

Abstract
Clinical biomarker tests that aid in making treatment decisions will play an important role in achieving personalized medicine for cancer patients. Definitive evaluation of the clinical utility of these biomarkers requires conducting large randomized clinical trials (RCTs). Efficient RCT design is therefore crucial for timely introduction of these medical advances into clinical practice, and a variety of designs have been proposed for this purpose. To guide design and interpretation of RCTs evaluating biomarkers, we present an in-depth comparison of advantages and disadvantages of the commonly used designs. Key aspects of the discussion include efficiency comparisons and special interim monitoring issues that arise because of the complexity of these RCTs. Important ongoing and completed trials are used as examples. We conclude that, in most settings, randomized biomarker-stratified designs (ie, designs that use the biomarker to guide analysis but not treatment assignment) should be used to obtain a rigorous assessment of biomarker clinical utility.
Improved understanding of cancer biology and advances in biotechnology bring us closer to the concept of personalized treatment of cancer. A key component of this new paradigm is development of biomarkers that can guide application of new and existing treatments. This requires a thorough understanding of the relationship between the biomarker and the treatment effect.
Traditionally, most randomized clinical trials (RCTs) focus on obtaining a reliable estimate of the average treatment effect in a broad patient population. Evaluation of biomarkers (and targeted therapies) often requires larger trials with more complex designs to provide a comprehensive assessment of the relationship between the biomarker and the treatment effect. However, in practice, clinical studies involve a delicate balance between the need for reliable evidence, the need to provide this evidence quickly, and feasibility. As we will discuss, achieving this balance in biomarker RCTs often requires a compromise between these competing considerations in both designing and monitoring these trials.
Biomarkers that are informative for clinical outcome can be broadly categorized as prognostic or predictive biomarkers. Prognostic biomarkers classify patients treated with standard therapies (including no treatment if that is standard) into subgroups with distinct expected clinical outcomes. The types of prognostic markers considered here are those for which the prognostic information has some implications for therapy decisions. For example, if the prognostic biomarker can identify a group of patients with very low risk of recurrence, additional treatment might not be considered, whereas higher-risk patients would be treated. Predictive biomarkers identify patients whose tumors are likely to be sensitive and/or resistant to a specific agent. For example, in advanced colorectal cancer, the benefit of cetuximab appears to be limited to patients with tumors that have the wild-type KRAS genotype (1). Note that biomarkers that predict toxicity to a certain agent are often treated as a separate type of biomarker. However, for the purpose of evaluating biomarker designs, we will consider toxicity biomarkers as a type of predictive biomarker (2).
The purpose of this commentary was to provide a comprehensive comparison of the commonly used biomarker RCT designs. Ongoing or recently completed trials are used throughout the discussion as illustrative examples. Issues related to interim monitoring of biomarker trials are also discussed because standard futility and superiority monitoring may be inadequate due to the multiple subgroups and hypotheses being considered.
Design Considerations for Biomarker Studies
Establishing clinical relevance of a biomarker test for guiding therapy decisions requires demonstrating that it can classify patients into distinct subgroups with different recommended management. Conventional RCTs (with no biomarker evaluation) only allow for estimation of the average treatment effect in the overall study population, and therefore, alternative designs must be considered to evaluate biomarker-guided therapy. We discuss three main types of biomarker RCT designs: biomarker-stratified designs, enrichment designs, and biomarker-strategy designs (3–8) (Figure 1).
Figure 1.
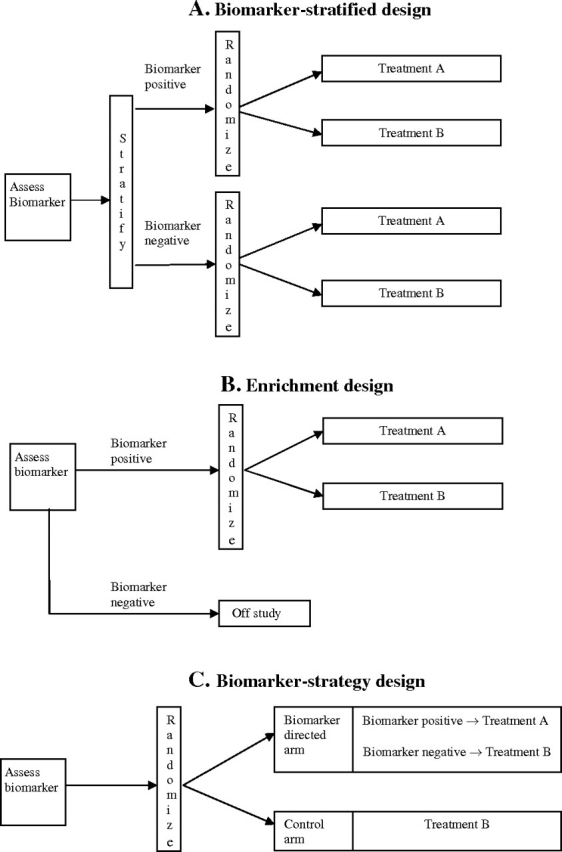
Biomarker designs. A) Biomarker-stratified design. All patients are randomly assigned regardless of biomarker status with the random assignment and analysis plan stratified by the biomarker status. Sometimes, a standard (nonstratified) randomization can be used (with the analysis plan stratified by the biomarker) when postrandomization biomarker evaluation is feasible. B) Enrichment design. The biomarker is evaluated on all patients, but random assignment is restricted to patients with specific biomarker values. C) Biomarker-strategy design. Patients are randomly assigned to an experimental treatment arm that uses the biomarker to direct therapy or to a control arm that does not. Some biomarker-strategy designs evaluate biomarkers only in patients randomly assigned to the biomarker-directed arm.
We assume throughout this presentation that the biomarker test to be evaluated in the RCT is fully specified and can effectively be treated as though it were a single measure. Additionally, we assume that discrete categories for the biomarker have been previously identified (eg, the cutoff value has been determined for a continuous biomarker to classify patients as biomarker-positive vs biomarker-negative). If this is not the case, other trials or retrospective analyses may need to be performed before a definitive RCT is initiated (9).
