

Abstract
The anion transporter 1 (ANTR1) from Arabidopsis thaliana, homologous to the mammalian members of the solute carrier 17 (SLC17) family, is located in the chloroplast thylakoid membrane. When expressed heterologously in Escherichia coli, ANTR1 mediates a Na+-dependent active transport of inorganic phosphate (Pi). The aim of this study was to identify amino acid residues involved in Pi binding and translocation by ANTR1 and in the Na+ dependence of its activity. A three-dimensional structural model of ANTR1 was constructed using the crystal structure of glycerol 3-phosphate/phosphate antiporter from E. coli as a template. Based on this model and multiple sequence alignments, five highly conserved residues in plant ANTRs and mammalian SLC17 homologues have been selected for site-directed mutagenesis, namely, Arg-120, Ser-124, and Arg-201 inside the putative translocation pathway and Arg-228 and Asp-382 exposed at the cytoplasmic surface of the protein. The activities of the wild-type and mutant proteins have been analyzed using expression in E. coli and radioactive Pi transport assays and compared with bacterial cells carrying an empty plasmid. The results from Pi- and Na+-dependent kinetics indicate the following: (i) Arg-120 and Arg-201 may be important for binding and translocation of the substrate; (ii) Ser-124 may function as a transient binding site for Na+ ions in close proximity to the periplasmic side; (iii) Arg-228 and Asp-382 may participate in interactions associated with protein conformational changes required for full transport activity. Functional characterization of ANTR1 should provide useful insights into the function of other plant and mammalian SLC17 homologous transporters.
Solute and metabolite transport proteins are essential for many physiological processes in an organism such as growth, nutrition, cell homeostasis, signal transduction, and stress responses. In plant chloroplasts, where photosynthesis takes place, the vast majority of characterized transporters are from the inner envelope membrane, whereas only a few have been identified in the thylakoid membrane (for recent reviews, see refs (1−3)).
Inorganic phosphate (Pi) is a solute essential for ATP synthesis during the photosynthetic light reactions, and it is supplied in the chloroplast stroma by several types of envelope transporters (for reviews, see refs (4) and (5)). The first thylakoid Pi transporter has been recently identified in Arabidopsis thaliana (Arabidopsis). This is anion transporter 1 (ANTR1)1, which has been initially localized to the chloroplast using labeling with the green fluorescent protein and fluorescent microscopy and most recently to the thylakoid membrane using peptide-specific antibodies (6,7). Its expression is restricted to photosynthetic tissues and follows a circadian rhythm (8,9). ANTR1 has been characterized as a Na+-dependent Pi transporter when expressed in Escherichia coli (E. coli) and as a H+-dependent Pi transporter when expressed in yeast (7,8). Therefore, this protein is also known as PHT4;1 (8). It is presumed to function as a symporter, but the nature of the cotransported ion in Arabidopsis chloroplasts has not yet been investigated. As for a role in planta, it has been proposed to export Pi produced during nucleotide metabolism in the thylakoid lumen (10) to the chloroplast stroma.
The ANTR, alias PHT4, family belongs to the ubiquitous major facilitator superfamily (MFS) and consists of six members in Arabidopsis, sharing 30−65% sequence identity (6). They display 29−34% sequence identity with the mammalian members of the solute carrier 17 (SLC17) family, including sialin, vesicular glutamate transporters (VGLUTs), and Na+/Pi cotransporters (NPTs) (6). Notably, ANTRs selectively transport Pi7,8, in contrast to SLC17 members, displaying a broad range of anionic substrates, such as sialic acid, glutamate, organic acids, Pi, and chloride (for a review, see ref (11) and references cited therein).
