg doxycline (Dox) (Bio-serve, Frenchtown, NJ). All experiments were conducted according to National Institutes of Health guidelines for animal care and use and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
In situ hybridization and Nissl staining. Mouse brains were dissected and flash frozen in 2-methylbutane. Sagittal cryostat sections (20 μm) were fixed and hybridized as described previously (Abel et al., 1997). [α-35S]-dATP-labeled, transgene-specific oligonucleotides were used to detect mRNA in CaMKIIα-R(AB) transgenic (5′-GCAGGATCCGCTTGGGCTGCAGTTGGACCT-3′) and tetO-R(AB) bitransgenic mice (5′-GGTGGGGGGTCCTCTAGATCCGCAGGATCCGC-3′). Slides were exposed to Kodak (Rochester, NY) Biomax autoradiographic film. For detection of Nissl bodies, sections on slides were stained with thionin.
PKA activity assays.
In vitro PKA activity assays were performed as described previously (Abel et al., 1997). Briefly, hippocampi, prefrontal cortex, temporal cortex, amygdalae, and cerebellum were dissected and homogenized in 300, 200, 300, 100, and 400 μl, respectively, of ice-cold extraction buffer [PBS containing the following (in mm): 1 EGTA, 1 EDTA, 0.1% Triton X-100, 5 DTT, 0.5 PMSF, and a mixture of proteases inhibitors]. For amygdalar extracts, amygdalae from two mice were pooled. The extracts were then diluted to a protein concentration of 2 mg/ml (Bradford, 1976) and kept in ice until use. PKA activity was assayed in duplicate at 30°C for 5 min as described previously (Clegg et al., 1987). To some reactions, cAMP (5 μm) and/or protein kinase inhibitor (40 μg/ml) were added. PKA activities (picomoles [γ-32P]ATP per minute per milligram of protein) were calculated relative to the background activity without added substrate.
Fear conditioning.
Fear conditioning experiments were performed in standard conditioning chambers (Med Associates, St. Albans, VT). Mice were handled for two consecutive days for 2 min each day. On the day of acquisition, all mice that were going to be subjected to extinction 24 h later or 4 weeks later, were placed into the conditioning chamber without a cue and received a 2 s, 1.5 mA footshock at 2 and 2.5 min after placement into the chamber as described previously (Lattal and Abel, 2004). Mice were removed from the chamber 30 s after the second shock. During the daily extinction sessions, mice were re-exposed to the conditioning context for 3 min (see Fig. 1) or 24 min (see Figs. 2, 5) in the absence of shock and freezing behavior [defined as complete immobility except for breathing (Fanselow, 1980)] was scored every 5 s for each mouse. In the experiments in which the role of PKA in fear memory formation in the adult was assessed, on the training day, mice were placed in the conditioning chamber and the cue (white noise) was activated from 2 to 2.5 min. The last 2 s of the cue were paired with a 1.5 mA footshock and after 30 additional seconds the mouse was returned to the home cage. Different sets of mice were used for testing contextual and cued conditioning. Conditioning was assessed by scoring freezing behavior. Contextual conditioning was assessed for five consecutive min in the chamber in which the mice were trained 1 h and 24 h after training. Different sets of mice were used for the 1 and 24 h retention tests. For testing cued conditioning, mice were placed in a novel context (another conditioning chamber with a smooth flat floor, altered dimensions, and a novel odorant) for 2 min (pre-CS) followed by exposure to the cue from 2 to 5 min (CS), 24 h after training.
Figure 1.

Greater fear extinction in transgenic mice with reduced PKA activity. A, Representative sagittal brain section of R(AB) transgenic mice hybridized with a probe specific to the transgene. As described previously (Abel et al., 1997), expression of CaMKIIα-R(AB) was detected in the hippocampus, cortex, amygdala, striatum, and olfactory bulb. B, To examine the effects of PKA inhibition on long-term retention of extinction across sessions, starting 24 h after contextual fear conditioning, CaMKIIα-R(AB) transgenics (n = 11) and wild-type littermates (n = 12) were re-exposed to the conditioning context for 3 min on 10 d (extinction), and freezing levels were measured. Mice were retested in the same context 21 d after the last extinction session to assess spontaneous recovery (retest; experimental day 31), and starting the following day they were subjected to additional extinction sessions for 4 d (re-extinction; experimental days 32, 33, 34, and 35). The retest and re-extinction trials were 3 min each. Error bars indicate SEM.
Figure 2.

Genetic inhibition of PKA facilitates learning of fear extinction. Given that PKA reduction promoted retention of extinction across short daily sessions, we examined the effects of PKA inhibition on the short-term development of extinction within long sessions, which are thought to favor extinction learning. Therefore, 24 h after contextual fear conditioning, CaMKIIα-R(AB) transgenics (n = 12) and wild-type littermates (n = 12) were re-exposed to the conditioning context in 24 min sessions for four consecutive days. Freezing is shown in blocks of 3 min for each 24 min daily session. Error bars indicate SEM.
