

Abstract
Alzheimer's amyloid precursor protein (APP) sorting and processing are modulated through signal transduction mechanisms regulated by protein phosphorylation. Notably, protein kinase C (PKC) appears to be an important component in signaling pathways that control APP metabolism. PKCs exist in at least 11 conventional and unconventional isoforms, and PKCα and PKCε isoforms have been specifically implicated in controlling the generation of soluble APP and amyloid-β (Aβ) fragments of APP, although identification of the PKC substrate phospho-state-sensitive effector proteins remains challenging. In the current study, we present evidence that chronic application of phorbol esters to cultured cells in serum-free medium is associated with several phenomena, namely: (i) PKCα down-regulation; (ii) PKCε up-regulation; (iii) accumulation of APP and/or APP carboxyl-terminal fragments in the trans Golgi network; (iv) disappearance of fluorescence from cytoplasmic vesicles bearing a green fluorescent protein tagged form of APP; (v) insensitivity of soluble APP release following acute additional phorbol application; and (vi) elevated cellular APP mRNA levels and holoprotein, and secreted Aβ. These data indicate that, unlike acute phorbol ester application, which is accompanied by lowered Aβ generation, chronic phorbol ester treatment causes differential regulation of PKC isozymes and increased Aβ generation. These data have implications for the design of amyloid-lowering strategies based on modulating PKC activity.
Keywords: amyloid β peptide; green fluorescent protein; amyloid precursor protein; phorbol-12,-14-di-butyrate; protein phosphorylation; trans Golgi network; vesicle incorporation
Alzheimer's amyloid precursor protein (APP) processing is complex, involving several phosphorylation-regulated pathways (Caporaso et al. 1992; Gandy and Greengard 1994; da Cruz e Silva and da Cruz e Silva 2003; Lee et al. 2003; Lanni et al. 2004; Vingtdeux et al. 2005). APP molecules traversing the non-amyloidogenic constitutive secretory pathway are cleaved within the amyloid-β (Aβ) domain by α-secretase (Weidemann et al. 1989; Esch et al. 1990) and the large APP ectodomain (designated soluble APPα; sAPPα) is released into the medium. Alpha-secretase cleavage of APP molecules within the Aβ sequence prevents amyloidogenesis. Alternatively, in the amyloidogenic pathway, APP is cleaved N terminally to Aβ by β-secretase (Seubert et al. 1993; Vassar et al. 1999), and the resulting amyloidogenic carboxyl terminal fragment (Gandy et al. 1992; Golde et al. 1992; Knops et al. 1992; Tamaoka et al. 1992) can subsequently undergo further proteolysis to yield Aβ (Haass et al. 1992; Shoji et al. 1992; Seubert et al. 1993). Stimulation of protein kinase C (PKC) by phorbol esters is one of the most robustly reproducible methods for activating sAPPα production (Buxbaum et al. 1992; Caporaso et al. 1992) and concomitantly inhibiting generation of the amyloidogenic Aβ fragment (Buxbaum et al. 1993; Gabuzda et al. 1993; Hung et al. 1993). Mechanistically, PKC has been shown to phosphorylate APP both in vitro (Gandy et al. 1988; Suzuki et al. 1992) and in vivo (Oishi et al. 1997) on Ser655 within the cytoplasmic domain (APP695 isoform numbering). However, phorbol ester stimulated release of sAPPα can occur even in the absence of the cytoplasmic tail (da Cruz e Silva et al. 1993), excluding an obligatory role for direct APP phosphorylation by PKC as a crucial reaction tightly linked to activation of α-secretase cleavage of APP and release of sAPPα. Alternatively, as PKC regulates formation of APP-bearing transport vesicles at the trans Golgi network (TGN) (Xu et al. 1995), one popular formulation is that PKC activates α-secretase cleavage of APP by enhancing delivery of APP out of the TGN and to the plasma membrane, where the ADAM (a disintegrin and metalloproteinase) family α-secretases ADAM-10 and ADAM-17 are concentrated. On the other hand, PKC has also been implicated in regulation of APP transcription (Trejo et al. 1994), an event that would likely increase generation of Aβ, thereby potentially neutralizing any therapeutic amyloid-lowering effect of chronic PKC activation. Therefore, we conducted the current study to determine how activation of α-secretase and enhancement of APP transcription might be integrated in the face of chronic exposure to a phorbol ester.
Protein kinases Cα, β, and ε have all been reported to play a role in APP processing (Benussi et al. 1998; Yeon et al. 2001; Zhu et al. 2001; Racchi et al. 2003). The specific PKC isoform(s) involved in APP metabolism vary according to the exact stage of the processing being considered, and, as noted above, PKC may regulate effects that oppose each other. For example, PKCε can stimulate basolateral endocytosis in intestinal epithelia, while PKCα can oppose this effect by stabilizing F-actin (Song et al. 2002). PKCα is thought to regulate APP secretion (Benussi et al. 1998), as cells transfected with antisense PKCα cDNA exhibited reduced sAPP production in response to phorbol esters (Racchi et al. 2003). Similarly, PKCε has also been implicated in APP processing (Yeon et al. 2001; Zhu et al. 2001). The use of isoform-specific PKC inhibitors indicates that PKCε is probably involved in both the activation of sAPP production and the suppression of Aβ production. In agreement with these findings, transfection with the cDNAs for either the α or the ε isoform of PKC produces corresponding increases in sAPP production, whereas the use of PKCδ fails to elicit a similar effect (Kinouchi et al. 1995). Also of note is the finding that down-regulation of PKCα and ε in SH-SY5Y neuroblastoma cells by the use of antisense cDNAs leads to an impairment in sAPPα-induction by phorbol esters (Yeon et al. 2001; Zhu et al. 2001). It is worth noting that the PKCα isoform has been observed to translocate from the cytosol to the plasma membrane via newly budded transport vesicles at the TGN, thus becoming ideally positioned to act upon potential PKC substrate effectors (Lanni et al. 2004). Interestingly, Alzheimer patients are reported to express lower levels of PKCε when compared with controls. PKCβI (Rossner et al. 2001) and PKCβII (Paola et al. 2000) may also be involved in APP processing, having been reported to modulate the intracellular levels of Aβ.