Biomarker-Stratified Designs
First, consider a situation in which there are two or more existing treatment options with no definitive evidence for one being preferred in a given population. In this situation, the most efficient trial design for evaluating biomarker utility is the biomarker-stratified design: All patients are randomly assigned regardless of biomarker status, but the analysis plan is centered on testing treatment effect dependence on biomarker status. For example, in a simple case with two treatments (A vs B) and two biomarker-defined subpopulations (biomarker positive vs biomarker negative), patients are randomly assigned to receive treatment A vs treatment B and their relative efficacy is evaluated in each of the two subpopulations (Figure 1, A). The biomarker-stratified design maximizes the advantage of randomization by providing unbiased estimates of benefit to risk ratios across different biomarker-defined subgroups and for the entire randomly assigned population. The precision with which treatment effects can be assessed in each of the biomarker-defined subgroups depends on the numbers of randomly assigned patients in each subgroup.
For predictive biomarkers, the biomarker-stratified design can assess whether the marker is useful in selecting the best among two or more treatments for a given patient.
Example 1. In the North Central Cancer Treatment Group (NCCTG)-0723 trial [Marker Validation for Erlotinib in Lung Cancer (MARVEL), Figure 2, A (10)], second-line advanced non–small cell lung cancer (NSCLC) patients were randomly assigned to a epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib, or to a multitargeted antifolate, pemetrexed. The trial was designed to evaluate whether epidermal growth factor receptor gene (EGFR) status, as measured by fluorescent in situ hybridization (FISH), can be used to guide treatment of these patients. The biological hypothesis was that EGFR status, as measured by FISH, predicts sensitivity to erlotinib. Consequently, in the FISH(+) subgroup, erlotinib was expected to be better than pemetrexed, and in the FISH(−) subgroup, pemetrexed was expected to be no worse than (and possibly better than) erlotinib.
Figure 2.
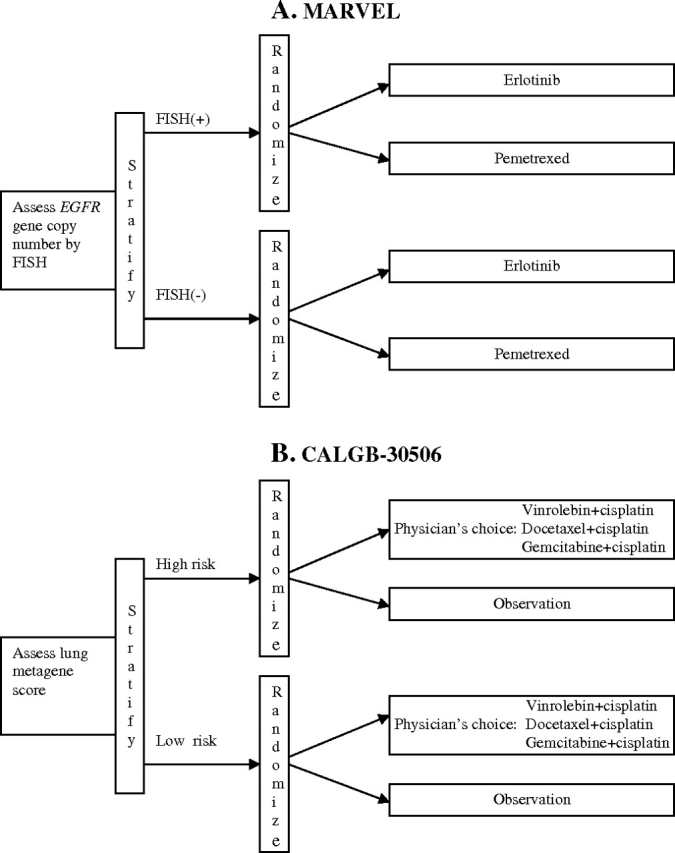
Examples of biomarker-stratified designs. A) The Marker Validation for Erlotinib in Lung Cancer (MARVEL) trial (10). Second-line advanced non–small cell lung cancer (NSCLC) patients were randomly assigned to erlotinib or pemetrexed with random assignment stratified by epidermal growth factor receptor gene (EGFR) status as measured by fluorescent in situ hybridization (FISH). B) The Cancer and Leukemia Group B (CALGB)-30506 trial (http://www.cancer.gov/clinicaltrials/CALGB-30506). Stage I NSCLC patients are randomly assigned to either chemotherapy or observation with random assignment stratified by risk group (high vs low) as defined by the Lung Metagene Score (11). Chemotherapy-arm patients receive physician choice of one of three prespecified chemotherapy regimens.
Prognostic biomarkers may or may not be useful for guiding therapy, in contrast to predictive biomarkers, which, by definition, are intended for use in selecting among treatments. If a prognostic biomarker separates patients into subgroups with increasing expected failure risk, and if it can be demonstrated that this separation can improve outcome by indicating more aggressive treatment strategies for the higher-risk group (or less aggressive treatment for the lower-risk group), then the prognostic marker has clinical utility for guiding therapy. However, if there are no effective alternative treatment strategies for the high-risk group (or less aggressive treatment strategies for the low-risk group), then the prognostic information is of limited clinical use. An example of a prognostic biomarker that is useful for guiding therapy in the adjuvant setting would be one which identifies a group of patients for whom risk of recurrence is so low that surgery alone is likely to be curative and adjuvant systemic therapy would therefore yield little benefit.
Example 2. A Cancer and Leukemia Group B trial, CALGB-30506 (http://www.cancer.gov/clinicaltrials/CALGB-30506), was designed to demonstrate benefit of adjuvant chemotherapy in stage I NSCLC patients, either overall or in a high-risk subpopulation defined by a genomic prognostic biomarker [Lung Metagene Score (11)]. Patients are stratified by their risk group (high vs low) and randomly assigned to either chemotherapy or observation (Figure 2, B).