Figure 5.

Transgenic reduction of PKA activity in the adult facilitates the extinction of contextual fear. A, TetO-R(AB) transgenic mice and wild-type littermates were raised on Dox to suppress R(AB) expression until they were 8 weeks old. Mice were then fear conditioned (FC) and removed from Dox 24 h later to induce R(AB) expression in the adulthood. After 28 d, mice were subjected to extinction. B, TetO-R(AB) transgenics (n = 21) and wild-type littermates (n = 19) were re-exposed to the conditioning context in 24 min sessions for three consecutive days. Freezing is shown in blocks of 3 min for each 24 min daily session. Error bars indicate SEM.
Statistical analysis.
Student's t tests were used to analyze PKA assays data and the behavioral data from experiments aimed to assess the role of PKA in memory formation in the adult. α was set to 0.05. Repeated-measures ANOVAs were used to analyze the effect of genotype and trials during extinction. Reliable interactions were explored using Fisher's tests.
Results
Postnatal transgenic inhibition of neuronal PKA activity in forebrain facilitates extinction
To determine whether extinction of contextual fear depends on PKA activity, we first used previously characterized transgenic mice that express a dominant-negative form of PKA, R(AB), under the control of the CaMKIIα promoter (Abel et al., 1997; Bourtchouladze et al., 1998; Rotenberg et al., 2000; Woo et al., 2000a, 2002, 2003). These CaMKIIα-R(AB) transgenic mice are a valuable tool to investigate the role of PKA in extinction of fear because PKA activity is selectively reduced in neurons of forebrain structures known to be relevant for fear extinction such as the amygdala, prefrontal cortex, and hippocampus (Delamater, 2004; Barad et al., 2006; Quirk et al., 2006) because of R(AB) expression from birth (Abel et al., 1997) (Fig. 1A). We found that CaMKIIα-R(AB) transgenic mice show normal memory formation for contextual fear conditioning in response to a stronger fear conditioning training protocol consisting of two massed high intensity footshocks (Woo et al., 2000b). Therefore, CaMKIIα-R(AB) transgenic mice and wild-type littermates received contextual fear conditioning with this protocol. During extinction, mice were re-exposed to the conditioning chamber for 3 min per day for 10 d and levels of freezing were scored (Fig. 1B, extinction). A single 3 min exposure per day to the conditioning context resulted in similar performance early in extinction, but the CaMKIIα-R(AB) transgenic mice showed enhanced extinction in later trials. A genotype by extinction session ANOVA revealed a reliable main effect of extinction session (F(9,189) = 8.7; p < 0.001) and a reliable interaction between genotype and session (F(9,189) = 2.0; p < 0.05). Additional analyses of the interaction demonstrated that although freezing was similar in wild-type and CaMKIIα-R(AB) transgenic mice during the first five sessions of extinction (F(1,21) < 1.0), CaMKIIα-R(AB) mice showed lower levels of freezing compared with wild-type mice during the final five sessions of extinction (F(1,21) = 5.5; p < 0.05), suggesting greater fear extinction. Importantly, CaMKIIα-R(AB) transgenic mice and wild-type littermates exhibited similar performance in a test for spontaneous recovery performed 21 d after extinction (t(21) < 1.0) (Fig. 1B, retest), indicating that the original fear memory remained intact. In subsequent daily re-exposures to the context (Fig. 1B, re-extinction), we observed a trend in which CaMKIIα-R(AB) transgenic mice showed greater re-extinction than wild-type controls [although neither the overall group difference F(1,21) = 3.1 nor the interaction (F(3,63) = 2.0) were reliable], supporting our observations during the initial extinction experiment. Together, these findings indicate that genetic inhibition of PKA facilitated the long-term retention of extinction across short extinction sessions.
Longer re-exposure to the context in the absence of the US is thought to favor development of extinction (Suzuki et al., 2004). We therefore further examined the effects of PKA inhibition on the development of extinction within long extinction sessions. We observed the same pattern of enhanced extinction within longer extinction sessions (Fig. 2) that we observed between brief extinction sessions (Fig. 1B). Twenty four hours after conditioning, CaMKIIα-R(AB) and wild-type mice received a 24 min extinction session for each of four consecutive days. CaMKIIα-R(AB) transgenic mice showed greater extinction over the course of the four 24 min sessions compared with wild-type controls (F(1,22) = 5.1; p < 0.05). Interestingly, at the beginning of each session, performance was similar for CaMKIIα-R(AB) transgenic mice and wild-type littermates, demonstrating recovery from the end of the previous session to the beginning of the next session and suggesting that reduction of neuronal PKA facilitated the development of extinction within the session.
Temporal regulation of the R(AB) transgene
The differences that emerged during extinction shown in Figures 1B and 2 suggest that PKA inhibition facilitated extinction. One caveat to this interpretation, however, is that because PKA was inhibited in CaMKIIα-R(AB) transgenic mice throughout postnatal development, it is difficult to isolate the effects of PKA inhibition directly on extinction, independent of effects on development or on initial conditioning. To demonstrate a direct role for PKA in extinction, we generated bitransgenic tetO-R(AB) mice in which R(AB) expression could be temporally regulated using the tetracycline responsive system.