Phorbol esters exert effects not only on APP processing, but also on APP expression. Phorbol 12-myristate 13-acetate was shown to increase APP mRNA levels by inducing known components of the transcription factor activator protein-1 (AP-1) and increasing protein-DNA binding activity to AP-1 sequences within the APP promoter (Trejo et al. 1994). It was suggested that the distal but not the proximal AP-1 recognition site binds nuclear proteins regulated by PKC, and that the AP-1 binding activity is likely to be composed of Jun-Jun homodimers rather than Jun-Fos heterodimers (Trejo et al. 1994). Thus, cellular mediators that activate PKC may up-regulate expression of the APP gene and consequently affect its production and processing.
Our present findings confirm that long-term exposure to phorbol ester leads to an increase in the levels of APP, and we find that this is associated with an up-regulation of PKCε but not other isoforms. Indeed, long-term exposure of cells to phorbol-12,-14-di-butyrate (PDBu) resulted in the down-regulation of the PKCα isoform. As PKCα principally acts on APP processing and not on APP expression, we propose that this down-regulation of PKCα led to APP retention in the TGN, thereby reducing incorporation of APP into visible vesicles. Consistent with this formulation, the levels of Aβ production were significantly increased when PKCα was down-regulated. The long-term effects of subjecting cells to aberrant PKCα and/or PKCε environments are discussed, and the implications for targeting PKC modulation as an amyloid-lowering therapy or a prophylactic for Alzheimer disease are presented.
Materials and methods
Sample preparation and immunoblotting
Monkey kidney (COS) cells were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum (Amador et al. 2004). Monolayers of COS cells at approximately 80% confluency were treated with 1 μM PDBu in serum-free DMEM (sf medium). Parallel experiments were carried out in complete, serum-containing DMEM (c medium). Cells were harvested every 2 h into 1% sodium dodecyl sulfate (SDS), samples were boiled, and the protein content of the cell lysates was determined by the bicinchoninic acid method (Pierce, Rockford, IL, USA). Samples were separated by electrophoresis through 7.5% polyacrylamide gels in Tris–glycine buffer containing 0.1% SDS–polyacrylamide gel electrophoresis). Proteins resolved by SDS–polyacrylamide gel electrophoresis were electrophoretically transferred to nitrocellulose membranes. Immunoblotting of the transferred proteins was performed by first blocking possible non-specific binding sites with non-fat dry milk in 10 mM Tris–HCl buffer (pH 8.0) containing 150 mM NaCl. Incubation with the primary antibody was carried out for periods from 2 h to overnight, depending upon the antibody used. Detection was achieved using the enhanced chemiluminescence detection system (Amersham Pharmacia, Piscataway, NJ, USA) or a colorimetric (Nitro-Blue Tetrazolium/5-Bromo-4-Chloro-3′-Indolyl Phosphate) system (Rebelo et al. 2004). For monitoring production of sAPPα, the conditioned media were collected and SDS was added to yield a final concentration of 1%. The media thus obtained are referred to as cumulative media (2, 4, 6, 8, and 10 h), and the volume loaded on the gel was normalized against the protein concentration of the corresponding cell lysate.
Immunodetection
Protein kinase C immunodetection was carried out using isozymespecific antibodies (Gibco, BRL, Invitrogen, Carlsbad, CA, USA) and the conditions specified by the manufacturer. As a control, a mouse monoclonal anti-tubulin antibody (Zymed, South San Francisco, CA, USA) was used to standardize protein loading. Other primary antibodies included the mouse monoclonal antibody 22C11 (Roche Diagnostics GmbH, Mannheim, Germany), which binds an epitope on the extracellular domain of APP, to detect APP and both sAPPα and sAPPβ, and the mouse monoclonal antibody 6E10 (Sigma-Aldrich), which recognizes the N-terminus of Aβ either within Aβ or within sAPPα. Secondary antibodies included horse-radish peroxidase-linked anti-mouse IgG (Amersham Pharmacia) and alkaline phosphatase conjugated anti-mouse IgG (Sigma-Aldrich).
Metabolic labeling and immunoprecipitation
For metabolic labeling, cells were plated at approximately 80% confluency onto six-well culture dishes. Cells were washed twice in methionine-free DMEM, and pre-incubated for 15 min in the same solution. Cells were maintained in the presence of methionine-free DMEM for 1 h supplemented with [35S]methionine (NEN-DuPont, Boston, MA, USA) during the `pulse' period. Cells were then `chased' in DMEM without serum (sf) for varying periods of time and processed for analysis. Procedures were carried out in the presence or absence of PDBu, as indicated. For studies addressing PKC-regulated APP processing, only the media corresponding to the last hour of the experimental conditions were collected. Subsequently, samples were immunoprecipitated with 6E10 (Sigma-Aldrich) after removal of cell debris by centrifugation (10 000 g). For experiments designed to measure secreted Aβ, media were collected after 1, 4, or 8 h and subjected to immunoprecipitation with 6E10. Immunoprecipitates were analyzed on 10–20% Tris/tricine gradient gels. Gels were treated with 2,5-diphenyloxazole/dimethyl sulfoxide and dried for fluorography. The dried gels were exposed to X-ray film, and the immunoprecipitated material was quantified as described below.
Quantification and statistical analysis
Immunoblots and autoradiograms were scanned and quantified using Quantity One densitometry software (Bio-Rad Laboratories, Hercules, CA, USA). Data are expressed as mean ± SEM of triplicate determinations, from at least three independent experiments. Statistical significance analysis was conducted by one-way Anova followed by the two-tail Student's t-test. Aβ detection from metabolically labeled immunoprecipitates was carried out using a Molecular Imager (Bio-Rad Laboratories).