Although biomarker-stratified designs often use randomization stratified by biomarker status (Figure 1, A), in theory, this is not necessary because the distribution of biomarker values is expected to be reasonably similar in the two treatment arms in moderate-to-large RCTs. Even if the distribution of biomarker status differed between the arms, this would not, in itself, invalidate estimates of the treatment effect within each biomarker subgroup; however, nontrivial differences in the distribution might adversely affect the sample sizes in the subgroups. If the biomarker can be evaluated retrospectively in a reliable way, then it is not necessary to obtain the biomarker status until the time of analysis. However, a key reason for evaluating biomarker status up front is to ensure that all randomly assigned patients have biomarker status determined. In particular, when the biomarker status is not evaluated up front, it is important that the study is carefully designed to anticipate a certain percentage of unavailable biomarker measurements to ensure that adequate numbers of patients are enrolled in the relevant biomarker subgroup(s) for a meaningful assessment of biomarker utility. For example, in the Iressa as a Second-Line Treatment for Advanced NSCLC (INTEREST) trial (12), in which the biomarker question was introduced retrospectively, tissue was available only for 374 out of 1466 randomly assigned patients resulting in only 174 patients used for evaluation of the biomarker question.
Enrichment Designs
In some settings, sufficiently convincing evidence is available to suggest that the potential treatment benefit is limited to a certain biomarker-defined patient subgroup. Whether or not such evidence exists, there could be a widely held perception that equipoise about the best treatment choice is present only in patients with certain biomarker values. In either case, it is not feasible to use a biomarker-stratified design, which requires random assignment of all the patients. In these situations, the clinical utility of the biomarker can be partially assessed by an enrichment trial design: the biomarker is evaluated on all patients but random assignment is restricted to patients with specific biomarker values (ie, biomarker-positive patients, Figure 1, B).
Example 3. CALGB-10603 (http://www.cancer.gov/clinicaltrials/CALGB-10603) uses a predictive biomarker to restrict eligibility to acute myeloid leukemia patients who have a documented FLT3 mutation [leading to constitutive activation of FLT3 kinase (13)] and then randomly assigns patients to a standard treatment or a standard treatment plus the FLT3 kinase inhibitor midostaurin. Patients without the FLT3 mutation are off-study.
Biomarker-Strategy Designs
The third type of biomarker design is the biomarker-strategy design: Patients are randomly assigned to an experimental treatment arm that uses the biomarker to determine therapy or to a control arm that does not. In its simplest version, patients in the control arm receive treatment B and patients in the experimental arm are treated with either treatment B or treatment A depending on their biomarker value (Figure 1, C).
Example 4. Excision repair cross-complementing 1 (ERCC1) gene expression has been suggested as a predictive biomarker associated with cisplatin resistance in NSCLC. In the ERCC1 trial, patients were randomly assigned to the control arm that received cisplatin+docetaxel or the biomarker-strategy arm that switched patients classified as cisplatin resistant to gemcitabine+docetaxel regimen while treating those nonresistant with standard cisplatin+docetaxel [Figure 3, A (14)].
Figure 3.

Examples of biomarker-strategy designs. A) The excision repair cross-complementing 1 (ERCC1) trial (14). Non–small cell lung cancer (NSCLC) patients were randomly assigned to the control arm that received cisplatin+docetaxel or the biomarker-strategy arm that switched patients classified as cisplatin-resistant to the gemcitabine+docetaxel regimen while treating those who were sensitive with cisplatin+docetaxel. B) The Tumor Chemosensitivity Assay (TCA) ovarian cancer study (15). Patients were randomly assigned to the biomarker-strategy arm that used a chemosensitivity assay that measured ATP levels in drug-treated cancer cells to choose from a panel of 12 chemotherapy regimens or to the control arm that received the physician's choice of chemotherapy.
It is possible that the biomarker-strategy experimental arm could guide decisions between three or more treatments (16). An example of this design is as follows.
Example 5. In the Tumor Chemosensitivity Assay Ovarian Cancer study, a luminescence assay that predicts chemosensitivity by measuring ATP levels in drug-treated cancer cells was performed on patients’ tumor cells to choose from a panel of 12 chemotherapy regimens in the biomarker-strategy arm. In the control arm, patients received their physicians’ choice of chemotherapy (15) (Figure 3, B).
The control arm could itself involve a random assignment of treatments (4). As an illustration, consider a modification of ERCC1 trial (Figure 3, A) that would randomly assign the patients in the control arm to either cisplatin+docetaxel or gemcitabine+ docetaxel, regardless of their ERCC1 status.
Combinations of the Biomarker Trial Designs
When several therapies targeting different molecular targets are being evaluated, use of multiple biomarkers is often required. In this case, the RCT can be based on a combination of the three biomarker designs.
Example 6. The Spanish Lung Cancer Group trial, SLCG0601, uses a combination of enrichment and biomarker-strategy designs [Figure 4, A (17)]. First, this trial design uses enrichment to restrict the stage IV NSCLC population to patients who have mutated EGFR (exon 19 or 21) in their tumors. Eligible patients are then randomly assigned to erlotinib or to a biomarker-strategy arm in which patients are assigned to either gemcitabine+cisplatin or docetaxel+cisplatin depending on methylation status of 14-3-3σ, a G2–M checkpoint control gene, in serum circulating DNA.
Example 7. In the Tarceva Italian Lung Optimization (TAILOR) study (http://www.cipomo.it/membri/documenti/protocolli/TAILORsinossi.doc), patients are assessed at the time of registration for 1) exon 19 or 21 EGFR mutations, 2) EGFR gene copy number by FISH, 3) EGFR protein expression by immunohistochemistry (IHC), and 4) KRAS mutation (Figure 4, B). Patients with exon 19 or 21 mutations are treated with erlotinib; patients without the mutation are randomly assigned to the erlotinib or docetaxel arms. By restricting the random assignment to patients without EGFR exon 19 or 21 mutations, this study uses a “reversed” enrichment approach that limits the random assignment to the subgroup that is less likely to benefit from erlotinib (erlotinib is expected to be more effective in patients with EGFR mutations). The randomized portion of the study uses a biomarker-stratified approach to compare erlotinib and docetaxel with respect to overall survival in 1) the subgroup of patients who are either KRAS(+) or [KRAS(−), EGFR FISH(−), and IHC(−)] and 2) the subgroup of patients with KRAS(−) and EGFR (FISH(+) or IHC(+)); docetaxel is expected to be better than erlotinib in subgroup 1, and the reverse is expected in subgroup 2.