One transgenic mouse line carried the tetracycline-responsive transcriptional transactivator (tTA) driven by the CaMKIIα promoter to restrict expression to forebrain neurons (line B) (Mayford et al., 1996b). The other transgenic mouse line carried R(AB) driven by the tetO-promoter (Mayford et al., 1996b). Crossing transgenic mice from both mouse lines gave rise to double transgenics [tetO-R(AB)] in which expression of R(AB) can be suppressed by the tetracycline analog Dox (Fig. 3A). To limit R(AB) expression to the adult, transgene expression was suppressed from conception to adulthood by delivering Dox in food at a dose of 40 mg/kg mouse chow (Bejar et al., 2002). Significant levels of R(AB) expression were found 4 weeks after Dox removal in the cortex, hippocampus, striatum, and amygdala (Fig. 3B). This time course of gene expression is similar to what has been described previously for other transgenes regulated by the tTA system (Bejar et al., 2002). Four weeks after Dox removal, PKA activity assays showed significant reduction of PKA activity in the temporal and prefrontal cortex, amygdala, and hippocampus from tetO-R(AB) transgenic mice (p values < 0.05) (Fig. 3C), consistent with the in situ hybridization analysis. Given these results, all subsequent experiments were performed in tetO-R(AB) transgenic mice and wild-type littermates raised on Dox, kept on Dox, and removed from Dox at 8 weeks of age for 4 weeks to allow for R(AB) expression.
Figure 3.

Generation of tetracycline-regulated R(AB) transgenic mice. A, Schematic of the generation of bitransgenic mice expressing R(AB) (modified from Mayford et al., 1996a). Two different transgenic lines are used: one expressing the tetracycline-responsive transcriptional transactivator (tTA) under the control of the CaMKIIα promoter (Mayford et al., 1996a) and another carrying the R(AB) cDNA under the control of tet operator sequences (tetO promoter). In mice bearing both transgenes, expression of R(AB) occurs in the absence of doxycycline (On). Administration of doxycycline in the food suppresses R(AB) expression (Off). B, Sagittal brain sections from bitransgenic R(AB) mice [referred to as tetO-R(AB)] raised on doxycycline containing diet (on Dox) did not express R(AB), as measured by in situ hybridization. TetO-R(AB) mice raised on doxycycline and removed from doxycycline food at 8 weeks of age for 28 d (off Dox as adult) showed a high level of R(AB) mRNA, as detected by in situ hybridization in hippocampus, cortex, striatum, and amygdala. The in situ probe did not hybridize with brain slices from WT littermates, demonstrating its specificity for the transgene. C, Levels of cAMP-stimulated PKA activity were determined in protein extracts from the hippocampus, temporal cortex, prefrontal cortex, amygdala, and cerebellum from tetO-R(AB) transgenics and wild-type littermates that had been raised on Dox and removed from Dox at 8 weeks of age for 28 d. Hippocampus: tetO-R(AB), n = 8; WT, n = 6; temporal cortex, prefrontal cortex, amygdala, and cerebellum: tetO-R(AB), n = 4; WT, n = 4. Error bars indicate SEM. *p < 0.05.
Genetic reduction of PKA in the adult is sufficient to impair formation of memory for contextual fear conditioning
It has been reported previously that postnatal transgenic expression of the dominant-negative form of PKA, R(AB), caused selective deficits in long-term memory for contextual fear conditioning (Abel et al., 1997; Bourtchouladze et al., 1998). To assess the role of neuronal PKA in formation of memory in the adult, tetO-R(AB) transgenic mice and wild-type littermates were raised and kept on Dox diet to suppress R(AB) expression throughout postnatal development. At 8 weeks of age, mice were removed from Dox to induce R(AB) expression and 4 weeks later mice were trained and tested in fear conditioning (Fig. 4A). To assess short-term memory, tetO-R(AB) transgenic mice and wild-type littermates were tested 1 h later in the conditioning context (Fig. 4B). TetO-R(AB) transgenic mice and wild-type littermates showed similar levels of freezing, indicating that inhibition of PKA did not impair their ability to learn and have normal short-term memory. To examine long-term memory for contextual fear conditioning, tetO-R(AB) transgenic mice and wild-type littermates were re-exposed to the conditioning context 24 h after training (Fig. 4C). Although both groups showed similar baseline levels of freezing during training, tetO-R(AB) transgenic mice exhibited a significant decrease in freezing compared with their wild-type littermates in the retention test (Fig. 4C). To test long-term memory for cued fear conditioning, tetO-R(AB) transgenic mice and wild-type littermates were presented the conditioning tone in a novel context 24 h after training (Fig. 4D). No differences were observed between groups either before (Fig. 4D, pre-CS) or after the tone (Fig. 4D, CS). Furthermore, tetO-R(AB) mice exhibited high levels of freezing to the tone, indicating that their ability to freeze is intact. No differences in the freezing levels measured immediately after delivering the shock during training were observed between tetO-R(AB) transgenic mice and wild-type littermates in any of the experiments (data not shown). Similar levels of freezing found in a control group of tetO-R(AB) transgenic mice and wild-type (WT) littermates raised on Dox and left on Dox during training and testing 24 h later in the conditioning context indicated that the long-term memory deficits observed were because of the acute inhibition of PKA [baseline: WT (n = 10), 2.0 ± 1.7%; TG (n = 10), 2.5 ± 1.1%; testing: WT (n = 10), 45.6 ± 5.7%; TG (n = 10), 37.9 ± 4.8%; p = 0.83]. Together, these data extend our previous finding that transgenic inhibition of PKA from birth impairs the establishment of contextual fear memories and demonstrate that neuronal reduction of PKA in the adult forebrain is sufficient to cause deficits in memory for contextual fear conditioning. These findings are consistent with previous studies of R(AB) transgenic mice, which have found deficits in contextual, but not cued fear conditioning (Abel et al., 1997).
