

Abstract
The absolute stereo structure of the natural product laurenditerpenol (1S, 6R, 7S, 10R, 11R, 14S, 15R) has been accomplished from eight plausible stereoisomers by its first asymmetric total synthesis in a highly convergent and flexible synthetic pathway. Six stereoisomers of laurenditerpenol were synthesized and evaluated for their biological activity.
The rapid proliferation of cells within a tumor mass quickly outpaces the capability of existing vasculature to supply oxygen and nutrients. In cancer patients, the extent of tumor hypoxia correlates with advanced disease stages and an overall poor prognosis.1 Presently, there is no drug that specifically targets hypoxic tumor cells. The transcription factor hypoxia-inducible factor 1 (HIF-1), a heterodimer of the bHLH-PAS proteins HIF-1α and HIF-1β/ARNT, functions as a master regulator of hypoxia-induced gene expression when mammalian cells are subjected to oxygen deprived conditions.2 Inhibition of HIF-1 production/function in animal models significantly reduces tumor growth,3 and small molecule HIF-1 inhibitors represent potential tumor hypoxia-selective anti-cancer drug leads.4
Bioassay-guided fractionation of the lipid extract of the red alga Laurencia intricata yielded a structurally novel bicyclic diterpene, laurenditerpenol 1.5 Laurenditerpenol was the first marine natural product found to inhibit hypoxia (1% O2)-induced HIF-1 activation and potently inhibited HIF-1 activation in T47D breast tumor cells by hypoxia with a IC50 value of 0.4 μM.5 Mitochondrial respiration studies suggest that laurenditerpenol is the first member of a structurally novel class of marine natural product-based mitochondrial inhibitors that block mitochondrial oxygen consumption and promote the degradation of HIF-1α protein under hypoxic conditions.5
The intriguing biological properties of laurenditerpenol with the structural complexity of the molecule including an unprecedented 7-oxabicyclo[2.2.1] heptane ring system and contiguous stereogenic centers at C1, C6 and C7 pose a significant synthetic challenge. Although previous spectroscopic studies permitted the identification of the structure of the natural product including the absolute configuration at C1 and the relative syn configuration of the 7-oxabicyclo[2.2.1]heptane ring methyl chiral centers at C11, C14, C15 as well as their trans relationship to C10, the configurations of the stereocenters at C6, C7 including the issue of absolute stereochemistry at C10, C11, C14 and C15 remained unknown. These uncertainties prompted us to initiate a total synthesis of laurenditerpenol to determine its absolute stereochemistry. We describe herein an inaugural total assignment of the absolute configuration of laurenditerpenol from one of eight possible stereoisomers of 1 (Figure 1) through the first asymmetric total synthesis.
Figure 1.

Originally proposed (1) and absolute stereochemical assignment (1c) of laurenditerpenol from eight possible stereoisomers
Our retrosynthetic analysis of the basic skeleton of laurenditerpenol is depicted in Scheme 1. This envisioned analysis is a result of preliminary studies with racemic materials that have eliminated many obvious construction strategies, leading to a rather straightforward sulfone-mediated C-C bond formation between bromide 4 and hindered bicyclic sulfone 5. This disconnection approach provides a flexible strategy to incorporate either enantiomer of sulfone 5, and the C7 configuration could be introduced by adopting appropriate stereoselective transformations. The double Michael/Diels-Alder addition of methyl crotonate to the furanone enolate can provide adduct (±)-6, and could be converted into olefin 3 with all the essential substituents and the correct relative configuration including a trans relationship between the methyl and carboxylic acid substituents.
Scheme 1.