Several prokaryotic MFS transporters, namely, glycerol 3-phosphate/phosphate antiporter (GlpT), lactose/H+ symporter (LacY), multidrug resistance protein D (EmrD), all three from E. coli, and the oxalate/formate antiporter (OxlT) from Oxalaobacter formigenes, have been crystallized (12−15). GlpT, LacY, and OxlT three-dimensional (3-D) structures have been obtained in the cytoplasmic-facing orientation, whereas the EmrD structure is available in an intermediate conformation. The current view is that the structure of most MFS members consists of two six-helix bundles connected by a large loop and with both termini facing the cytoplasm. A single substrate-binding site, which has alternating access to either side of the membrane (rocker switch), has been proposed to mediate solute transport by GlpT and LacY (16,17). Among SLC17 proteins, this topology has been validated for VGLUT2, using antibodies against various epitopes (18). It is likely that many other structural and functional features of GlpT and LacY are valid for SLC17 members.
In this study we have identified five potentially important amino acids for the function of Arabidopsis ANTR1 in Pi transport, based on multiple sequence alignments (MSAs) with other plant ANTRs and mammalian SLC17 homologues and homology modeling using GlpT as a template. Site-directed mutagenesis combined with activity assays in transformed E. coli cells verified the importance of these residues for Pi transport. Our findings may have relevance for elucidation of the mechanism of transport by SLC17-type of transporters.
Experimental Procedures
Site-Directed Mutagenesis and Expression in E. coli
The pTrcHisC plasmid (Invitrogen, U.K.) was used for expressing the His6-Xpress-ANTR1-FLAG fusion protein in E. coli TOP10F′ cells (Invitrogen, U.K.) as previously described (7). The mutants were prepared by oligonucleotide-directed amino acid-specific mutagenesis using the Stratagene Quikchange II kit (La Jolla, CA). The oligonucleotides used are listed in Supporting Information Table S1. The PCR products were subcloned into the plasmid containing the wild-type ANTR1 (WT) cDNA and sequenced to verify the substitutions.
Transport Assay
Cells transformed with WT or mutant cDNA as well as control (carrying an empty plasmid) cells were assayed as previously described (7). Briefly, cells were precultivated overnight in standard Terrific broth medium, containing 70 mM potassium phosphate buffer (TB, pH 7.5) supplemented with 50 μg/mL ampicillin, and reinoculated to an optical density (A600) of 0.6 in fresh TB medium, supplemented with 50 μg/mL ampicillin and 100 μM isopropyl 1-thio-β-d-galactopyranoside (IPTG) for induction for 4 h at 25 °C. The cells were then harvested by centrifugation, resuspended to an A600 of 0.3 in TB medium prepared without the addition of Pi, and starved for 1 h. Cells were harvested by centrifugation and washed with 25 mM Tris−succinate buffer (pH 6.5). Pi uptake was assayed by the addition of 1 μL of [32P]orthophosphate (50 mCi/mmol; 1 mCi = 37 MBq; Perkin-Elmer, USA) to a final concentration of 100 μM in 20 μL aliquots, each containing 2 mg (wet weight) of cells in Tris−succinate buffer (pH 6.5), supplemented with 3% glucose, in the presence of 25 mM NaCl or choline chloride. The suspension was incubated for 3 min at 25 °C. Pi transport was terminated by the addition of 1 mL of ice-cold 25 mM Tris−succinate buffer, pH 6.5, and rapid filtration under vacuum (7). After three washes with the same ice-cold buffer, the radioactivity retained on the filter was determined by liquid scintillation spectrometry. Results are the average of two to three independent experiments performed in triplicate ± SD.
For determination of the apparent Pi transport affinity (Km) and maximal rate (Vmax), uptake of 32Pi was carried out for 3 min using a range of concentrations between 0 and 200 μM in the presence of 25 mM NaCl. For the analysis of Na+-dependent kinetics, the cells were incubated for 3 min with 100 μM Pi and the indicated concentrations of NaCl (0−25 mM). Osmolarity was kept constant with choline chloride. Results were analyzed using the Michaelis−Menten equation by nonlinear regression (Prism 5, GraphPad software). Km and Vmax are means of two to three independent experiments performed in triplicate ± SE.