Figure 4.
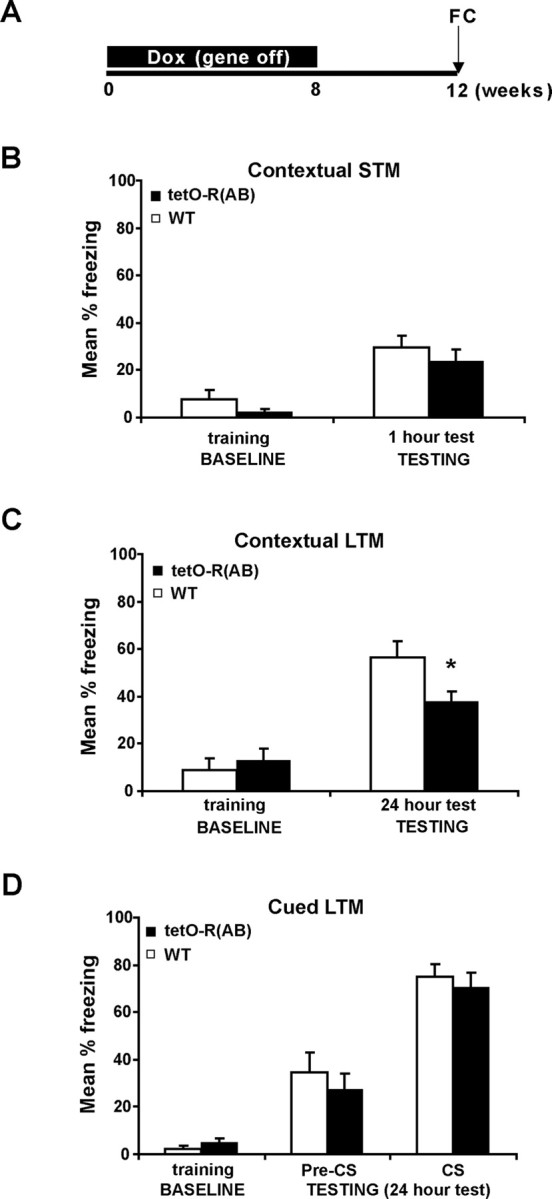
Genetic inhibition of PKA in the adult impairs memory for contextual fear conditioning. A, TetO-R(AB) transgenic mice and wild-type littermates were raised on Dox-containing food to suppress R(AB) expression throughout development and until they became 8 weeks old. They were removed from Dox to induce R(AB) expression for 4 weeks before being trained and tested in fear conditioning (FC). B, Baseline freezing behavior in tetO-R(AB) transgenic mice (n = 8) and wild-type littermates (n = 8) (training, baseline) and during a test of short term memory for contextual fear conditioning (testing, 1 h test). C, Baseline freezing levels are shown for tetO-R(AB) transgenic mice (n = 13) and wild-type littermates (n = 11) (training, baseline) and during a test of long term memory (LTM) for contextual fear conditioning (testing, 24 h test). D, Baseline levels of freezing for tetO-R(AB) transgenic mice (n = 10) and wild-type littermates (n = 9) (training, baseline) and during a test of long term memory for cued fear conditioning performed 24 h later in a novel context (testing, 24 h test). Baseline freezing is that exhibited during the 2.28 s previous to the delivery of the footshock during fear conditioning. Error bars indicate SEM. *p < 0.05.