aRetrosynthetic analysis of laurenditerpenol
The synthetic venture commenced with an “anion assisted” Diels-Alder reaction/sequential Michael addition of lithium enolate 7 with methyl crotonate which remarkably produced 6-exo ketoester (±)-6 as a single diastereomer (Scheme 2).6 The assignment of 6-exo configuration was made based on NMR experiments as shown in the Scheme 2. Racemic ketoester (±)-6 was protected as the thioketal and hydrolyzed into acid (±)-9. With the ambiguity in the relative configuration of the bicyclic moiety, we decided to alter our synthesis to obtain both the (+)- and (−)-bicyclic intermediates of 5. Seeking a higher level of convergence, the best resolution of racemic acid (±)-9 has been achieved by converting it into N-acyloxazolidinone diastereomers (−)-10 and (+)-10 employing standard coupling conditions PivCl/Et3N/LiCl in 82% yield.7 The crystal structure of oxazolidinone (−)-108 confirmed the soundness of NMR experiments analysis on ketoester (±)-6 and established the absolute configuration of the 7-oxabicyclo[2.2.1]heptane ring system. With completely characterized diastereomers (−)-10 and (+)-10 in hand, each isomer was separately converted into its corresponding enantiomeric sulfone in a four step reaction sequence. Thus, reductive elimination of the thioketal functionality followed by reductive removal of the chiral auxiliary with LiBH4 provided alcohol (−)-11. Direct iodination of alcohol (−)-11 with TPP/I2/imidazole9 followed by sulfonylation with PhSO2Na produced sulfone (−)-5. Following the same reaction sequence sulfone (+)-5 was also prepared from oxazolidinone (+)-10.
Scheme 2. aSynthesis of chiral 7-oxabicyclo[2.2.1]-heptane ring system.

aReagents and conditions: a) LDA, Et2O:cyclohexane, −78 °C, then methyl crotonate, 69%; b) i. Ethanedithiol, p-TsOH, benzene, reflux; ii. LiOH, aq. MeOH, 92%, two steps; c) TEA, trimethylacetyl chloride, LiCl, (S)-(−)-4-benzyl-2-oxazolidinone, CH2Cl2, rt, separation of diastereomers by silica gel chromatography: (−)-10, 40%, (+)-10, 42%; d) i. Raney Ni, EtOH, reflux, 98%; ii. LiBH4, THF, 0 °C, (−)-11, 95%, (+)-11 96%; e) Ph3P, I2, imidazole, CH2Cl2, rt, (−)-12, 94%, (+)-12, 92%; f) PhSO2Na, DMF, 60 °C, (−)-5, 90%, (+)-5, 91%. ORTEP representation of the N-acyloxazolidinone (−)-10 (H atoms are omitted for clarity)
Following the synthetic strategy, an acid catalyzed ketalization of R-(+)-pulegone accompanied by double bond migration introduced the C6 stereogenic center to produce diequatorial ketal 13 in high diastereoselectivity.10 Regioselective allylic bromination of ketal 13 with NBS employing Yamanaka conditions11 afforded bromide 4 in moderate yields, while classical radical bromination disappointingly produced an inseparable plethora of products. Stereoselective alkylation of allyl bromide 4 with sulfone (−)-5 was successfully achieved in 98% yield with high diastereoselectivity (dr 95:5). Direct hydrogenation12 of olefin 3 proved to be unsuccessful in our hands, while stereoselective hydroxylation of the olefin moiety occurred under routine conditions employing a hydroboration/oxidation13 sequence. The resultant anti-Markovnikov alcohol 14 was formed as expected while introducing the C7 methyl stereocenter. As anticipated, the hydroxy sulfone 14 was produced with excellent regio- and stereoselectivity which could be explained in terms of electrophilic borane addition by the Felkin-Anh-Houk paradigm (Figure A).14 Sulfinylation of 14 with PhSSPh/Bu3P15 followed by reductive elimination afforded methyl sulfone 15 in excellent yield. Deketalization of 15 with PdCl2(CH3CN)2/acetone16 efficiently produced ketosulfone 2 in 92% yield with no epimerization at C6 whereas longer reaction times under the same conditions, or other acid catalyzed deketalization conditions17 resulted in C6 epimerization in minor quantities. X-ray crystallographic analysis18 of ketosulfone 2 revealed 7R configuration at the C7 methyl center, and supported the evolution of C7 configuration as anticipated by the Felkin-Anh-Houk model of electrophilic hydroboration.
Having established the absolute configuration of ketosulfone 2, reductive desulfonylation19 as originally planned was accomplished using Na-Hg in MeOH to produce ketone 16 with traces of the C6 epimerized product.20 Regioselective enolization of 2 with lithium diisopropylamide at −78 °C followed by addition of PhSeCl produced a mixture of diastereomers,20 which was further subjected to oxidative elimination to afford α,β-unsaturated ketone 18.21 Regioselective reduction of theα,β-unsaturated ketone with CeCl3-NaBH422 produced chromatographically separable C1 epimers, ent-1g and 1a in a 2:3 ratio. Thus we have synthesized two stereoisomers of laurenditerpenol in a single pot, one with the desired 1S configuration, and one is the enantiomer of probable eight isomers (Figure 1).