Production and subcellular localization of APP-GFP fusion proteins
Fusion constructs expressing human APP695-cDNA fused to the green fluorescent protein (GFP) were prepared using standard recombinant DNA techniques. In brief, human APP695 cDNA was amplified by the PCR using specific oligonucleotide primers designed to remove the APP stop codon. The amplified fragment was then subcloned into the pEGFP-N1 mammalian expression vector (Clontech Laboratories Inc, Mountain View, CA, USA) and fully sequenced prior to use (da Cruz e Silva et al. 2004). Constructs bearing the Swedish mutation were also prepared. Human APP695–GFP fusion constructs were transfected into COS-7 cells previously plated onto coverslips pre-treated with 100 μg/mL poly-ornithine. The cells were treated with phorbol esters as described above and the intracellular localization of APP–GFP was determined using epifluorescence microscopy. For co-localization studies Texas Red-labeled anti-syntaxin 6 antibody (BD Biosciences, San Jose, CA, USA) was used as a TGN marker, and an anti-clathrin antibody (ICN Immunobiologicals, Aurora, OH, USA) was used as a marker of clathrin-coated vesicles.
Northern blot analysis of amyloid precursor protein total RNA
COS-7 cells were exposed for 8 h to 1 μM PDBu or vehicle in sf or c medium, as described above. Cells were lysed using TRI REAGENT (Sigma-Aldrich), and total RNA was isolated according to manufacturer's instructions. Mass-normalized aliquots (15 μg) were separated by formaldehyde gel electrophoresis and transferred to nitrocellulose filters. The blot was hybridized with a [32P]-labeled APP cDNA probe (25 ng, 1 × 106 cpm/ng), as described in the MTN Blot User Manual (BD Biosciences, Clontech). The APP probe used (756 bp) was obtained by AgeI/BamHI restriction enzyme digestion of APP751 cDNA, and labeled with [α-32P]dCTP (Amersham Pharmacia, GE Healthcare, Little Chalfont, UK) using the High Prime DNA Labelling Kit (Roche Diagnostics GmbH), followed by purification through NucTrap Probe Purification Columns (Stratagene, La Jolla, CA, USA and Alfagene). Hybridizing RNAs were detected using a Molecular Imager (Bio-Rad Laboratories).
Immunofluorescence
COS-7 cells grown on coverslips and transfected with APP695–GFP, as described above, were fixed with 4% p-formaldehyde, permeabilized with methanol for 2 min and blocked in 3% bovine serum albumin in phosphate-buffered saline for 1 h. Co-localization studies were carried out with incubation in primary antibody to PKCα or PKCε (Gibco, BRL) and sequentially with syntaxin 6 or clathrin, respectively. Alexa 350 and Texas Red-conjugated antibodies (Molecular Probes, Eugene, OR, USA and BD Biosciences) were added for 2 h at 22°C. Between each stage of preparation, cells were carefully washed with phosphate-buffered saline. Visualization was carried out using a Zeiss LSM 510-Meta confocal microscope (Carl Zeiss Microimaging GmbH, Jena, Germany), and a 63×/1.4 oil immersion objective. Argon laser lines of 405 and 488 nm were used to excite Alexa Fluor 350 and enhanced GFP, respectively, and a 561 nm Diode Pumped Solid State laser was used to excite Texas Red. Microphotographs were acquired in a sole section in the z-axis (xy-mode) and represent a mean of 16 scans.
Results
PKC isozyme expression profiles following exposure to phorbol ester
Long-term exposure to the phorbol ester PDBu is a standard treatment used to down-regulate PKC. Thus, COS-7 cells were exposed to PDBu for increasing periods of time, and levels of PKC expression were monitored. Two distinct conditions were tested, whereby cells were either incubated in medium without serum (sf medium) or in medium with serum (c medium).
We observed that PKCα levels were down-regulated under both conditions (Fig. 1a and b). In contrast, PKCε was down-regulated only when cells were incubated in c medium (Fig. 1b), and PKCε was up-regulated when the experiment was carried out in sf medium (Fig. 1a).
Fig. 1.

PKC isozyme expression profiling of COS cells. The expression of the main PKC isozymes was assessed in COS-7 cell lysates by immunoblotting with PKC isoform specific antibodies, following exposure to PDBu for the indicated periods (0, 2, 4, 6, 8, and 10 h). (a) Cells were incubated in serum-free medium (sf medium). (b) Cells were incubated in complete, serum containing, medium (c medium). Tubulin expression levels were monitored as a control. (c) The effect of removal of serum alone (− PDBu) for 2 h on the levels of PKCα and PKCε was monitored. Antibodies to both isoforms were used simultaneously. (d) Levels of PKCα and PKCε in rat cortex homogenates (hm) were compared with COS cells under basal conditions (−) and following 8 h exposure to PDBu (+).
Interestingly, under conditions where both PKC isozymes were down-regulated (c medium), their rates of decline were different. PKCα down-regulation was approximately linear with time, with decreases of 0.50 at 2 h, 0.30 at 4 h, and 0.15 at 6 h (Fig. 1b). For PKCε, the decreases were approximately 0.33 at 2 h, and ~0.20–0.23 for the remainder of the experiment (Fig. 1b). Removal of serum per se at this time point (2 h) had no effect on the expression levels of PKCα or PKCε (Fig. 1c). The relevance of both isoforms in neuronal systems is established by evidence that both are expressed in rat cortex (Fig. 1d). Other PKC isozymes (namely PKCβ and PKCζ,) were also monitored, but their levels were barely detectable and did not appear to change with the treatments tested. Tubulin expression was used as a control and also remained relatively unchanged throughout the experiments.
Morphological analysis of APP distribution following chronic PDBu treatment
As noted, regardless of whether cells were incubated in sf medium or c medium, PKCα was down-regulated following long-term exposure to phorbol esters. The effect on intracellular APP distribution under these conditions was monitored using APP fused to GFP (Fig. 2). Cells were analyzed both qualitatively (Fig. 2a and b) and quantitatively (Fig. 2d). For quantitative analysis, 30 cells in triplicate were counted and scored for fluorescent TGN and cytoplasmic vesicular structures. Syntaxin 6 was used as a TGN marker in co-localization studies to confirm TGN scoring (Fig. 2c). Following long-term PDBu exposure, two significant qualitative effects were observed: an increase in the intensity and apparent volume of the fluorescent TGN structure and a dramatic decrease in the number of fluorescing cytoplasmic vesicles. The number of cells with fluorescent vesicles dropped drastically from 70% to 15% (Fig. 2d). Similar results were obtained regardless of whether cells were incubated in sf or c medium.