Figure 4.
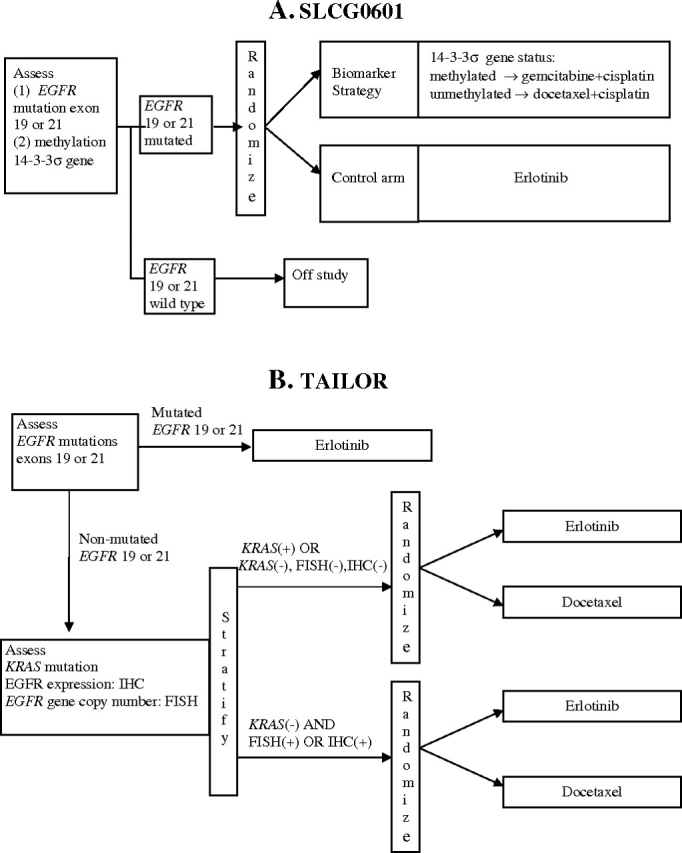
Examples of combination designs. A) The Spanish Lung Cancer Group (SLCG) 0601 trial (17). This trial uses a combination of enrichment and biomarker-strategy designs. First, enrichment is used to restrict the stage IV non–small cell lung cancer (NSCLC) population to patients who have mutated epidermal growth factor receptor (EGFR) genes (exon 19 or 21) in their tumors. Eligible patients are then randomly assigned to a control arm (erlotinib) or to a biomarker-strategy arm in which patients are assigned either to gemcitabine+cisplatin or to docetaxel+cisplatin depending on 14-3-3σ gene methylation status. B) The Tarceva Italian Lung Optimization (TAILOR) study (http://www.cipomo.it/membri/documenti/protocolli/TAILORsinossi.doc). NSCLC patients are assessed at the time of registration for 1) exon 19 or 21 EGFR mutations, 2) EGFR gene copy number by fluorescent in situ hybridization (FISH), 3) EGFR protein expression by immunohistochemistry (IHC), and 4) KRAS mutation. Patients with EGFR exon 19 or 21 mutations are treated with erlotinib; patients without the mutation are randomly assigned to the erlotinib or docetaxel arms, with random assignment stratified by EGFR gene copy number, EGFR protein expression, and KRAS mutation status.
Many of the trials using biomarker designs include important subgroups of patients whose treatment is not determined by random assignment. For example, in the Program for the Assessment of Clinical Cancer Trials’ Trial Aligning Individualized Options for Treatment, PACCT-1 TAILORx (18), in addition to randomized evaluation of the need for chemotherapy in the intermediate genomic risk breast cancer patients (enrichment design), the study includes a low-risk arm treated with hormonal therapy and a high-risk arm treated with a chemohormonal regimen. However, results from the nonrandomized components of such a trial must be interpreted with caution because the evidence obtained from nonrandomly selected patient subgroups is less reliable than that obtained from the main randomized component (19).
Statistical and Practical Considerations for Biomarker Trials
Inefficiency of Biomarker-Strategy Design
The biomarker-strategy design seems to address the relevant question by comparing the new personalized treatment strategy arm based on the biomarker to the standard-approach arm that does not consider the biomarker. However, the statistical properties of the biomarker-strategy design are problematic. In the biomarker-strategy approach, a certain (potentially nontrivial) proportion of study patients would receive, by design, the same treatment on either arm. For example, in the ERCC1 trial, 57% of the biomarker-strategy arm patients were assigned to the same cisplatin+docetaxel regimen as those in the control arm (14). Including patients receiving the same treatment in both arms in a randomized comparison will dilute the between-arm treatment difference and reduce the statistical power to reject null hypotheses as compared with an enrichment- or biomarker-stratified design (see Appendix 1). Therefore, use of the biomarker-strategy approach could, in some cases, lead to either missing a valuable biomarker or to an unacceptable and unnecessary delay in its evaluation (because of increased sample size required to maintain adequate power).
Another issue with the biomarker-strategy design is that a positive study cannot distinguish between a successful treatment selection strategy and a situation in which some of the treatment options on the experimental arm are better than the control arm therapy in all patients. For example, suppose a study described by Figure 1, C, demonstrated a benefit for the experimental arm compared with the control arm. Then, in theory, it is possible that the biomarker is totally useless and treatment A is better than treatment B overall, not just in the biomarker-positive subpopulation. By contrast, the biomarker-stratified design allows one to address the optimal treatment for all subpopulations: An adequately sized design that randomly assigns patients to treatment A or treatment B stratified by the biomarker value (Figure 1, A) will provide rigorous evidence for determining the best treatment in the biomarker-positive and biomarker-negative subgroups.