The 1H NMR spectroscopic data and the physical parameters (TLC and specific rotation) of the stereoisomer 1a did not match with an authentic sample of natural product 1 whereas the ent-1g 1H NMR spectrum was closely similar. 1H NMR spectral data analyses of these C-1 epimers were clearly characteristic of the natural product, which in turn reduced the synthetic maneuver, required to identify the correct stereoisomer of laurenditerpenol. 1H NMR spectrum of 1a showed the H2 proton resonance at δ 5.39 ppm as a broad singlet and the H1 proton appeared at 4.03 ppm as a broad multiplet, whereas the 1H NMR spectrum of ent-1g showed the H2 resonance at 5.64 ppm as a doublet (J = 4.0 Hz) and H1 at 4.11 ppm as a broad singlet. Comparison of this spectral data with reported spectral data of similar systems,23 we concluded that 1,6-syn-cyclohex-2-en-1-ol and 1,6-anti-cyclohex-2-en-1-ol systems exhibit these characteristic 1H NMR signal difference in chemical shift values.24 Based on this characterization and comparison of the H2 and H1 proton chemical shift values of natural product 1 with ent-1g, it is clear that laurenditerpenol must possess a 1,6-syn- cyclohex-2-en-1-ol system within its structure. This analysis eliminates all of the remaining 1,6-anti-cyclohex-2-en-1-ol isomers 1b, 1e, 1f and leaves 1,6-syn- cyclohex-2-en-1-ol isomers. Thus the first successful synthesis of stereoisomers of laurenditerpenol 1a and ent-1g has limited the possibility of the natural product from its eight stereoisomers 1a–1h to three stereoisomers 1c, 1d, and 1h (Figure 1).
At this stage we advanced to synthesize the remaining target stereoisomers with sulfone (−)-5. Following the developed synthetic technology, we altered the reaction sequence to maneuver the synthesis of both the C7 stereoisomers. Thus sulfone 3 was desulfonylated to culminate the observed stereoinduction in the hydroboration reaction due to phenylsulfone functionality, which in turn facilitates to access both the 7R and 7S hydroxy compounds 20a and 20b respectively. Since these diastereomers are chromatographically difficult to separate, the mixture was further subjected to sulfinylation to provide the corresponding phenylsulfides 21a and 21b as a chromatographically separable mixture of diastereomers. Reductive elimination of phenylsulfide 21a followed by deketalization afforded ketone 16.
The desired C1–C6 syn stereoselectivity was achieved by epimerization of ketone 16 to 22, and the diastereomers were separated by silica gel flash chromatography.25 Dehydrogenation of ketone 22 to enone 23 using a two-step protocol followed by regioselective reduction under Luche conditions produced stereoisomers of laurenditerpenol 1d and ent-1f in 2:3 ratio as a chromatographically separable mixture of isomers. As anticipated, stereoisomer 1d closely matched the 1H NMR spectral data of the natural product, but the carbon values varied moderately, and the TLC Rf did not match with an authentic sample of the natural product. Following the same synthetic transformations, stereoisomer 1c also synthesized from phenylsulfide 21b. The 1H and 13C NMR spectra of synthetic 1c were identical with natural product 1. Moreover, synthetic 1c and authentic natural product 1 exhibited identical behavior on TLC confirming the absolute configuration as drawn.26
The synthetically produced isomers of laurenditerpenol were evaluated for their ability to inhibit hypoxia-induced HIF-1 activation in T47D breast tumor cells.27 Synthetic laurenditerpenol 1c is very similar in potency (IC50 0.82 μM) to the original compound isolated from L. intricata (Table 1).28 At concentrations as high as 30 μM, compound 1c neither inhibited luciferase expression from the pGL3-control reporter, nor inhibited T47D cell proliferation/viability under assay conditions. Therefore, the inhibition of hypoxia-induced HIF-1 activation (pHRE3-TK-luc) by 1c was selective and independent of any effect on T47D cell viability. Inversion of configuration at C7 1d was associated with a 76% drop in potency, relative to synthetic laurenditerpenol 1c. Inversion of configuration at C1, C6 and C7 essentially deactivated ent-1g.
Table 1.