Fig. 2.

Intracellular localization of APP following down-regulation of PKCα. COS-7 cells were transfected with APP695–GFP, incubated for 8 h either in sf or c medium (both in the absence or presence of PDBu), and monitored for subcellular APP distribution. (a) Comparison of APP subcellular localization when cells were incubated in sf medium under control conditions and in the presence of PDBu. (b) Comparison of APP subcellular localization when cells were incubated in c medium under control conditions and in the presence of PDBu. (c) Co-localization of transfected APP695–GFP with the TGN/Golgi marker syntaxin 6. (d) Comparison of the number of cells exhibiting intensely fluorescent TGN or containing a large number of fluorescent cytoplasmic vesicles in sf medium following PDBu exposure. Data are expressed as mean ± SEM of triplicate determinations of three independent experiments. Statistical significance analysis was conducted by Anova followed by the two-tail Student's t-test. Statistical significance differences against control determinations (− PDBu) are presented as ***p < 0.001; ****p < 0.0001. G, Golgi; V, Vesicles.
Co-localization of APP with PKC isozymes and organelle markers
Absence of APP–GFP incorporation into budding TGN vesicles following chronic exposure to PDBu was confirmed by confocal microscopy (Fig. 3). As expected, the PKCα immunoreactivity also decreased although syntaxin 6 immunoreactivity was maintained. Merged images confirmed that, under basal conditions, APP–GFP, PKCα, and syntaxin 6 were especially co-localized at the TGN (Fig. 3, white speckles). However, following chronic PDBu exposure, syntaxin 6 immunoreactive vesicles (red) were apparently devoid of APP–GFP fluorescence.
Fig. 3.

Co-localization of APP, PKCα and syntaxin 6. COS-7 cells transfected with APP695–GFP were analyzed for co-localization with PKCα and syntaxin 6. Observations were carried out under control conditions (panel a) and in the presence of PDBu for 8 h (panel b). PKCα was detected using a primary rabbit antibody (Gibco, BRL), and a secondary anti-rabbit Alexa Fluor 350 (Molecular Probes and BD Biosciences). Syntaxin 6 was detected using a primary mouse antibody, and a secondary anti-mouse Texas Red-conjugated antibody (Molecular Probes and BD Biosciences). Visualization was carried out with a Zeiss confocal microscope.
Contrary to the results observed for PKCα in sf medium, chronic exposure to PDBu led to increased PKCε immunoreactivity which co-localized in the juxta-plasma membrane region identified by clathrin immunoreactivity (red) (Fig. 4).
Fig. 4.

Co-localization of APP, PKCε, and clathrin. COS-7 cells transfected with APP695–GFP were analyzed for co-localization with PKCε and clathrin. Observations were carried out under control conditions (panel a) and in the presence of PDBu for 8 h (panel b). PKCε was detected using a primary rabbit antibody (Gibco, BRL), and a secondary anti-rabbit Alexa Fluor 350 (Molecular Probes and BD Biosciences). Clathrin (ICN Immunobiologicals) was detected using a primary goat antibody, and a secondary anti-goat Texas Red-conjugated antibody (Molecular Probes and BD Biosciences). Visualization was carried out with a Zeiss confocal microscope.
Acute PDBu application to cells chronically exposed to PDBu
Previous studies have demonstrated increases in sAPPα secretion following short-term exposure of cells to PDBu. This increase is likely to have several contributing factors, including enhanced vesicle budding from the TGN (Xu et al. 1995). Hence, cells labeled with [35S]methionine were exposed to PDBu for increasing periods of time, and then subjected to a subsequent 1 h exposure to PDBu. The conditioned media were collected, immunoprecipitated with 6E10, and tested for sAPPα levels. A typical induction was evident when cells were not pre-incubated with PDBu. With only a 2 h pre-incubation, the response was already highly compromised (Fig. 5a). When pre-incubation periods with PDBu were increased to 4 h or more, subsequent acute stimulation with a spike of additional PDBu caused none of the typical stimulated release in sAPPα (Fig. 5a). Hence, the 4.5-fold increase in sAPPα for the APP751/770 isoform, evident with the 1 h PDBu spike in the absence of PDBu pre-incubation (Fig. 5a, 0 time point), was highly diminished by 4 h pre-incubation with PDBu. In summary, acute stimulation of the α-secretase pathway during long-term exposure to PDBu was considerably compromised. The failure of cells to respond by further increasing sAPP production correlated with down-regulation of PKCα (Fig. 1), and is consistent with the notion that PKCα in particular controls the α-secretase pathway, probably by modulating budding of APP transport vesicles at the TGN (Fig. 2).
Fig. 5.

Effect of chronic PDBu exposure on sAPP secretion. (a) Regulated sAPP secretion. COS cells were subjected to a pre-treatment with PDBu (PDBu PT) from 0 to 8 h, followed by a 1 h exposure to PDBu stimulation (PDBu St), using serum-free medium. The 1 h medium from pulse chased cells was collected, and secreted sAPPα was immunoprecipitated with the antibody 6E10. Immunoprecipitates were separated by SDS–polyacrylamide gel electrophoresis and sAPPα was detected by autoradiography. (b) Cumulative sAPP secretion. Immunoblot analysis of sAPP accumulating in the extracellular milieu at 2 h intervals. COS-7 cells maintained in sf medium or in c medium were exposed to PDBu. The conditioned medium was separated on SDS–polyacrylamide gel electrophoresis and immunoblotted with the antibody 22C11. Total sAPP (sAPPt) is indicated. (c) Quantitative representation in fold increase of sAPPt produced in the absence or presence of PDBu, for cells maintained in sf or c medium. Data are expressed as mean ± SEM of triplicate determinations of three independent experiments. Statistical significance analysis was conducted by Anova followed by the two-tail Student's t-test. Statistical significance differences against control determinations (− PDBu) are presented as *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. (d) Comparative immunoblot analysis of sAPPt and sAPPα produced by cells when exposed to PDBu medium in sf or c medium.