SDS−PAGE and Western Blotting
To verify expression levels of the WT and mutant FLAG-tagged proteins, E. coli cells induced with IPTG for 4 h were harvested by centrifugation. The cells were then resuspended and lysed for 30 min on ice in 20 mM Tris-HCl (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride, 1% (w/v) Triton X-100, 1% dodecyl maltoside, protease inhibitor cocktail (Sigma), and 1 mg/mL lysozyme (Sigma), followed by sonication. Thereafter, the cell lysate was incubated for 2 h under gentle agitation at 4 °C, followed by centrifugation for 30 min at 20000g, to remove cell debris and vesicles. Equivalent amounts of protein lysate (1 A600 unit per lane) were separated in 12% (w/v) polyacrylamide−SDS gels and transferred to PVDF membranes (Millipore, Bedford, MA). Thereafter, the membranes were probed with primary anti-FLAG M2 monoclonal antibodies (Sigma), followed by incubation with secondary anti-mouse horseradish peroxidase-conjugated antibodies and chemiluminescent substrate kit (GE Healthcare, U.K.). To quantify the protein expression levels in various mutants, densitometry analysis of the protein band signals on blots was performed using the ImageJ software (NIH, Bethesda, MD). Where indicated, an ANTR1-specific antibody was also used to verify that the anti-FLAG antibody detected signal is the ANTR1 protein, as previously described (7).
Multiple Sequence Alignments
To identify functionally important amino acid residues in the ANTR1 sequence, a conservation analysis was performed based on four different MSAs calculated using MUSCLE (19). The first MSA was obtained using the amino acid sequences of the six ANTR members from Arabidopsis. The second and third MSAs were obtained from a homology search performed at the ARAMEMNON plant membrane protein database (20) with criteria of 36% (7 sequences) and 20% (13 sequences) identity, respectively. The fourth MSA was obtained from a BLAST search at the UniProt database (fragments excluded) with an E-value <10−40 (42 eukaryotic sequences, including mammalian SLC17 homologues).
Homology Modeling and Substrate Docking
The ANTR1 model was constructed based on the structure of GlpT (PDB ID: 1PW4) (12). An initial alignment with the template was performed using ClustalW for amino acid residues 84−512 from ANTR1 and revealed 18% identity. Of the missing 83 residues in the N-terminus, region 1−59 is a transit peptide (7). Three other structures were also evaluated as potential templates, displaying the same or lower sequence identity with ANTR1, namely, OxlT (18%), EmrD (17%), and LacY (13%). This information and the fact that GlpT transports Pi determined us to choose it as template for homology modeling of ANTR1.
Next, we have performed a transmembrane helix prediction at the ARAMEMNON database, which applies 17 different prediction methods. Most methods predicted 9−11 transmembrane segments (TM) for ANTR1. One prediction, however, indicated 12 TMs, the HmmTop_v2 (21), which is consistent with the consensus prediction for three other ANTRs at ARAMEMNON database. This also corresponds with the 12 TM topology of crystallized MFS members (including the template) and was therefore used to improve the initial sequence alignment between ANTR1 and GlpT. To improve the quality of regions showing lower conservation in the alignment, such as the large loop between the two six-helix domains, we have performed small adjustments based on the MSA of ANTR1 with the other Arabidopsis ANTRs and the one with eukaryotic homologues at the UniProt database. The final pairwise alignment used for homology modeling of ANTR1 is shown in Figure 1.
Figure 1.

Sequence alignment and secondary structure comparison between ANTR1 from Arabidopsis and the template protein GlpT from E. coli. The sequence alignment was obtained using a combination of transmembrane helix prediction, multiple sequence alignments, and minor manual adjustments. The identical (*), conserved (:), and semiconserved (.) amino acids are indicated above the alignment. The five residues from ANTR1 selected for mutagenesis and functional characterization are highlighted on a dark background. The 12 transmembrane helices (H regions) and connecting loops (—), as detected in the crystal structure of GlpT (12), are indicated below the alignment.