IC50 values of synthetic stereoisomers of laurenditerpenol.
| Isomer | Hypoxia (16 h) pHRE3-TK-luc[a] |
|---|---|
| 1c | 0.82 μM |
| 1d | 3.4 μM |
| ent-1g | >30 μM |
Values were obtained fromT47D cell-based reporter assay
In summary, the goal of assigning the absolute configuration of laurenditerpenol has been accomplished from eight plausible stereoisomers by its first asymmetric total synthesis in a highly convergent manner. The flexibility of the current strategy to deliver either configuration at each stereocenter allows construction of all stereoisomers of this valuable natural product for further biological evaluation. An asymmetric variant of the current total synthesis and the structure activity relationships of all the isomers and the rational design of other novel HIF-1 analogues for chemical biology studies are in progress and the results will be reported in due course.
Supplementary Material
Scheme 3. aFragment coupling and synthesis of keto-sulfone 2 (X = SO2Ph).

aReagents and conditions; a) (CH2OH)2, CSA, benzene, reflux, 24 h, 73% (dr 92:8); b) NBS, Yb(OTf)3, TMSCl, CH2Cl2, 0 °C, 15 min, 48%; c) (−)-5, n-BuLi, HMPA, THF, −40 °C, 40 min, then 4, 2 h, 98% (dr 95:5); d) i. BH3.S(CH3)2, THF, 10 h, ii. H2O2, NaOH, 0 °C, 87%; e) i. PhSSPh, (n-Bu3)P, toluene, rt, 94%; ii. Raney Ni, EtOH, reflux, 90%; f). PdCl2(CH3CN)2, acetone, rt, 30 min, 92%. In ORTEP representation of one of the two crystallographically independent molecules of 2 (H atoms and cocrystallized ether molecules are omitted for clarity).
Scheme 4. aSynthesis and absolute configuration of stereoisomers of laurenditerpenol 1a and ent-1g.

aReagents and conditions: a) Na-Hg (10%), MeOH, rt, 68%; b) i. LDA, PhSeCl, THF, −78 °C, ii. Py, H2O2, CH2Cl2, H2O, 0 °C, 78%; c) CeCl3, NaBH4, MeOH, 0 °C, 72%.
Scheme 5. aTotal synthesis of laurenditerpenol.

aReagents and conditions: a) Na-Hg; MeOH, rt, 6h, 78%; b) i. BH3.DMS, THF followed by H2O2, NaOH; 0 °C, 86%; c) (PhS)2, n-Bu3P, toluene, rt, 90% (21a:21b 3:1); d) i. Raney Ni, EtOH, reflux, 94%; 92%; ii. PdCl2(CH3CN)2, acetone, rt, 30 min, 92%; 90%; e) KOH, MeOH, rt, 12 h, 30% epimerization; 35% epimerization; f) i. 21a/21b, LDA, −78°C, HMPA, THF, 1 h; PhSeCl, 2 h; ii. H2O2, THF, 30 min, 72%; 68%; g) CeCl3, NaBH4, MeOH, 0 °C, 84% (2:3 1d:ent-1f); 80% (2:3 1c:ent-1e). The representative yields in italics are corresponds to the 7S isomer.
Acknowledgments
We thank Dr. Blake E. Watkins for his technical help. This investigation was conducted in a facility constructed with support from research facilities improvement program grant number C06 Rr-14503-01 from the National Centre for Research Resources, National Institutes of Health. Support provided by NIH-NCI CA98787 (DGN).
Footnotes
Supporting Information Available: Spectral characterization data of selected compounds (1H, 13C NMR, HRMS,) are available free of charge via Internet at http://pubs.acs.org.