Cumulative production of total sAPP following PKCα down-regulation
Although APP-regulated cleavage cannot be stimulated following PKCα down-regulation, sAPP is still being produced as is evident from Fig. 5b. Under the experimental conditions tested (−/+ PDBu), sAPPt (total sAPP) accumulated in the extracellular medium over time, albeit at different rates. For cells grown in sf medium, during the initial time points (2 and 4 h) continued PDBu exposure resulted in approximately three and four times more sAPP accumulating in the media, respectively (Fig. 5b and c), whereas in the absence of PDBu only a twofold increase was obtained. At these earlier time points, PKCα was not yet completely down-regulated for sf and c media, and thus PDBu still activated PKC, resulting in the observed increases in sAPPt production. Interestingly, with continued PDBu exposure, the increased accumulation of sAPPt in the conditioned media per unit time levels off, with fold-increases reaching seven-to eightfold by 8–10 h, respectively (Fig. 5b and c). In the absence of PDBu, the rate of sAPPt production is maintained throughout the experiment, and the accumulated levels of sAPPt approach those obtained with PDBu, rising to fiveand sevenfold by 8 and 10 h (Fig. 5c). For cells maintained in c medium, the increases are more modest, resulting in a maximum fourfold sAPPt increase by 10 h. From the data presented above, it is evident that although PKCα is down-regulated and that the fluorescent signal from holo APP–GFP (hAPP) is no longer incorporated into visible vesicular structures, sAPP is still produced and secreted. Thus, the media collected were tested for both sAPPt and sAPPα (Fig. 5d). It appears that in sf medium sAPPt increases with time up to the 10 h time point, whereas sAPPα increases initially but remains constant from 6 to 10 h, suggesting that there is a tendency toward relatively more amyloidogenic processing as PKCα is down-regulated.
PKCα independent APP processing and Aβ production
To address amyloidogenic processing, COS-7 cells were labeled with [35S]-methionine and incubated in sf and c media, with and without PDBu, and the medium was immunoprecipitated with 6E10. The resulting immunoprecipitates were analyzed for sAPPα (not shown) and Aβ (Fig. 6a). As discussed above, PDBu produced increases in sAPPα for cells maintained in sf and c media, although the increases were greater in sf medium. Enhanced sAPPα production is often accompanied by decreased Aβ production under acute exposure to PDBu. When the experiment was carried out in c medium, Aβ could not be detected; however, when sf medium was used, an increase in Aβ production was clearly visible (Fig. 6a). Hence, increased Aβ production occurred even when PKCα was down-regulated. Similar increases were obtained if cells were transfected with the Swedish APP mutant (Fig. 6b). However, given the unexpected concurrent increases in sAPPα and Aβ, levels of endogenous hAPP were also monitored (see below).
Fig. 6.
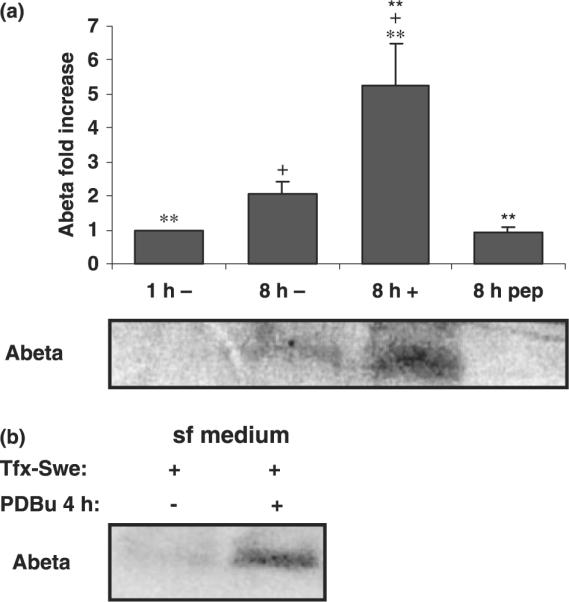
Effect of long-term PDBu exposure on Aβ production. (a) Metabolic labeling experiments were carried out in COS cells maintained in sf or c medium in the absence or presence of PDBu. Media from 8 h were collected, immunoprecipitated with antibody 6E10, and subjected to SDS–polyacrylamide gel electrophoresis analysis. Antibody specificity was tested by including the Aβ peptide for a control immunoprecipitation. Detected Aβ is indicated and quantified. Data are expressed as mean ± SEM of triplicate determinations from at least three independent experiments. Statistical significance analysis was conducted by Anova followed by the Tukey test. Statistical significance symbols used for statistically significant differences were +p < 0.05 (between 8 h minus and plus PDBu determinations); **p < 0.01 (between 1 h minus and 8 h plus PDBu determinations); **p < 0.01 (between 8 h plus PDBu determinations, with or without Aβ peptide competition). (b) The procedure was repeated for cells transfected with APPSwe cDNA (Tfx-Swe). PDBu exposure times were reduced to 4 h since at this time point APP expression levels remain largely unaltered.
Given that increases in α and β cleavages for APP were both detected, intracellular levels of APP were monitored. Further, when experiments were carried out in sf medium, PKCε levels increased. Hence, cell lysates of COS cells cultured in sf or c medium and exposed to PDBu were subjected to immunoblot analysis. COS cells exposed to phorbol esters in sf medium showed a significant increase in intracellular levels of APP (Fig. 7a and b). Both the more abundant APP751/770 isoform and the less abundant APP695 isoform expressed in COS cells increased in a dose-dependent manner with time of exposure to PDBu. Maximal levels of APP expression were obtained with 1 μM PDBu for 8 h, when a greater than twofold increase could be detected (Fig. 7b). In contrast, when the experiment was carried out in c medium, no increases could be detected in the intracellular levels of hAPP (Fig. 7a). In this situation, PKCε is not up-regulated (Fig. 1). Thus, it seems reasonable to conclude that increased PKCε levels resulted in increased intracellular APP levels, perhaps because of a role for PKCε in APP transcription. This conclusion is also supported by a direct correlation between the concentration of PDBu added and increased intracellular levels of hAPP (Fig. 7b). Consequently, northern blot analysis was carried out on cells exposed to PDBu for long periods of time in sf (Fig. 7c) or c (data not shown) medium. The results showed that under conditions where PKCε was up-regulated, APP mRNA steady state levels were also increased.