Transgenic reduction of PKA activity in the adult facilitates fear extinction
To examine the acute role of PKA in extinction in the adult and to avoid interference of R(AB) expression during acquisition of fear conditioning, tetO-R(AB) transgenic mice and wild-type littermates were raised and kept on Dox diet to suppress R(AB) expression. At 8 weeks of age, mice still fed Dox were fear conditioned and 24 h later were removed from Dox food to induce R(AB) expression (Fig. 5A). Four weeks after Dox withdrawal, tetO-R(AB) transgenic mice and wild-type littermates were re-exposed to the conditioning context for 24 min over three consecutive days and freezing in the context was measured (Fig. 5B). Similar to what we observed in the CaMKIIα-R(AB) transgenic mice, tetO-R(AB) transgenic mice exhibited greater extinction over the course of the first two 24 min sessions compared with wild-type controls. A genotype by 3 min session block ANOVA revealed reliable group by block interactions during the first 2 d of extinction sessions (F(7,266) values > 2.2; p values < 0.05). Additional analyses of the interactions revealed that although there were no group differences during the first 3 min of each daily extinction session, there were differences during the final 3 min of each session (t(38) values > 2.0; p values <0.05). The similar levels of freezing observed at the beginning of the second and third sessions suggest that spontaneous recovery was equivalent in tetO-R(AB) and wild-type mice, consistent with the idea that the development of extinction within a session was facilitated by PKA inhibition. In the tetO-R(AB) mice, neuronal PKA was reduced during systems consolidation, the process by which storage of contextual memories become increasingly dependent on other brain regions such as the cortex (Frankland and Bontempi, 2005). However, our observations that transgenic mice and wild-type controls performed similarly during the beginning of long extinction trials (Fig. 5B) suggest that R(AB) expression did not interfere with systems consolidation or with retrieval of remote memories. Together, these experiments demonstrate that the inhibition of neuronal PKA in the adult forebrain is sufficient to facilitate fear extinction and suggest that not only the fear responses associated with recent but also remote fear memories can be more rapidly extinguished by inhibition of PKA.
Discussion
We examined the role of PKA in extinction of context-evoked fear using two distinct yet complementary transgenic approaches that allowed us to reduce PKA activity selectively in forebrain neurons. Both approaches revealed a facilitative effect of PKA inhibition on the development of extinction. Such an effect was observed both across extinction sessions with daily brief exposures (3 min) to the conditioning context in the absence of shock (Fig. 1A), as well as within extinction sessions during daily long exposures (24 min) to the context in the absence of shock (Fig. 2, 5B). These findings suggest that PKA inhibition affects both the short-term development of extinction within a session as well as the retention of extinction across sessions, depending on the duration of the extinction session. Spontaneous recovery was observed in both cases, either the next day, which occurred with long extinction sessions (Figs. 2, 5B), or 21 d later, which occurred with short extinction sessions (Fig. 1B). Our demonstrations of spontaneous recovery support the idea that the behavioral changes that occurred during extinction did not result from erasure of the original memory, but instead reflected new learning, distinct from the learning that occurred during the acquisition of fear conditioning (Lattal and Abel, 2004; Rescorla, 2004; Power et al., 2006). Indeed, the studies presented here provide the first genetic evidence that fear extinction is itself a learning process with its own molecular requirements.
In what brain regions does PKA activity act to modulate extinction? The transgenic expression of R(AB) in our experiments reduced PKA activity in the prefrontal cortex, hippocampus, and amygdala, three structures that are important in extinction. Although we cannot be sure which structure mediated these extinction effects, we do know that the CaMKIIα promoter drives expression in excitatory forebrain neurons (Mayford et al., 1996a), suggesting that inhibitory projections within or between these structures [such as between the medial prefrontal cortex (mPFC) and the amygdala] likely were not responsible for the effects of PKA inhibition on extinction. Further, although there also is evidence that stimulation of the mPFC can enhance both the short-term and long-term development of extinction, most studies generally show that the mPFC is most critical for the consolidation or long-term retention of the extinction memory (Milad and Quirk, 2002) (for review, see Quirk et al., 2006). The short-term enhancements in extinction that we observed within a long extinction session did not result in long-term enhancements in extinction, suggesting that if the mPFC was involved, its role was limited to the short-term development of extinction, which seems unlikely given the results from previous studies (Quirk et al., 2006). Studies of the hippocampus have shown that permanent lesions do not affect extinction per se (Wilson et al., 1995; Frohardt et al., 2000; Ji and Maren, 2005) and inactivation of the dorsal hippocampus before extinction training attenuates, but does not entirely block, the development of extinction, suggesting that the hippocampus is not essential for the acquisition of extinction (Corcoran et al., 2005). There are numerous studies showing that pharmacological manipulations of various signaling pathways in the amygdala before extinction training lead to facilitation or blockade of extinction, suggesting a critical role for this structure in the development of extinction (for review, see Barad et al., 2006). Thus, the amygdala may be a likely site of action for the effects of transgenic PKA inhibition on extinction that were observed in our experiments. Future experiments examining the role of PKA in the extinction of cue-induced freezing may provide a way to examine more selectively the role of PKA within the amygdala during extinction.