References
- 1.Tatum JL, Kelloff GJ, Gillies RJ, Arbeit JM, Brown JM, Chao KSC, Chapman JD, Eckelman WC, Fyles AW, Giaccia AJ, Hill RP, Koch CJ, Krishna MC, Krohn KA, Lewis JS, Mason RP, Melillo G, Padhani AR, Powis G, Rajendran JG, Reba R, Robinson SP, Semenza GL, Swartz HM, Vaupel P, Yang D, Croft B, Hoffman J, Liu G, Stone H, Sullivan D. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol. 2006;82:699–757. doi: 10.1080/09553000601002324. [DOI] [PubMed] [Google Scholar]
- 2.(a) Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nature Reviews Drug Discovery. 2003;2:803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]; (b) Semenza GL. Targeting HIF-1 for cancer therapy. Nature Reviews Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 3.(a) Maxwell PH, Dach GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Puch CW, Ratcliffe PJ. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8104–8109. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, Dewhirst MW. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]; (c) Ryan HE, Lo J, Johnson RS. HIF-1a is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1a is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]; (e) Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. The hypoxia-inducible factor-1a is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- 4.(a) Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer and Metastasis Reviews. 2007;26:341–352. doi: 10.1007/s10555-007-9059-x. [DOI] [PubMed] [Google Scholar]; (b) Semenza GL. Development of novel therapeutic strategies that target HIF-1. Expert Opinion on Therapeutic Targets. 2006;10:267–280. doi: 10.1517/14728222.10.2.267. [DOI] [PubMed] [Google Scholar]
- 5.Mohammed KA, Hossain CF, Zhang L, Bruick RK, Zhou YD, Nagle DG. Laurenditerpenol, a New Diterpene from the Tropical Marine Alga Laurencia intricata that Potently Inhibits HIF-1 Mediated Hypoxic Signaling in Breast Tumor Cells. J Nat Prod. 2004;67:2002–2007. doi: 10.1021/np049753f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Caine D, Collison RF. Reactions of the lithium dienolate of 2,5-dimethyl-3(2H)-furanone with unsaturated compounds. Synlett. 1995:503–504. [Google Scholar]; (b) Caine DS, Paige MA. Reactions of a 3(2H)-furanone lithium enolate with 4-halocrotonates. Synlett. 1999:1391–1394. [Google Scholar]; (c) Lipshutz BH. Five-membered heteroaromatic rings as intermediates in organic synthesis. Chem Rev. 1986;86:795–820. [Google Scholar]
- 7.(a) Evans DA, Chapman KT, Bisaha J. Asymmetric Diels-Alder cycloaddition reactions with chiral α,β-unsaturated N-acyloxazolidinones. J Am Chem Soc. 1988;110:1238–1256. [Google Scholar]; (b) Liao L-a, Zhang F, Yan N, Golen JA, Fox JM. An efficient and general method for resolving cyclopropene carboxylic acids. Tetrahedron. 2004;60:1803–1816. [Google Scholar]
- 8.CCDC 656780 contains the supplementary crystallographic data for compound (−)-10. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.”
- 9.Lange GL, Corelli N. Synthesis of the sesquiterpenoid lactarane skeleton by a radical cyclobutylcarbinyl/cyclopropylcarbinyl fragmentation sequence. Tetrahedron Lett. 2007;48:1963–1965. [Google Scholar]
- 10.Miller D, Bilodeau F, Burnell RH. Stereoselective syntheses of isomers of 3,7-dimethylnonadecane, a sex pheromone of the alfalfa blotch leafminer (Agromyza frontella (Rondani)) Can J Chem. 1991;69:1100–1106. [Google Scholar]
- 11.Yamanaka M, Arisawa M, Nishida A, Nakagawa M. An intriguing effect of Yb(OTf)3-TMSCl in the halogenation of 1,1-disubstituted alkenes by NXS: selective synthesis of allyl halides. Tetrahedron Lett. 2002;43:2403–2406. [Google Scholar]
- 12.Hydrogenation with Pd/C-EtOAc or MeOH or EtOH, at H2 atm or 80 psi for 16 h did not furnish any required product. Hydrogenation with Wilkinson’s catalyst also found to be unfavorable.
- 13.(a) Kabalka GW, Shoup TM, Goudgaon NM. Sodium perborate: a mild and convenient reagent for efficiently oxidizing organoboranes. J Org Chem. 1989;54:5930–5933. [Google Scholar]; (b) Stephan E, Brossat M, Lecomte V, Bouit PA. Synthesis of the 11b-hydroxymethyl-androst-4-en-3,17-dione. Tetrahedron. 2006;62:3052–3055. [Google Scholar]
- 14.Paddon-Row MN, Rondan NG, Houk KN. Staggered models for asymmetric induction: attack trajectories and conformations of allylic bonds from ab initio transition structures of addition reactions. J Am Chem Soc. 1982;104:7162–7166. [Google Scholar]
- 15.(a) Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981:1–28. [Google Scholar]; (b) Paquette LA, Guevel R, Sakamoto S, Kim IH, Crawford J. Convergent Enantioselective Synthesis of Vinigrol, an Architecturally Novel Diterpenoid with Potent Platelet Aggregation Inhibitory and Antihypertensive Properties. 1. Application of Anionic Sigmatropy to Construction of the Octalin Substructure. J Org Chem. 2003;68:6096–6107. doi: 10.1021/jo0301301. [DOI] [PubMed] [Google Scholar]
- 16.(a) Chanu A, Safir I, Basak R, Chiaroni A, Arseniyadis S. Enantioselective Total Synthesis of 1-epi-Pathylactone A. Org Lett. 2007;9:1351–1354. doi: 10.1021/ol070207y. [DOI] [PubMed] [Google Scholar]; (b) Lipshutz BH, Pollart D, Monforte J, Kotsuki H. Palladium(II)-catalyzed acetal/ketal hydrolysis/exchange reactions. Tetrahedron Lett. 1985;26:705–708. [Google Scholar]
- 17.(a) Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 2. 1991. p. 473. [Google Scholar]; (b) Omura K. Iodine oxidation of a-tocopherol and its model compound in alkaline methanol: unexpected isomerization of the product quinone monoketals. J Org Chem. 1989;54:1987–1990. [Google Scholar]
- 18.CCDC 656779 contains the supplementary crystallographic data for compound 2. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.”