Fig. 7.

Effect of PDBu exposure on expression levels of APP. (a) Immunoblot analysis of holoAPP (hAPP) expression levels in COS-7 cells, using mouse monoclonal antibody 22C11. Cells were grown in the absence or presence of PDBu for up to 10 h without serum (sf medium) and with serum (c medium). Tubulin expression levels were monitored as a control. (b) PDBu dose effect on hAPP expression levels. Data are expressed as mean ± SEM. (c) Northern blot analysis of APP mRNA levels following long-term exposure to PDBu (8 h).
Effect of PKCα down-regulation on Aβ production
Given that increased Aβ production resulted from long-term exposure to PDBu of cells incubated in sf medium, and that this was accompanied by increased intracellular levels of hAPP and PKCα down-regulation, we monitored whether the latter alone could contribute to increased Aβ production. Thus, APP cDNA carrying the Swedish mutation was transiently transfected into COS cells and therefore not under the control of the endogenous APP promoter. The Swedish mutant shows an approximately 10-fold increase in catalytic efficiency for the APP : β-secretase reaction. Additionally, experiments were carried out at a total time of 4 h PDBu exposure (Fig. 6b). At this time, PKCα was considerably down-regulated, with only low levels of protein being detected (Fig. 1), and intracellular levels of endogenous APP were only marginally increased (Fig. 7a). Under these conditions, Aβ production was dramatically increased (Fig. 6b). Taken together, we conclude that our data are most consistent with the formulation that down-regulation of PKCα results in decreased TGN budding and a consequent retention of APP in the TGN, leading to increased Aβ production.
Discussion
Various reports have indicated that PKC plays important roles at several stages of APP processing, including sAPPα production, perhaps by controlling APP exit from the TGN. However, the phorbol ester sensitive molecular players involved in regulated APP shedding have remained elusive (Ikin et al. 2007). Up to now, most studies addressing regulated secretion have primarily employed acute PKC activation protocols. In our hands, both PKCα and PKCε were down-regulated by exposing to PDBu in c medium following long-term exposure of COS cells to phorbol esters. However, in sf medium, we observe up-regulation of PKCε and a strong down-regulation of PKCα. Elucidation of the molecular basis for the differential behavior of the PKC isozymes will require additional investigation.
In our hands, down-regulation of PKCα alone is sufficient to abolish the ability of additional PDBu to stimulate sAPPα production. However, with increasing periods of exposure to PDBu, sAPPα production plateaus for cells maintained in sf medium, while sAPPt continues to increase. In fact, direct measurement of Aβ shows that its production is dramatically increased as PKCα is down-regulated. Our results thus demonstrate roles for PKCα both in enhancing sAPPα generation and in diminishing amyloidogenic APP processing. This predicts the corollary that PKCα down-regulation almost completely inhibits stimulation of sAPPα production and leads to increased Aβ production (Fig. 6 and 7), and our data bear this out. Consistent with this role for PKCα, fibroblasts from Alzheimer patients exhibit down-regulation of PKCα and defective metabolism (Bergamaschi et al. 1995).
Mechanistically, PKCα down-regulation also affects the subcellular localization of APP, leading to its apparent accumulation in the TGN. Simultaneously, a dramatic decrease was observed in detectable APP-containing cytoplasmic vesicles, perhaps resulting from a diminution in budding of these vesicles at the TGN (Fig. 3) (Bergamaschi et al. 1995; Rossner et al. 2001), but we cannot formally exclude the possibility that these vesicles are endosomes that disappear because PKC activity is involved in endocytosis (Prevostel et al. 2000; Becker and Hannun 2003).
Most Aβ1–40 and Aβ1–42 peptides are believed to be generated in the TGN or in the endocytic pathway. Given that long-term exposure to phorbol esters appears to stimulate Aβ production to a greater extent than sAPPt secretion (Figs 5 and 6), one can conclude that PKCα down-regulation favors amyloidogenic APP processing. That is, increased Aβ production is not solely the result of increased APP mRNA expression, as discussed below, but is also a consequence of altered processing. Indeed, the results observed following transfection of an APP cDNA clearly illustrate this point (Fig. 6b).
Given the documented effects of PDBu on the AP-1 promoter which result in increased APP mRNA levels (Trejo et al. 1994), we addressed how long-term exposure to PDBu might affect APP transcription. A significant increase in the intracellular levels of hAPP was observed for cells maintained in sf medium (Fig. 7). Additionally, cells exposed to increasing concentrations of PDBu for increasing periods of time revealed a direct relationship between the PDBu concentration used and the observed increase in intracellular levels of hAPP. Thus, PDBu may be stimulating the APP promoter. Consistent with this model, both PKCα and PKCε have been reported to regulate transactivation of the AP-1 site in the APP promoter (Uberall et al. 1994). Further interplay between APP and PKCε expression are suggested in a recent study by Liron et al. (2007) in which they show that APP over-expression results in a specific and significant down-regulation of PKCε, with no change in the levels of other PKC isozymes.