Limitations of Enrichment Design
The enrichment design may seem to be an attractive alternative to the biomarker-strategy design. Moreover, it has been shown that limiting random assignment to biomarker-positive patients is generally more efficient compared with the standard approach of randomly assigning and analyzing all patients together (20). However, this improvement in efficiency (for the enrichment design) is relative to the overall comparison that disregards the biomarker status. The same efficiency comparison does not apply to situations in which the competing design is the biomarker-stratified design. In addition, the biomarker-stratified design avoids a limitation of the enrichment design that one must be confident that the biomarker can identify the subpopulation of patients who benefit with reasonable accuracy; if the targeted therapy actually benefits all patients equally regardless of biomarker status, then enrolling only biomarker-positive patients will slow trial accrual, increase expense, and unnecessarily limit the size of the indicated patient population. If the targeted therapy truly benefits some subset of patients, but the biomarker used for enrichment does not correctly identify that group, then a beneficial therapy could mistakenly be abandoned.
Efficacy vs Effectiveness and Biomarker Trial Designs
An ongoing methodological debate in the clinical trial community centers on the differences between estimating treatment “efficacy” (the biological effect under ideal conditions) and “effectiveness” (the effect achieved when the treatment is used in broad clinical practice) (21). This issue may appear relevant in the present setting because one could argue that the biomarker-strategy design provides a more realistic estimate of effectiveness than the biomarker-stratified design by naturally accounting for compliance and biomarker measurement issues and for the fact that components of the biomarker-directed therapy will be the standard treatment in a certain proportion of the patients. However, in most situations, the biomarker-stratified design can provide all necessary information for assessing effectiveness in a more efficient way by estimating the treatment effect in all relevant biomarker subpopulations, possibly including the subpopulation of subjects without available biomarker values (see below). The effect of noncompliance is automatically incorporated into the estimates from a biomarker-stratified design. Therefore, even if the effectiveness question is of primary interest, the biomarker-stratified design should be used when feasible.
Missing Biomarker Status
An important practical consideration in biomarker RCTs is that biomarker measurements will usually not be available for some fraction of patients. This unavailability may happen for logistical reasons (eg, specimens not submitted), technical reasons (eg, insufficient amount of specimen, inadequate specimen quality, or assay failure), or clinical reasons (eg, tumor inaccessible or too small to be biopsied). Ideally, the proportion of patients with unavailable biomarker status should be kept small. The study protocol should provide an estimate for this unavailability rate (for sample size calculation) and clearly specify how these patients will be treated and analyzed. In biomarker-strategy designs, patients with unavailable biomarker status are often taken off study. When this is done only in the strategy arm (as was done in ERCC1 trial), concerns may arise about bias being introduced because the strategy-arm patients may no longer be comparable to the control-arm patients. Even if the unavailability rate is not high, situations in which the unavailability is related to prognosis may point to a potential problem with study interpretation and generalizability. For example, in Grignon et al (22), patients with available p53 status tended to have higher Gleason scores and higher clinical stage. Therefore, one may want to collect follow-up and prognostic variable data on patients with unavailable biomarker status.
Compliance Issues
In any design in which knowledge of the biomarker status may affect compliance to the randomized treatment, patient or physician access to the biomarker values can impair interpretability of the study (this may be particularly pertinent to designs in which the biomarker is measured but is not used in guiding treatment, as is the case for the biomarker-stratified design or sometimes the control arm of the biomarker-strategy design). If this is a concern, then it is advisable to withhold the biomarker status from each patient until the study endpoint is reached for that patient (eg, recurrence for trials with a disease-free survival endpoint). In theory, an alternative approach is to ascertain the biomarker status retrospectively after the study is completed. However, this works only when the status can be reliably determined retrospectively and estimates of the biomarker positivity and unavailability rates are available for use in sample size calculations. Even if biomarker status is withheld, there are still situations in which biomarker status may correlate with clinicopathologic features of the patient that might influence treatment preference. For example, EGFR mutation status correlates with Asian ethnicity, female gender, and adenocarcinoma histology (23). These features are prognostic, and there may also be a bias toward treating these patients with an EGFR inhibitor. Therefore, noncompliance and its associated biases may not be entirely avoidable even if biomarker measurements are withheld.
Practical Considerations Favoring the Biomarker-Strategy Design
There are two practical considerations that may favor the biomarker-strategy design over the biomarker-stratified design. First, not all biomarker strategies can be evaluated in a biomarker-stratified design. Since the biomarker-stratified design involves randomizing patients between all possible treatments, testing strategies with a large number of treatment options is not practical. Moreover, some of the possible treatments may not be appropriate for some of the biomarker subgroups. (This contingency can sometimes be addressed in a biomarker-stratified design by limiting the randomization options in each subgroup to the subset of acceptable treatments for that subgroup.) Second, biomarker evaluation is necessary in the biomarker-strategy design only in the patients randomly assigned to the biomarker-strategy arm. In addition to the economic advantage of limiting the number of potentially expensive biomarker assessments, this approach indirectly solves logistical issues related to the access of control-arm patients to their biomarker status. However, this consideration alone could not generally justify use of the biomarker-strategy design in all situations (because of its statistical deficiencies). The ultimate consideration for the design choice is whether the existing evidence on the optimal treatment for patients with certain biomarker values upsets the equipoise required for randomly assigning these patients in the biomarker-stratified design.
A summary of the key advantages and disadvantages for the three biomarker designs is presented in Table 1. To guide design of new trials and to assist interpretation of existing studies, the table also lists the main research questions that can and cannot be answered by each of the designs. Results of ongoing and future trials will additionally clarify the practical advantages and disadvantages of the designs.
Table 1.