- 19.Dabby RE, Kenyon J, Mason RF. The basic reductive fission of sulfones. J Chem Soc. 1952:4881–4882. [Google Scholar]
-
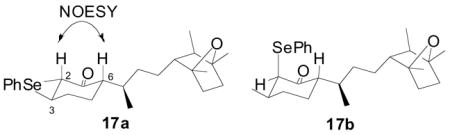
20.Ketone 16 was separated from traces of epimerized diastereomers by column chromatography. In order to confirm the configuration of 16 at the C6 stereocenter, a diastereomeric mixture of selenides were separated by silica gel flash column chromatography to afford the 2,3-anti isomer 17a and the 2,3-syn isomer 17b in 2:3 ratio. Observation of NOESY correlations between H2 and H6 in the 2,3-anti isomer 17a confirmed its 6S configuration.
- 21.Kim JH, Lim HJ, Cheon SH. A facile synthesis of (6S,1′S)-(+)-hernandulcin and (6S,1′R)-(+)-epihernandulcin. Tetrahedron. 2003;59:7501–7507. [Google Scholar]
- 22.(a) Luche JL. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J Am Chem Soc. 1978;100:2226–2227. [Google Scholar]; (b) Luche JL, Gemal AL. Lanthanoids in organic synthesis. 5. Selective reductions of ketones in the presence of aldehydes. J Am Chem Soc. 1979;101:5848–5849. [Google Scholar]; (c) Luche JL, Rodriguez-Hahn L, Crabbe P. Reduction of natural enones in the presence of cerium trichloride. J Chem Soc, Chem Commun. 1978:601–602. [Google Scholar]
- 23.(a) Serra S, Brenna E, Fuganti C, Maggioni F. Lipase-catalyzed resolution of p-menthan-3-ol monoterpenes: preparation of the enantiomer-enriched forms of menthol, isopulegol, trans- and cis-piperitol, and cis-isopiperitenol. Tetrahedron: Asymmetry. 2003;14:3313–3319. [Google Scholar]; (b) Takao K, Tsujita T, Hara M, Tadano K. Asymmetric Total Syntheses of (+)-Cheimonophyllon E and (+)-Cheimonophyllal. J Org Chem. 2002;67:6690–6698. doi: 10.1021/jo0203140. [DOI] [PubMed] [Google Scholar]
- 24.Spectroscopic analysis and mechanistic implications of these kind of systems are under progress and the detailed results will be explored elsewhere in future.
- 25.To improve the epimerized product yield, several basic conditions were attempted. Either similar yield or decomposed product(s) especially with metal hydrides in aprotic solvents was obtained. It was convinced at this stage to use this quick and processible reaction-albeit to provide the material for exploration.
- 26.See supporting information for complete set of spectral data; Regarding the rotation value: Variation in rotation value from reported 1 and synthesized 1c may be due to non-identical experimental conditions. When we recorded the rotation of our authentic sample and synthetic sample under identical experimental conditions, rotation value is identical.
- 27.For experimental details see the supporting information.
- 28.It is critical to note that due to the poor solubility of laurenditerpenol in the formulation for biological evaluation, the original IC50 values varied between separate concentration-response studies, and the average IC50 value (0.4 μM) from a series of independent experiments was ultimately reported. This variation in potency is within the range of values observed in the original studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.