In conclusion, although short-term phorbol ester treatment inhibits Aβ production, long-term exposure favors Aβ production. Consistent with previous reports (Jolly-Tornetta and Wolf 2000), PKCα appears to be involved in phorbol ester-regulated sAPPα secretion. However, PKCα down-regulation does not inhibit Aβ production. Further, the data herein support (but do not establish) the notion that PKCα down-regulation leads to failure of APP packaging into nascent vesicles budding from the TGN or failure of the budding process per se. Nonetheless, even in the absence of detectable APP-bearing vesicles, APP is processed, and high levels of sAPP and Aβ are still produced. PKCε, on the other hand, does not appear to be involved in regulating proteolytic cleavage of APP to an important degree, but rather appears to play a key role in modulating APP expression. Hence, PKCα and PKCε are differentially involved in APP metabolism. As discussed in the introduction, transfection with PKCα or PKCε isoforms leads to increased sAPP production (Kinouchi et al. 1995). However, given our data, it seems reasonable to assume that those increases have different origins. In the first case, increased sAPP secretion is likely because of enhanced APP metabolism stimulated by PKCα, whereas, in the case of PKCε, the increase is probably because of increased APP expression. These differential effects and time courses related to PKC isozyme biology must be taken into account when developing therapeutic drugs aimed at lowering Aβ generation by targeting PKC mediating signaling.
Acknowledgements
This study was supported by the EU VI Framework Program (cNEUPRO and APOPIS), by the FCT (REEQ/1025/BIO/2005, POCTI/CBO/34349/1999, POCTI/NSE/40682/2001, POCI/BIA-BCM/58469/2004 and POCI/SAU-OBS/57394/2004) of the Portuguese Ministry of Science and Technology, and by CBC, Universidade de Aveiro. SG was supported by USPHS P01 AG10491 and the P50 AG 005138 Mt Sinai Alzheimer's Disease Research Center (to Mary Sano). PG was supported by USPHS PO1 AG09464.
Abbreviations used
- ADAM
a disintegrin and metalloproteinase
- AP-1
activator protein-1
- APP
amyloid precursor protein
- Aβ peptide
amyloid-β peptide
- c medium
complete medium
- COS
monkey kidney
- DMEM
Dulbecco's modified Eagle's medium
- GFP
green fluorescent protein
- hAPP
holo APP
- PDBu
phorbol-12,-14-di-butyrate
- PKC
protein kinase C
- sAPP
soluble APP
- sAPPt
total sAPP
- SDS
sodium dodecyl sulfate
- sf medium
serum-free medium
- TGN
trans Golgi network
References
- Amador FC, Henriques AG, da Cruz ESOA, da Cruz ESEF. Monitoring protein phosphatase 1 isoform levels as a marker for cellular stress. Neurotoxicol. Teratol. 2004;26:387–395. doi: 10.1016/j.ntt.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Becker KP, Hannun YA. cPKC-dependent sequestration of membrane-recycling components in a subset of recycling endosomes. J. Biol. Chem. 2003;278:52747–52754. doi: 10.1074/jbc.M305228200. [DOI] [PubMed] [Google Scholar]
- Benussi L, Govoni S, Gasparini L, Binetti G, Trabucchi M, Bianchetti A, Racchi M. Specific role for protein kinase C alpha in the constitutive and regulated secretion of amyloid precursor protein in human skin fibroblasts. Neurosci. Lett. 1998;240:97–101. doi: 10.1016/s0304-3940(97)00894-x. [DOI] [PubMed] [Google Scholar]
- Bergamaschi S, Binetti G, Govoni S, Wetsel WC, Battaini F, Trabucchi M, Bianchetti A, Racchi M. Defective phorbol ester-stimulated secretion of beta-amyloid precursor protein from Alzheimer's disease fibroblasts. Neurosci. Lett. 1995;201:1–5. doi: 10.1016/0304-3940(95)12168-4. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc. Natl Acad. Sci. USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc. Natl Acad. Sci. USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Ramabhadran TV, Greengard P. Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc. Natl Acad. Sci. USA. 1992;89:3055–3059. doi: 10.1073/pnas.89.7.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz e Silva EF, da Cruz e Silva OA. Protein phosphorylation and APP metabolism. Neurochem. Res. 2003;28:1553–1561. doi: 10.1023/a:1025630627319. [DOI] [PubMed] [Google Scholar]
- da Cruz e Silva OA, Iverfeldt K, Oltersdorf T, Sinha S, Lieberburg I, Ramabhadran TV, Suzuki T, Sisodia SS, Gandy S, Greengard P. Regulated cleavage of Alzheimer beta-amyloid precursor protein in the absence of the cytoplasmic tail. Neuroscience. 1993;57:873–877. doi: 10.1016/0306-4522(93)90031-a. [DOI] [PubMed] [Google Scholar]
- da Cruz e Silva OAB, Vieira SI, Rebelo S, da Cruz e Silva EF. A model system to study intracellular trafficking and processing of the Alzheimer's amyloid precursor protein. Neurodegener. Dis. 2004;1:196–204. doi: 10.1159/000080986. [DOI] [PubMed] [Google Scholar]
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, Busciglio J, Yankner BA. Inhibition of beta-amyloid production by activation of protein kinase. C. J. Neurochem. 1993;61:2326–2329. doi: 10.1111/j.1471-4159.1993.tb07479.x. [DOI] [PubMed] [Google Scholar]
- Gandy S, Greengard P. Processing of Alzheimer A beta-amyloid precursor protein: cell biology, regulation, and role in Alzheimer disease. Int. Rev. Neurobiol. 1994;36:29–50. doi: 10.1016/s0074-7742(08)60302-5. [DOI] [PubMed] [Google Scholar]
- Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc. Natl Acad. Sci. USA. 1988;85:6218–6221. doi: 10.1073/pnas.85.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy SE, Bhasin R, Ramabhadran TV, Koo EH, Price DL, Goldgaber D, Greengard P. Alzheimer beta/A4-amyloid precursor protein: evidence for putative amyloidogenic fragment. J. Neurochem. 1992;58:383–386. doi: 10.1111/j.1471-4159.1992.tb09322.x. [DOI] [PubMed] [Google Scholar]
- Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J. Biol. Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- Ikin AF, Causevic M, Pedrini S, et al. Evidence against roles for phorbol binding protein Munc13-1, ADAM adaptor Eve-1, or vesicle trafficking phosphoproteins Munc18 or NSF as phospho-state-sensitive modulators of phorbol/PKC-activated Alzheimer APP ectodomain shedding. Mol. Neurodegener. 2007;2:23. doi: 10.1186/1750-1326-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly-Tornetta C, Wolf BA. Regulation of amyloid precursor protein (APP) secretion by protein kinase calpha in human ntera 2 neurons (NT2N) Biochemistry. 2000;39:7428–7435. doi: 10.1021/bi0003846. [DOI] [PubMed] [Google Scholar]
- Kinouchi T, Sorimachi H, Maruyama K, Mizuno K, Ohno S, Ishiura S, Suzuki K. Conventional protein kinase C (PKC)-alpha and novel PKC epsilon, but not -delta, increase the secretion of an N-terminal fragment of Alzheimer's disease amyloid precursor protein from PKC cDNA transfected 3Y1 fibroblasts. FEBS Lett. 1995;364:203–206. doi: 10.1016/0014-5793(95)00392-m. [DOI] [PubMed] [Google Scholar]
- Knops J, Lieberburg I, Sinha S. Evidence for a nonsecretory, acidic degradation pathway for amyloid precursor protein in 293 cells. Identification of a novel, 22-kDa, beta-peptide-containing intermediate. J. Biol. Chem. 1992;267:16022–16024. [PubMed] [Google Scholar]
- Lanni C, Mazzucchelli M, Porrello E, Govoni S, Racchi M. Differential involvement of protein kinase C alpha and epsilon in the regulated secretion of soluble amyloid precursor protein. Eur. J. Biochem. 2004;271:3068–3075. doi: 10.1111/j.1432-1033.2004.04240.x. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liron T, Seraya CB, Ish-Shalom M, Souroujon MC, Neumann D. Overexpression of amyloid precursor protein reduces epsilon protein kinase C levels. Neuroscience. 2007;146:152–159. doi: 10.1016/j.neuroscience.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Oishi M, Nairn AC, Czernik AJ, Lim GS, Isohara T, Gandy SE, Greengard P, Suzuki T. The cytoplasmic domain of Alzheimer's amyloid precursor protein is phosphorylated at Thr654, Ser655, and Thr668 in adult rat brain and cultured cells. Mol. Med. 1997;3:111–123. [PMC free article] [PubMed] [Google Scholar]
- Paola D, Domenicotti C, Nitti M, et al. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem. Biophys. Res. Commun. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- Prevostel C, Alice V, Joubert D, Parker PJ. Protein kinase C(alpha) actively downregulates through caveolae-dependent traffic to an endosomal compartment. J. Cell Sci. 2000;113:2575–2584. doi: 10.1242/jcs.113.14.2575. [DOI] [PubMed] [Google Scholar]
- Racchi M, Mazzucchelli M, Pascale A, Sironi M, Govoni S. Role of protein kinase Calpha in the regulated secretion of the amyloid precursor protein. Mol. Psychiatry. 2003;8:209–216. doi: 10.1038/sj.mp.4001204. [DOI] [PubMed] [Google Scholar]
- Rebelo S, Henriques AG, da Cruz e Silva EF, da Cruz e Silva OA. Effect of cell density on intracellular levels of the Alzheimer's amyloid precursor protein. J. Neurosci. Res. 2004;76:406–414. doi: 10.1002/jnr.20091. [DOI] [PubMed] [Google Scholar]
- Rossner S, Mendla K, Schliebs R, Bigl V. Protein kinase Calpha and beta1 isoforms are regulators of alpha-secretory proteolytic processing of amyloid precursor protein in vivo. Eur. J. Neurosci. 2001;13:1644–1648. doi: 10.1046/j.0953-816x.2001.01525.x. [DOI] [PubMed] [Google Scholar]
- Seubert P, Oltersdorf T, Lee MG, et al. Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature. 1993;361:260–263. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Song JC, Rangachari PK, Matthews JB. Opposing effects of PKCalpha and PKCepsilon on basolateral membrane dynamics in intestinal epithelia. Am. J. Physiol. Cell Physiol. 2002;283:C1548–C1556. doi: 10.1152/ajpcell.00105.2002. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Nairn AC, Gandy SE, Greengard P. Phosphorylation of Alzheimer amyloid precursor protein by protein kinase C. Neuroscience. 1992;48:755–761. doi: 10.1016/0306-4522(92)90264-3. [DOI] [PubMed] [Google Scholar]
- Tamaoka A, Kalaria RN, Lieberburg I, Selkoe DJ. Identification of a stable fragment of the Alzheimer amyloid precursor containing the beta-protein in brain microvessels. Proc. Natl Acad. Sci. USA. 1992;89:1345–1349. doi: 10.1073/pnas.89.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo J, Massamiri T, Deng T, Dewji NN, Bayney RM, Brown JH. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer's beta-amyloid precursor protein gene. J. Biol. Chem. 1994;269:21682–21690. [PubMed] [Google Scholar]
- Uberall F, Kampfer S, Schubert C, Doppler W, Grunicke HH. Role of protein kinase C in ras-mediated fos-expression. Adv. Enzyme Regul. 1994;34:257–268. doi: 10.1016/0065-2571(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the trans-membrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Hamdane M, Gompel M, et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis. 2005;20:625–637. doi: 10.1016/j.nbd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- Xu H, Greengard P, Gandy S. Regulated formation of Golgi secretory vesicles containing Alzheimer beta-amyloid precursor protein. J. Biol. Chem. 1995;270:23243–23245. doi: 10.1074/jbc.270.40.23243. [DOI] [PubMed] [Google Scholar]
- Yeon SW, Jung MW, Ha MJ, Kim SU, Huh K, Savage MJ, Masliah E, Mook-Jung I. Blockade of PKC epsilon activation attenuates phorbol ester-induced increase of alpha-secretase-derived secreted form of amyloid precursor protein. Biochem. Biophys. Res. Commun. 2001;280:782–787. doi: 10.1006/bbrc.2000.4181. [DOI] [PubMed] [Google Scholar]
- Zhu G, Wang D, Lin YH, McMahon T, Koo EH, Messing RO. Protein kinase C epsilon suppresses Abeta production and promotes activation of alpha-secretase. Biochem. Biophys. Res. Commun. 2001;285:997–1006. doi: 10.1006/bbrc.2001.5273. [DOI] [PubMed] [Google Scholar]