Three models were created using ICM 3.5 from Molsoft Inc. (22), and the model with the lowest energy value and the least number of outliers in the Ramachandran plot was chosen as the final structure. The PROCHECK results (23) showed no large errors in this structure, as based on the following: 83.1% of the residues in allowed regions, 13.7% in additional allowed regions, 1.6% in generously allowed regions, and 1.6% in disallowed regions. Conservation analysis was performed using ConSurf (24,25). Visual analysis and manipulation of the model was done using ICM. The obtained model structure is available at the Protein Model DataBase (26) under PMDB ID: PM0076247.
As for the homology modeling, ICM 3.5 was employed for substrate docking. By using the built-in pocket finder, the binding pocket was identified. Here, a ball of a water molecule size was rolled around the protein structure to define its surface; thereafter, pockets with large enough volume and depth were identified. Residues involved in the pocket were defined as those that had any contact with the surface. The pocket we chose for docking prediction is located inside the transportation pathway shown in Figure 2. Even though the binding pocket was quite large, we performed three completely unbiased docking calculations with flexible ligand and side chains in the pocket, while the rest of the protein was treated as rigid. The docking conformation with the lowest total binding energy was treated as the best conformation.
Figure 2.

Homology model of ANTR1. (A) Ribbon structure of ANTR1 (side view) based on homology modeling using GlpT as a template. The transmembrane segments are labeled 1−12. The main cavity situated between the two domains is shown in blue. The side chains of the five residues selected for site-directed mutagenesis are indicated as follows: Arg-120, Ser-124, and Arg-201 (red); Arg-228 and Asp-382 (blue). (B) Close-up view of the putative phosphate-binding pocket, as determined from docking predictions. The positions of Arg-120, Ser-124, and Arg-201 are shown. The figure was created using ICM Molsoft.
Results
Homology Modeling, Substrate Docking, and Conservation Analysis
The 3-D structure of any member of the Arabidopsis Pi transporter (ANTR) family is presently unknown. The crystal structure of GlpT in a cytoplasmic-open conformation (12) was chosen as template to model the thylakoid member of this family (ANTR1), as described in Experimental Procedures. The modeled structure consists of two six-TM-containing domains, connected by a large loop and forming a central cavity open toward the cytoplasm (Figure 2A and Supporting Information Figure S1). The TMs forming the cavity are the same as in GlpT, namely, TM1, -2, -4, -5, and the lower part of TM6 from the first domain and TM8, -10, and -11 from the other domain. The diameters of the cavity are the following: 9 Å at the bottom (periplasmic side), 6 Å in the middle, and 5 Å at the entrance (cytoplasmic side). The sequences of GlpT and ANTR1 are more similar in the TMs forming the cavity than in those outside the cavity (TM3, -7, -9, and -12) (Figure 1). When the model was analyzed using ConSurf, the inner core of the ANTR1 structure was found to be highly conserved (Supporting Information Figure S2), indicating confidence in the pairwise alignment. TM1 contains eight residues that are identical in the two proteins followed by TM11 with six identical residues. Least identity is found in TM5 with only three residues, Glu-227, Arg-228 (mutated in this study), and Gly-242. The main cavity is the putative translocation pathway, expected to contain residues involved in substrate binding and translocation (Figure 2A).
Docking predictions into the putative translocation pathway of ANTR1 were performed for Pi and also for glutamate. The best binding position for Pi was found to be located about 6 Å up from the bottom of the cavity, close to residue Arg-120 (Figure 2B). A distinct location, namely, at the bottom of the cavity, at the level of residue Arg-201, was preferred by the relatively large ligand glutamate (data not shown). Interestingly, a docking study in a GlpT-based homology model of VGLUT1 revealed positioning of the glutamate substrate across the cavity and with two sites of similar binding energy (27), one central and one lower in the cavity, in level with Arg residues corresponding to Arg-120 and Arg-201 of ANTR1, respectively. The docking of Pi in VGLUT1 was similar to the one in the present study. The explanation for the discrepancy in positioning of glutamate could be the fact that the bottom of the cavity is 3 Å wider in ANTR1 as compared to the VGLUT1 model.