How does PKA act to facilitate extinction? There are two very general ways that extinction may be facilitated: some aspect of the original memory may be weakened, or some aspect of the extinction memory may be strengthened. Many studies have shown that extinction can occur through a temporary weakening of the original memory, either through a depression of the CS–US association, or through a depression of the representation of the CS or US itself (Pavlov, 1927; Rescorla and Cunningham, 1977; Robbins, 1990; reviewed in Rescorla, 2004; Schmajuk and Larrauri, 2006). At a theoretical level, the loss of conditioned behavior after an extinction session is often attributed to a deficit in the reconsolidation of the original memory. Behaviorally, enhancements in extinction and impairments in reconsolidation are functionally equivalent: both result in a loss of conditioned responding. However, one complication from our experiments in attributing the effects of PKA inhibition on extinction to deficits in reconsolidation is that short-term extinction was enhanced, suggesting that changes in performance occurred quickly. Thus, the time course of this effect suggests a rapid process, such as a within-session change in attention to the CS, which may occur in extinction (Schmajuk and Larrauri, 2006). Our observations of spontaneous recovery after PKA inhibition are consistent with many other studies of extinction showing that any effect on the original associative learning was temporary.
The alternative to some aspect of the original memory being weakened is that some aspect of the extinction memory was strengthened as a direct result of PKA inhibition. The idea that PKA inhibition enhanced new learning is counterintuitive because the effects of PKA inhibition on memory are often attributed to a failure to sufficiently activate transcription factors, leading to deficits in memory (Abel et al., 1997). However, PKA inhibition also can result in changes in phosphatase activation (Genoux et al., 2002; Mansuy, 2003), which may be critical for the development of extinction. One prevailing theory about the molecular mechanisms underlying learning and memory is that kinases and phosphatases oppose each other to regulate signaling pathways, synaptic efficacy, and learning and memory (Lisman, 1989). Generally, kinases are considered positive modulators of learning and memory whereas phosphatases are thought of as constraints for these processes (Genoux et al., 2002). However, it has been hypothesized recently that whereas excitatory responses to a conditioned stimulus (like those triggered by fear acquisition) may be orchestrated by kinases, inhibitory responses (like those triggered by fear extinction) may be supported by phosphatases (Myers and Davis, 2002). Evidence that extinction may be mediated by phosphatases came recently from work showing that the Ca2+-dependent phosphatase calcineurin is rapidly activated in the amygdala in mice after extinction training, and that extinction is blocked by pharmacological inhibition of calcineurin (Lin et al., 2003a,b). Given that PKA counteracts calcineurin and protein phosphatase 1 (Genoux et al., 2002; Mansuy, 2003), the facilitatory effect of PKA inhibition on extinction may be attributable to partial removal of an inhibitory constraint imposed by PKA on calcineurin signaling during extinction.
Consistent with this interpretation, transgenic mice overexpressing type 1 adenylyl cyclase with increased PKA activity levels in forebrain neurons, also exhibited slower extinction, indicating that increased PKA activity impaired fear extinction (Wang et al., 2004). Moreover, subchronic delivery of rolipram in rats has been shown to slow extinction of conditioned fear (Monti et al., 2006). Similarly, McNally et al. (2005) found that administration of the cAMP analog 8-Br-cAMP impaired extinction, although PKA activation by Sp-cAMPs had no effect. Additional indirect genetic evidence for this negative role of the cAMP–PKA pathway in extinction comes from cannabinoid receptor 1 (CB1) knock-out mice, which showed impaired extinction (Marsicano et al., 2002). Because activation of CB1 receptors leads to adenylyl cyclase inhibition, it is possible that endocannabinoids may subserve extinction through inhibition of adenylyl cyclase.
These data together with our findings suggest that PKA, although required for fear memory formation, may be a molecular constraint for fear extinction, but the specific ways in which PKA may act on inhibitory processes during extinction remains to be determined. The basic demonstration in our experiments that the same genetic manipulations that impair initial memory formation may facilitate subsequent extinction is consistent with many other studies showing behavioral, systems, and molecular differences between initial learning and extinction (for review, see McNally et al., 2005; Bouton et al., 2006; Lattal et al., 2006). However, other studies have found a number of important similarities between initial learning and extinction, suggesting that a complete picture of extinction memory formation and consolidation will need to include multiple mechanisms. Understanding these mechanisms may help translate basic research to clinical settings, where extinction is a commonly used therapeutic intervention for psychiatric disorders involving fear (Bouton et al., 2001; Rothbaum and Schwartz, 2002; Richardson et al., 2004; Barad, 2005; Davis et al., 2006; Milad et al., 2006).
Footnotes
This work was supported by grants from the National Institutes of Health and the Human Frontiers Science Program (T.A., C.I., E.R.K., K.M.L.). T.A. is a David and Lucile Packard Foundation Fellow. E.R.K. is also supported by grants from Howard Hughes Medical Institute. We thank Tom Deuel, Kyle Lapidus, Alan Young, and Mark Barad for their participation in the early stages of this project. We thank Mark Mayford for the CaMKIIα-tTA mouse line B.