Comparison of the key features of the biomarker designs
| Feature | Biomarker-stratified design | Enrichment design | Biomarker-strategy design, with biomarker assessment in the control arm | Biomarker-strategy design, without biomarker assessment in the control arm |
| Questions design can answer | What is the best treatment in each biomarker-defined subgroup? What is the best treatment in the overall study population? |
What is the best treatment in the biomarker-positive patients? | Is the biomarker-directed treatment strategy better than the control treatment in the overall study population? (direct assessment) | Is the biomarker-directed treatment strategy better than the control treatment in the overall study population? (direct assessment) |
| Is the biomarker-directed treatment strategy better than the control in the overall study population? (indirect assessment) | What is the best treatment in the biomarker-positive subgroup? Is the biomarker prognostic? |
What is the best treatment in the biomarker-positive subgroup? (indirect assessment) Is the biomarker prognostic? (indirect assessment) |
||
| Is the biomarker prognostic? Predictive? | ||||
| Questions design cannot answer | What is the best treatment in the biomarker-negative subgroup? | What is the best treatment in the biomarker-negative subgroup? | What is the best treatment in the biomarker-negative subgroup? | |
| Is the biomarker prognostic? Predictive? | Is the biomarker predictive? | Is the biomarker predictive? | ||
| Advantages | Provides efficient assessment of relative treatment efficacy in each biomarker-defined subgroup and in the whole group | If the assumption that the biomarker reliably identifies the group likely to benefit from the experimental therapy is true, then the design provides an efficient test of efficacy of the experimental treatment in that subgroup, particularly if the biomarker positivity rate is low | Can be used for evaluation of complex biomarker-guided treatment strategies with a large number of treatment options or biomarker categories | Biomarker assessment is limited to the biomarker-directed arm (resource consideration) No issues associated with withholding the biomarker status from the control-arm patients Compliance not influenced by patient knowledge of the biomarker status in the control arm Can be used for evaluation of complex biomarker-guided treatment strategies with a large number of treatment options or biomarker categories |
| Disadvantages | The design is not feasible for evaluation of biomarker strategies with a large number of treatment options | If the experimental therapy is beneficial in a subgroup but the biomarker does not correctly identify this subgroup, a promising therapy may be missed | A positive trial does not prove the utility of the biomarker because the experimental treatment may be better than the control treatment for all patients regardless of biomarker status | A positive trial does not prove the utility of the biomarker because the experimental treatment may be better than the control treatment for all patients regardless of biomarker status |
| A positive trial does not prove the utility of the biomarker because the relative treatment efficacy may be the same in the unevaluated biomarker-negative patients | Inefficiency | Inefficiency |
Interim Monitoring
Interim monitoring for efficacy and futility is a critical component of any RCT design (24). Below, we discuss considerations related to interim monitoring, both in general and in the context of biomarker trials.
Interim Monitoring for Trials Without Biomarker Evaluations
In an RCT that does not use biomarkers, interim monitoring is relatively simple and can be based on values of the treatment-effect estimator in the study population at prespecified times. Most RCTs are designed to show that the experimental therapy (A) is better than the control therapy (B). Interim monitoring plans for such superiority trials typically include a superiority (efficacy) monitoring rule that allows stopping for early convincing evidence that the experimental arm A is better than the control arm B with respect to some relevant clinical outcome. Common superiority monitoring rules [eg, the O'Brien-Fleming boundary (25)] require very strong evidence that arm A is better than arm B (eg, a P value < .0005) for stopping in the first half of the trial and use a less stringent criterion for stopping in the second half of the trial (24). In addition to being monitored for superiority, an RCT should be monitored for lack of benefit (futility). For RCTs that are designed to show that a new therapy (A) is better than the control (B), there is generally no need to provide the same degree of evidence that B is better than A to stop the study for futility as needed to stop for superiority (showing that A is better than B). In the second half of the study, common futility rules often recommend stopping the trial unless a minimal positive trend in favor of the new therapy is observed. If the experimental therapy is at least as toxic as the control treatment, futility monitoring should commence earlier than half way through the study. In some situations, an indication that arm A is no worse than arm B may still be clinically relevant (eg, when a new agent with a more favorable toxicity profile is compared to the standard active chemotherapy). In this case, a more conservative futility boundary that allows the study to continue unless the new therapy appears worse is appropriate (26).
Noninferiority trials, in contrast to superiority trials, are designed to show that a standard treatment (B) may be replaced by a lesser treatment (A). For example, the goal might be to determine whether a standard chemotherapy can be substituted by a new less toxic agent without loss of efficacy. There are two ways for a noninferiority trial to be stopped early: 1) when it becomes clear that A is inferior to B or 2) when it becomes clear that A is noninferior to B. The first way is more critical because it minimizes possibility of patients not getting an effective standard therapy. However, if it becomes clear that A is not inferior to B, there still may be scientific value in continuing the study to refine the understanding of the risk to benefit ratio while patients on both arms are receiving an apparently effective therapy. Thus, a more conservative interim monitoring (or possibly no interim monitoring) is appropriate in this case.
Interim Monitoring for Biomarker Trials
The monitoring rules described in the above section are also appropriate for straightforward implementations of the biomarker-strategy and enrichment designs that focus on comparing the overall efficacy between the randomized arms. For example, the biomarker-strategy trials and enrichment trials (examples 3–6) can use the standard superiority and futility interim monitoring.
For the biomarker-stratified designs, the situation can become more complex because there may be multiple potentially overlapping patient subgroups and/or multiple hypotheses under consideration. To protect patient interests, it may be necessary to stop the trial (or some of its components) before all of the study objectives are definitively addressed. Conventional monitoring rules that are based on the observed treatment effect in the overall randomized population may often not be sensitive enough for timely stopping based on biomarker subgroup–specific trends in treatment effect. Ideally, the monitoring rule should be able to stop the study in the subgroup(s) for which the therapeutic question has been answered while continuing the subgroups that have open questions.
Example 1 (MARVEL) continued. Interim monitoring in the EGFR FISH(+) subgroup used standard superiority and futility boundaries for testing that erlotinib was better than pemetrexed. If either boundary is crossed in the FISH(+) subgroup, accrual to that subgroup is stopped. In the FISH(−) subgroup, a symmetric superiority boundary was used to stop accrual to this cohort only for strong evidence of difference (in either direction) between the two arms.
Example 2 (CALGB-30506) continued. The trial uses the following monitoring plan: 1) stop the entire study if the chemotherapy superiority boundary for the overall population is crossed, 2) stop accrual to the high-risk subgroup only if the superiority boundary for only the high-risk subgroup is crossed, 3) stop the entire study if the futility boundary for the high-risk subgroup is crossed, and 4) stop accrual to the low-risk subgroup if a conservative futility boundary specified for the low-risk group is crossed.
Example 7 (TAILOR) continued. The primary analysis of this study is based on first testing for a treatment-by-biomarker interaction in the randomized population overall. However, separate futility (and possibly superiority) monitoring should be implemented in each of the two biomarker subgroups.