Four MSAs of ANTR1 with Arabidopsis ANTRs, plant homologues at the ARAMEMNON database (>36% and >20% identity) and eukaryotic homologues at the UniProt database (E-value <10−40) were created. A complete sequence alignment of ANTR1 with homologues found at UniProt database is shown in Supporting Information Figure S3. Using information gained from the docking predictions, position in the structure, and type of amino acid residue, the 25 most conserved residues in all MSAs were classified in four groups, namely, binding/translocation, exposed, closed, and other (Table 1). The side chains of these residues have been highlighted in various colors in the ANTR1 model structure (Supporting Information Figure S4). “Binding/translocation” is a group of three charged or hydrophilic amino acid residues (Arg-120, Ser-124, and Arg-201) located inside the cavity and predicted to be involved in binding and translocation of the substrate and/or coupling ion. “Exposed” group consists of two charged residues, Arg-228 and Asp-382, which are both exposed at the cytoplasmic surface in the modeled conformation and believed not to be directly involved in translocation. “Closed”’ includes residues located inside a small cavity at the periplasmic side, which is not open to substrates in the modeled conformation. Finally, the “other” group contains residues that are believed to be solely important for the stability of the protein structure. Seventeen out of the 25 amino acids (68%) are located in TM1, -2, -4, -7, and -8. When mapping them onto the model structure, many were found situated in the cavity, supposed to be the translocation pathway of ANTR1 (Supporting Information Figure S4).
Table 1. Classification of the 25 Most Conserved Residues in ANTR1 Homologuesa.
| amino acid residue | ANTR 1−6 | ARAMEMNON, >36% identity | ARAMEMNON, >20% identity | UniProt 10−40 cutoff | TM no. |
|---|---|---|---|---|---|
| binding/translocation | |||||
| R120 | : | * | *-1 | *-3 | 1 |
| S124 | * | * | *-1 | 1 | |
| R201 | * | * | : | *-3 | 4 |
| exposed | |||||
| R228 | * | * | * | *-1 | 5 |
| D382 | * | * | * | * | 8 |
| closed | |||||
| G142 | * | * | * | *-2 | 2 |
| W343 | * | * | * | 7 | |
| P345 | * | * | *-1 | *-2 | 7 |
| Y347 | * | * | *-1 | :-1 | 7 |
| other | |||||
| R102 | * | * | : | *-3 | 1 |
| F148 | * | * | *-1 | 2 | |
| Y152 | * | * | *-1 | 2 | |
| G160 | * | * | *-1 | * | 2 |
| G205 | * | * | * | *-1 | 4 |
| P213 | *-1 | * | * | 4 | |
| W222 | * | * | *-1 | *-3 | 4−5 |
| G242 | * | * | * | * | 5 |
| F263 | * | * | * | * | 6 |
| G270 | * | * | *-2 | 6 | |
| A325 | * | * | *-1 | 7 | |
| P366 | * | * | * | 8 | |
| G378 | *-1 | * | *-1 | *-3 | 8 |
| G439 | * | * | *-1 | * | 10 |
| G463 | *-1 | * | *-1 | 11 | |
| G475 | *-1 | * | * | 11 |
Four multiple sequence alignments of ANTR1 with Arabidopsis ANTRs, plant homologues at ARAMEMNON database (>36% and >20% identity), and eukaryotic homologues at UniProt database (E-value <10−40) were created using MUSCLE. The listed amino acid residues are the ones found most conserved in these alignments and are classified in four groups based on their position in the ANTR1 model structure. Asterisks (*) represent an identical amino acid residue, *-1 means identical with one exception, colon (:) means similar properties, and empty box means neither identical nor similar properties. TM, transmembrane segment of ANTR1.