References
- Abel and Lattal, 2001.Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11:180–187. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- Abel et al., 1997.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Barad, 2005.Barad M. Fear extinction in rodents: basic insight to clinical promise. Curr Opin Neurobiol. 2005;15:710–715. doi: 10.1016/j.conb.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Barad et al., 2006.Barad M, Gean PW, Lutz B. The role of the amygdala in the extinction of conditioned fear. Biol Psychiatry. 2006;60:322–328. doi: 10.1016/j.biopsych.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Bejar et al., 2002.Bejar R, Yasuda R, Krugers H, Hood K, Mayford M. Transgenic calmodulin-dependent protein kinase II activation: dose-dependent effects on synaptic plasticity, learning, and memory. J Neurosci. 2002;22:5719–5726. doi: 10.1523/JNEUROSCI.22-13-05719.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze et al., 1998.Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Bouton, 1993.Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99. doi: 10.1037/0033-2909.114.1.80. [DOI] [PubMed] [Google Scholar]
- Bouton et al., 2001.Bouton ME, Mineka S, Barlow DH. A modern learning theory perspective on the etiology of panic disorder. Psychol Rev. 2001;108:4–32. doi: 10.1037/0033-295x.108.1.4. [DOI] [PubMed] [Google Scholar]
- Bouton et al., 2006.Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60:352–360. doi: 10.1016/j.biopsych.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Bradford, 1976.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Byrne and Kandel, 1996.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain et al., 2002.Cain CK, Blouin AM, Barad M. L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci. 2002;22:9113–9121. doi: 10.1523/JNEUROSCI.22-20-09113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota et al., 2005.Cammarota M, Bevilaqua LR, Barros DM, Vianna MR, Izquierdo LA, Medina JH, Izquierdo I. Retrieval and the extinction of memory. Cell Mol Neurobiol. 2005;25:465–474. doi: 10.1007/s10571-005-4009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg et al., 1987.Clegg CH, Correll LA, Cadd GG, McKnight GS. Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]
- Corcoran et al., 2005.Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis et al., 2006.Davis M, Myers KM, Chhatwal J, Ressler KJ. Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx. 2006;3:82–96. doi: 10.1016/j.nurx.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamater, 2004.Delamater AR. Experimental extinction in Pavlovian conditioning: behavioural and neuroscience perspectives. Q J Exp Psychol B. 2004;57:97–132. doi: 10.1080/02724990344000097. [DOI] [PubMed] [Google Scholar]
- Fanselow, 1980.Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Fanselow and Poulos, 2005.Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- Frankland and Bontempi, 2005.Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Frohardt et al., 2000.Frohardt RJ, Guarraci FA, Bouton ME. The effects of neurotoxic hippocampal lesions on two effects of context after fear extinction. Behav Neurosci. 2000;114:227–240. doi: 10.1037//0735-7044.114.2.227. [DOI] [PubMed] [Google Scholar]
- Genoux et al., 2002.Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418:970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- Ji and Maren, 2005.Ji J, Maren S. Electrolytic lesions of the dorsal hippocampus disrupt renewal of conditional fear after extinction. Learn Mem. 2005;12:270–276. doi: 10.1101/lm.91705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh and Bernstein, 2003.Koh MT, Bernstein IL. Inhibition of protein kinase A activity during conditioned taste aversion retrieval: interference with extinction or reconsolidation of a memory? NeuroReport. 2003;14:405–407. doi: 10.1097/00001756-200303030-00021. [DOI] [PubMed] [Google Scholar]
- Lattal and Abel, 2001.Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21:5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal and Abel, 2004.Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc Natl Acad Sci USA. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal et al., 2006.Lattal KM, Radulovic J, Lukowiak K. Extinction: does it or doesn't it? The requirement of altered gene activity and new protein synthesis. Biol Psychiatry. 2006;60:344–351. doi: 10.1016/j.biopsych.2006.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al., 2003a.Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003a;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al., 2003b.Lin CH, Yeh SH, Lu HY, Gean PW. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci. 2003b;23:8310–8317. doi: 10.1523/JNEUROSCI.23-23-08310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman, 1989.Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy, 2003.Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Maren and Quirk, 2004.Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marsicano et al., 2002.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mayford et al., 1996a.Mayford M, Baranes D, Podsypanina K, Kandel ER. The 3′-untranslated region of CaMKII alpha is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci USA. 1996a;93:13250–13255. doi: 10.1073/pnas.93.23.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford et al., 1996b.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996b;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McNally et al., 2005.McNally GP, Lee BW, Chiem JY, Choi EA. The midbrain periaqueductal gray and fear extinction: opioid receptor subtype and roles of cyclic AMP, protein kinase A, and mitogen-activated protein kinase. Behav Neurosci. 2005;119:1023–1033. doi: 10.1037/0735-7044.119.4.1023. [DOI] [PubMed] [Google Scholar]