Note that when the superiority boundary in the biomarker-positive subgroup is crossed (in a biomarker-stratified design), some of the monitoring rules above recommend stopping the biomarker-positive subgroup and continuing the biomarker-negative subgroup (eg, in CALGB-30506 and MARVEL). In studies that are designed to establish treatment benefit either in the overall study population or in the subgroup of biomarker-positive patients and that are not sized for a separate definitive evaluation in the biomarker-negative subgroup (eg, CALGB-30506), continuing the biomarker-negative subgroup after the biomarker-positive subgroup has been stopped may require adjusting the analysis plan. For studies with rapid accrual and/or low event rates (eg, the adjuvant setting like CALGB-30506) in which crossing a superiority boundary (especially in a subgroup) is likely to occur after completion of accrual, this potential adjustment may be just a minor issue: The results for the biomarker-positive patients can be released immediately, and the overall comparison can be performed after additional follow-up (assuming that the release of the biomarker-positive results does not affect how the biomarker-negative patients on the trial are treated or followed). However, in a more advanced disease setting, the biomarker-positive subgroup may be stopped for benefit before the study accrual is completed. In this case, it may be useful to consider increasing target accrual for the biomarker-negative subgroup to better understand the biomarker's ability to identify patients who benefit from the new therapy (7).
Another issue is whether the entire study should be stopped if the biomarker-positive subgroup is stopped for futility. Once absence of a treatment effect in the biomarker-positive subgroup has been accepted, expecting a treatment effect in the biomarker-negative subgroup would generally refute the underlying biological rationale, thus suggesting that the entire study should be stopped (especially in studies that are not powered for a separate evaluation of the biomarker-negative subgroup, eg, CALGB-30506).
Summary
We have reviewed common phase III biomarker RCT designs and have shown that in most settings the biomarker-strategy and enrichment designs do not provide complete information on the relationship between the treatment effect and the biomarker. (A possible exception is the use of the enrichment designs in development of targeted agents.) Therefore, when possible, the biomarker-stratified designs should be used to obtain a rigorous assessment of biomarker clinical utility. However, proper implementation of the biomarker-stratified designs requires special interim monitoring rules to balance scientific and ethical considerations.
Funding
This work was done as an official duty by US Federal employees.
Appendix 1: Efficiency of the Enrichment and Biomarker-Stratified Designs Relative to the Biomarker-Strategy Design
The following presentation uses a time-to-event endpoint that is typical of phase III RCTs for cancer. We first derive an approximate formula for the sample size needed in each of the biomarker designs. It is assumed that all random assignments use 50:50 patient allocation.
Appendix Table 1.
Sample sizes* needed to achieve 90% power at a .025 one-sided significance level for a hazard ratio of 0.7 in the biomarker-positive subgroup (assuming 3 years of accrual and 2 years of follow-up)
| Biomarker positive, % |
||||||
| Design | 100 | 80 | 60 | 50 | 40 | 30 |
| Strategy | 392 | 600 (613) | 1008 (1089) | 1440 (1568) | 2220 (2450) | 3880 (4356) |
| Enrichment or stratified | 392 | 490 | 654 | 784 | 980 | 1307 |
Given in parentheses are the sample size estimates based on the approximate formula for the biomarker-strategy design (Dstrategy).
Consider the biomarker-strategy design in Figure 1, C. Let p denote the proportion of patients who have a biomarker-positive status (ie, patients with known biomarker status who are biomarker-positive), and θ (<1) denote the target hazard ratio (treatment A vs treatment B) in this biomarker-positive population. The overall hazard ratio between experimental and control arms can be approximated by 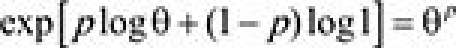 (assuming that [1] in the biomarker-negative population, the hazard ratio is one and [2] there is no prognostic effect of the marker under treatment B). The required number of events, Dstrategy, needed to achieve power (1−β) at significance level α is approximately
(assuming that [1] in the biomarker-negative population, the hazard ratio is one and [2] there is no prognostic effect of the marker under treatment B). The required number of events, Dstrategy, needed to achieve power (1−β) at significance level α is approximately 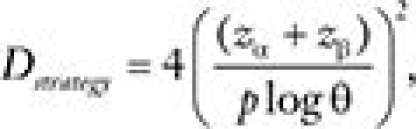 , where zγ denotes the γ quantile of a standard normal distribution.
, where zγ denotes the γ quantile of a standard normal distribution.
For the enrichment design comparing treatments A and B in the biomarker-positive patients, the number of events (for the same power and significance level) is 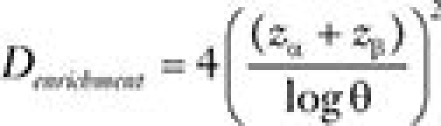 . This means that Dstrategy/Denrichment =1/p2 is the ratio of the number of events required in the biomarker-strategy design relative to the enrichment design. The above calculation does not take into account that in the enrichment design only the biomarker-positive patients are randomly assigned, and therefore, 1/p times more patients will need to be screened (have their biomarker assessed) for a given number of randomly assigned patients. Thus, a more appropriate comparison is between the number of patients needed to be randomly assigned in the biomarker-strategy design (Nstrategy) and the number of patients needed to be screened for the enrichment design (Nenrichment). Therefore, for a simple case where prognostic value of biomarker is ignored, the ratio of required sample sizes to achieve the same power can be approximated by:
. This means that Dstrategy/Denrichment =1/p2 is the ratio of the number of events required in the biomarker-strategy design relative to the enrichment design. The above calculation does not take into account that in the enrichment design only the biomarker-positive patients are randomly assigned, and therefore, 1/p times more patients will need to be screened (have their biomarker assessed) for a given number of randomly assigned patients. Thus, a more appropriate comparison is between the number of patients needed to be randomly assigned in the biomarker-strategy design (Nstrategy) and the number of patients needed to be screened for the enrichment design (Nenrichment). Therefore, for a simple case where prognostic value of biomarker is ignored, the ratio of required sample sizes to achieve the same power can be approximated by:  .
.