Figure 3 shows an MSA of ANTR1 with selected members of the SLC17 family, all from human except NPT4. We used the rat sequence of NPT4 because the human sequence lacks 77 amino acid residues in TM4 and TM5, as compared to its orthologues and paralogues. Highlighted are the five residues classified as “binding/translocation” and “exposed” in Table 1. These residues are located in TM1, -4, -5, and -8, as based on the alignment with GlpT (Figure 1) and its 3-D structure (12). All five residues except Ser-124 were found identical in SLC17 members. Ser-124 residue is replaced with Gly in VGLUTs and with Asn in NPT1. VGLUT1, VGLUT2, and NPT1 have been shown in heterologous system to transport Pi in a Na+-dependent manner (28−30) but with 10−100-fold lower affinities as compared to ANTR1. This information and the fact that there are so far no reports on Pi transport by sialin, NPT3, NPT4, and VGLUT3 indicate that a Na+-dependent Pi transport may not be the primary function for SLC17 members. In GlpT, Ser-124 is replaced with an Ala residue (Figure 1). Since GlpT transports Pi in exchange for glycerol 3-phosphate via a Na+-independent mechanism, a potential role for Ser-124 in binding of the cotransported Na+ ion is more likely than one in Pi binding. Arg-120, Arg-228, and Asp-382 were found identical in ANTR1 and GlpT, implying a general role for these residues in the MFS-type of transport mechanism. Residue Arg-201 is only conserved in the SLC17 family and may therefore play a more specific role during transport.
Figure 3.

Conservation of mutated residues in ANTR1 and mammalian SLC17 homologues. The amino acid sequences of Arabidopsis ANTR1 (UniProt O82390), sialin from human (Q9NRA2), vesicular glutamate transporters VGLUT1−3 from human (Q9P2U7, Q9P2U8, Q8NDX2), and Na+/Pi cotransporters NPT1 and NPT3 from human (Q14916 and O00624) and NPT4 from rat (Q8CJH9) were aligned using MUSCLE. The five mutated residues are indicated in dark boxes. Conservation is marked in ClustalX style, where asterisks (*) indicate an identical residue, colon (:) represents a similar (conserved) amino acid property, and period (.) corresponds to a semiconserved residue.
Site-Directed Mutagenesis, Expression, and Activity Characterization of the Wild-Type and Mutant ANTR1 Proteins
On the basis of findings from the homology protein modeling and conservation analysis, we selected five amino acid residues, namely, Arg-120, Ser-124, Arg-201, Arg-228, and Asp-382, for generating site-directed mutant proteins. These mutants were used to study the importance of the selected residues for the function of ANTR1 in Pi transport and its Na+ dependency. For this purpose, the WT and mutated His6-Xpress-ANTR1-FLAG constructs were cloned into the pTrcHisC vector and expressed in E. coli upon IPTG induction. Western blotting analyses of protein lysate from transformed cells using an anti-FLAG peptide antibody revealed the expression of ANTR1 (Mr of 56 kDa) in WT and mutant cells but not in the control cells (Figure 4 inset). Densitometry analysis revealed a drastic increase in the expression of R228E, R228K, and D382A mutants, ranging between 160% and 200% relative to the WT levels. It is not clear at present the reason for the increased accumulation of these mutants. Nevertheless, for the other mutants, the protein expression levels did not appear to be largely altered, ranging between 80% and 120% relative to the WT levels, indicating that the corresponding mutations did not dramatically perturb the protein structure and stability. The same pattern of protein expression was obtained when using an anti-ANTR1 peptide-specific antibody (data not shown).
Figure 4.