- Milad and Quirk, 2002.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Monti et al., 2006.Monti B, Berteotti C, Contestabile A. Subchronic rolipram delivery activates hippocampal CREB and Arc, enhances retention and slows down extinction of conditioned fear. Neuropsychopharmacology. 2006;31:278–286. doi: 10.1038/sj.npp.1300813. [DOI] [PubMed] [Google Scholar]
- Myers and Davis, 2002.Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Pavlov, 1927.Pavlov I. Conditioned reflexes, an investigation of the physiological activity of the cerebral cortex. London: Oxford UP; 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps and LeDoux, 2005.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Power et al., 2006.Power AE, Berlau DJ, McGaugh JL, Steward O. Anisomycin infused into the hippocampus fails to block “reconsolidation” but impairs extinction: the role of re-exposure duration. Learn Mem. 2006;13:27–34. doi: 10.1101/lm.91206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk et al., 2006.Quirk GJ, Garcia R, Gonzalez-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rescorla, 2001.Rescorla RA. Experimental extinction. In: Mowrer RR, Klein S, editors. Handbook of contemporary learning theories. Hillsdale: Erlbaum; 2001. pp. 119–154. [Google Scholar]
- Rescorla, 2004.Rescorla RA. Spontaneous recovery. Learn Mem. 2004;11:501–509. doi: 10.1101/lm.77504. [DOI] [PubMed] [Google Scholar]
- Rescorla and Cunningham, 1977.Rescorla RA, Cunningham CL. The erasure of reinstated fear. Anim Learn Behav. 1977;5:386–394. [Google Scholar]
- Richardson et al., 2004.Richardson R, Ledgerwood L, Cranney J. Facilitation of fear extinction by d-cycloserine: theoretical and clinical implications. Learn Mem. 2004;11:510–516. doi: 10.1101/lm.78204. [DOI] [PubMed] [Google Scholar]
- Robbins, 1990.Robbins SJ. Mechanisms underlying spontaneous recovery in autoshaping. J Exp Psychol Anim Behav Processes. 1990;16:235–249. [Google Scholar]
- Rodrigues et al., 2004.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rotenberg et al., 2000.Rotenberg A, Abel T, Hawkins RD, Kandel ER, Muller RU. Parallel instabilities of long-term potentiation, place cells, and learning caused by decreased protein kinase A activity. J Neurosci. 2000;20:8096–8102. doi: 10.1523/JNEUROSCI.20-21-08096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbaum and Schwartz, 2002.Rothbaum BO, Schwartz AC. Exposure therapy for posttraumatic stress disorder. Am J Psychother. 2002;56:59–75. doi: 10.1176/appi.psychotherapy.2002.56.1.59. [DOI] [PubMed] [Google Scholar]
- Schmajuk and Larrauri, 2006.Schmajuk NA, Larrauri JA. Experimental challenges to theories of classical conditioning: application of an attentional model of storage and retrieval. J Exp Psychol Anim Behav Process. 2006;32:1–20. doi: 10.1037/0097-7403.32.1.1. [DOI] [PubMed] [Google Scholar]
- Siwicki and Ladewski, 2003.Siwicki KK, Ladewski L. Associative learning and memory in Drosophila: beyond olfactory conditioning. Behav Processes. 2003;64:225–238. doi: 10.1016/s0376-6357(03)00137-2. [DOI] [PubMed] [Google Scholar]
- Suzuki et al., 2004.Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro et al., 2003.Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CaMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Tonegawa et al., 2003.Tonegawa S, Nakazawa K, Wilson MA. Genetic neuroscience of mammalian learning and memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:787–795. doi: 10.1098/rstb.2002.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson et al., 2006.Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2004.Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- Wilson et al., 1995.Wilson A, Brooks DC, Bouton ME. The role of the rat hippocampal system in several effects of context in extinction. Behav Neurosci. 1995;109:828–836. doi: 10.1037//0735-7044.109.5.828. [DOI] [PubMed] [Google Scholar]
- Woo et al., 2000a.Woo NH, Duffy SN, Abel T, Nguyen PV. Genetic and pharmacological demonstration of differential recruitment of cAMP-dependent protein kinases by synaptic activity. J Neurophysiol. 2000a;84:2739–2745. doi: 10.1152/jn.2000.84.6.2739. [DOI] [PubMed] [Google Scholar]
- Woo et al., 2000b.Woo NH, Duffy SN, Scharf M, Abel T, Nguyen PV. Rescue of hippocampal LTP in PKA mutant mice by specific temporal patterns of synaptic activity. Soc Neurosci Abstr. 2000b;26:132.3. [Google Scholar]
- Woo et al., 2002.Woo NH, Abel T, Nguyen PV. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur J Neurosci. 2002;16:1871–1876. doi: 10.1046/j.1460-9568.2002.02260.x. [DOI] [PubMed] [Google Scholar]
- Woo et al., 2003.Woo NH, Duffy SN, Abel T, Nguyen PV. Temporal spacing of synaptic stimulation critically modulates the dependence of LTP on cyclic AMP-dependent protein kinase. Hippocampus. 2003;13:293–300. doi: 10.1002/hipo.10086. [DOI] [PubMed] [Google Scholar]