The biomarker-stratified design randomly assigns both biomarker-positive and biomarker-negative patients. Thus, to compare treatments A and B among biomarker-positive patients, the sample size required by the biomarker-stratified design for detecting a given effect size in the biomarker-positive subgroup is identical to that required by an enrichment design (sample size equal all patients screened) targeting the same effect size.
The table below tabulates sample sizes needed for the different biomarker designs as a function of the proportion of biomarker-positive patients. Both exact sample sizes (obtained by simulations) and approximate sample sizes (obtained from the formula derived above) are presented (approximate numbers are in parentheses). Although the formula provides a relatively rough approximation to the exact sample size, it can be used as a simple way to compare the designs in a given setting.
From the cost analysis perspective, it should be mentioned that although the enrichment and biomarker-stratified designs have the same biomarker testing expenses, the cost of on-study treatment and follow-up is higher for the biomarker-stratified design.
Footnotes
The authors take full responsibility for the collection, analysis, or interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
References
- 1.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2004;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 2.Mandrekar SJ, Grothey A, Goetz MP, Sargent DJ. Clinical trial designs for prospective validation of biomarkers. Am J Pharmacogenomics. 2005;5(5):317–325. doi: 10.2165/00129785-200505050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Pusztai L, Hess KR. Clinical trial design for microarray predictive marker discovery and assessment. Ann Oncol. 2004;15(12):1731–1737. doi: 10.1093/annonc/mdh466. [DOI] [PubMed] [Google Scholar]
- 4.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23(9):2020–2027. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 5.Therasse P, Carbonnelle S, Bogaerts J. Clinical trials design and treatment tailoring: general principles applied to breast cancer research. Crit Rev Oncol Hematol. 2006;59(2):98–105. doi: 10.1016/j.critrevonc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Buyse M. Towards validation of statistically reliable biomarkers. Eur J Cancer Suppl. 2007;5(5):89–95. [Google Scholar]
- 7.Simon R. The use of genomics in clinical trial design. Clin Cancer Res. 2008;14(19):5984–5993. doi: 10.1158/1078-0432.CCR-07-4531. [DOI] [PubMed] [Google Scholar]
- 8.Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J Clin Oncol. 2009;27(24):4027–4034. doi: 10.1200/JCO.2009.22.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ransohoff DF. The process to discover and develop biomarkers for cancer: a work in progress. J Natl Cancer Inst. 2008;100(20):1419–1420. doi: 10.1093/jnci/djn339. [DOI] [PubMed] [Google Scholar]
- 10.Wakelee H, Kernstine K, Vokes E, et al. Cooperative group research efforts in lung cancer 2008: focus on advanced-stage non-small-cell lung cancer. Clin Lung Cancer. 2008;9(6):346–351. doi: 10.3816/CLC.2008.n.050. [DOI] [PubMed] [Google Scholar]
- 11.Potti A, Mukherjee S, Petersen R, et al. A genomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N Engl J Med. 2006;355(6):570–580. doi: 10.1056/NEJMoa060467. [DOI] [PubMed] [Google Scholar]
- 12.Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372(9652):1809–1818. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- 13.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 14.Cobo M, Isla D, Massuti B, et al. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: a phase III trial in non-small-cell lung cancer. J Clin Oncol. 2007;25(19):2747–2754. doi: 10.1200/JCO.2006.09.7915. [DOI] [PubMed] [Google Scholar]
- 15.Cree IA, Kurbacher CM, Lamont A, et al. A prospective randomized controlled trial of tumour chemosensitivity assay directed chemotherapy versus physician's choice in patients with recurrent platinum-resistant ovarian cancer. Anticancer Drugs. 2007;18(9):1093–1101. doi: 10.1097/CAD.0b013e3281de727e. [DOI] [PubMed] [Google Scholar]
- 16.Rosell R, Vergnenegre A, Fournel P, et al. Pharmacogenetics in lung cancer for the lay doctor. Targeted Oncol. 2008;3(3):161–171. [Google Scholar]
- 17.Rosell R, Taron M, Sanchez JJ, et al. Setting the benchmark for tailoring treatment with EGFR tyrosine kinase inhibitors. Future Oncol. 2007;3(3):277–283. doi: 10.2217/14796694.3.3.277. [DOI] [PubMed] [Google Scholar]
- 18.Sparano JA. TAILORx: trial assigning individualized options for treatment (Rx) Clin Breast Cancer. 2006;7(4):347–350. doi: 10.3816/CBC.2006.n.051. [DOI] [PubMed] [Google Scholar]
- 19.Ransohoff DF. Bias as a threat to the validity of cancer molecular-marker research. Nat Rev Cancer. 2005;5(2):142–149. doi: 10.1038/nrc1550. [DOI] [PubMed] [Google Scholar]
- 20.Simon R, Maitournam A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clin Cancer Res. 2004;10(20):6759–6763. doi: 10.1158/1078-0432.CCR-04-0496. [DOI] [PubMed] [Google Scholar]
- 21.Piantadosi S. Clinical Trials: A Methodologic Perspective. New York, NY: Wiley; 1997. [Google Scholar]
- 22.Grignon DJ, Caplan R, Sarkar FH, et al. p53 status and prognosis of locally advanced prostatic adenocarcinoma: a study based on RTOG 8610. J Natl Cancer Inst. 1997;89(2):158–165. doi: 10.1093/jnci/89.2.158. [DOI] [PubMed] [Google Scholar]
- 23.Tsao AS, Tang XM, Sabloff B, et al. Clinicopathologic characteristics of the EGFR gene mutation in non-small cell lung cancer. J Thorac Oncol. 2006;1(3):231–239. doi: 10.1016/s1556-0864(15)31573-2. [DOI] [PubMed] [Google Scholar]
- 24.Jennison C, Turnbull BW. Group Sequential Methods With Applications To Clinical Trials. Boca Raton, FL: Chapman & Hall/CRC; 2000. [Google Scholar]
- 25.O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35(3):549–556. [PubMed] [Google Scholar]
- 26.Freidlin B, Korn EL. Monitoring for lack of benefit: a critical component of a randomized clinical trial. J Clin Oncol. 2009;27(4):629–633. doi: 10.1200/JCO.2008.17.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]