Comparative effects of site-directed mutations on Pi uptake and protein expression levels. The transport activity and ANTR1 protein abundance were measured in E. coli cells transformed with an empty vector (control) and cells expressing the wild-type ANTR1 (WT) or mutant protein and are shown as a percentage of the WT levels. The transport activity of 100 μM 32Pi was measured for 3 min in 25 mM Tris−succinate (pH 6.5) in the presence of 25 mM NaCl. Protein expression was determined by quantification of the ANTR1 Western blots. The bars are means ± SD. Inset: Representative Western blot of protein lysate (1 A600 unit per lane) from E. coli cells with an anti-FLAG peptide antibody. The loaded samples correspond to the strains in the plot. The equal loading of the proteins in each well of the gel was visually monitored by Coomassie staining of duplicate gels (data not shown). The mass of ANTR1 is approximately 56 kDa, indicated to the right of the blot.
The levels of Pi uptake into E. coli cells were determined at 3 min in the presence of 25 mM NaCl (Figure 4) and represented relative to the WT levels (100% = 21.6 ± 1.1 nmol of Pi (mg of protein)−1). Pi uptake in the control cells under the same conditions yielded 35% of the maximal level accumulated in WT cells and is attributed to the activity by various E. coli Pi transporters (31). In the presence of choline chloride, WT cells took up 6.9 ± 0.3 nmol of Pi (mg of protein)−1, whereas all mutants and control cells displayed uptake levels below 5 nmol of Pi (mg of protein)−1 (data not shown).
All mutants had much lower transport activity than protein expression relative to the WT levels. Most mutants displayed reduced transport activity although their expression levels were similar to the WT levels (Figure 4). This indicates that those mutations specifically affected their ability to transport Pi. Hence, to characterize these residues for their role in substrate binding or translocation, we have measured kinetic parameters of the mutants by measuring the initial rates of Pi uptake over a range of Pi concentrations (Supporting Information Figure S5) and subtracting the corresponding control values. As R120E, R201E, R228E, and D382N had very poor activity, these could not be included for the kinetic study. The apparent Km, Vmax, and corrected Vmax values (calculated based on protein expression levels) have been summarized in Table 2.
Table 2. Apparent Kinetic Parameters for Pi Transport of Wild-Type ANTR1 and Mutantsa.
| mutation | Km (μM) | Vmax (nmol (mg of protein)−1 h−1) | corrected Vmaxb (nmol (mg of protein)−1 h−1) | corrected Vmax/Km (nmol (mg of protein)−1 h−1 μM−1) |
|---|---|---|---|---|
| WT | 77.1 ± 20.7 | 329.0 ± 35.8 | 4.3 | |
| R120K | 161.0 ± 57.9 | 255.0 ± 47.7 | 250.0 | 1.6 |
| S124A | 44.8 ± 16.5 | 50.9 ± 6.2 | 47.1 | 1.1 |
| S124T | 109.0 ± 40.9 | 303.0 ± 51.9 | 254.6 | 2.4 |
| R201K | 214.0 ± 75.5 | 141.0 ± 28.2 | 100.7 | 0.5 |
| R228K | 103.0 ± 28.0 | 453.0 ± 55.1 | 274.5 | 2.5 |
| D382A | 57.2 ± 9.7 | 253.0 ± 15.7 | 156.2 | 2.8 |
| D382E | 46.2 ± 17.9 | 101.0 ± 13.2 | 96.2 | 2.1 |
The uptake of various concentrations of 32Pi was carried out for 3 min in 25 mM Tris−succinate buffer (pH 6.5) containing 25 mM NaCl. The values obtained for wild-type ANTR1 (WT) and mutant cells were subtracted by those in the control cells. Apparent maximum velocity (Vmax) and Km values were determined by fitting the data to the Michaelis−Menten equation using the nonlinear regression program Prism 5. Vmax/Km ratio is a measure of transport efficiency. Data shown are means of two to three experiments ± SE.
Vmax was adjusted to the protein expression levels.
The estimated Km for ANTR1 in the present study is in close agreement with the previously reported value (78.7 ± 34 μM (7)). However, the Vmax differed significantly from the previously reported value (161 ± 28 nmol (mg of protein)−1 h−1). Since Km is an intrinsic property of the transporter whereas Vmax can vary with the protein expression levels and experimental conditions, the results were considered to be reliable and used for evaluation of the mutants. In the last column of Table 2, the ratio between the respective corrected Vmax and Km is shown and used to directly compare the efficiency of Pi transport by the various mutated proteins. A value of 4.3 was determined for the WT protein and values ranging between 0.5 and 2.8 in the mutants with measurable activity.
Residue Arg-120 may be found in TM1 inside the cavity, close to the Pi docking position (Figure 1B). Its substitution with Glu reduced Pi uptake at 3 min below the control levels (Figure 3). Its substitution with the smaller Lys resulted in 40% lower Pi uptake activity as compared to the one obtained for the WT. This could be explained by the 2-fold increase in Km, resulting in a nearly 3-fold decrease in transport efficiency of the mutant (Table 2). These results indicate that Arg-120 may be an important residue for Pi binding and associated conformational changes.
Ser-124 may be located in TM1 at the bottom of the cavity in the cytoplasmic-open conformation (Figure 1). This residue could function as a transient binding site for Pi and/or coupling ions. Its substitution with Ala, which is nonpolar and lacks an OH group at the β-carbon, resulted in reduction of the Pi uptake down to the control levels (Figure 4). A reduction by 7-fold was determined for the corrected Vmax value, resulting in a 4-fold decrease in transport efficiency (Table 2). These results indicate a critical role of this residue in the conformational changes associated with the translocation rather than in binding of the substrate itself.
The S124T mutant, preserving the OH group at the β-carbon, displayed only slightly lower uptake levels than WT cells (Figure 4). Km increased while the corrected Vmax decreased by 1.5-fold (Table 2). The data obtained for S124A and S124T show that the presence of an OH group at the β-carbon is necessary for Pi transport activity. Furthermore, the data also indicate that Ser and Thr may have slightly different impact on the helix packing since the S124T mutant displayed nearly 2-fold lower transport efficiency for Pi.
Residue Arg-201 may be found in TM4 inside the cavity, where it may participate in Pi binding. The R201E mutant displayed levels of Pi uptake at 3 min below the control levels (Figure 4). The Arg-201 substitution with Lys resulted in 45% lower uptake as compared to WT and had drastic effects on the transport kinetics, since Km increased whereas Vmax decreased by 3-fold. Among all mutants used in this study, R201K was the one with the lowest transport efficiency, i.e., nearly 9-fold lower than WT, confirming the crucial importance of this residue for Pi binding and translocation.
The highly conserved residue Arg-228 may be located in TM5 at the outer surface of the protein, far from the predicted Pi binding site. Its substitution with Glu resulted in a drastic reduction of uptake at 3 min below the control levels, as in the case of R120E and R201E mutations (Figure 4). Lys substitution of Arg-228 resulted in slightly higher uptake levels at 3 min as compared to WT, consistent with its higher relative expression levels (Figure 4). Only a weak impact on the kinetic parameters and nearly 2-fold lower Pi transport efficiency were observed for this mutation. Thus, although further experimentation may be required, Arg-228 appears to be important for a full Pi transport activity.
Residue Asp-382, located at the exit of TM8, may be involved in interaction with positively charged residues and/or with coupling ions. However, substitution of this residue with Ala did not affect the uptake at 3 min, while it only slightly decreased Km and reduced by 2-fold Vmax, without dramatically changing the transport efficiency (Vmax/Km). Its substitution with Glu lowered by 2-fold the transport efficiency, mainly due to more than 3-fold reduction in Vmax. The uptake at 3 min was reduced by 45% in the D382E and below the control levels in the D382N mutant. These data indicate a weak participation of Asp-382 in interactions involved in protein conformational changes required for transport.
To test the possibility that the observed changes in Vmax are due to an effect of mutations on the binding of coupling ions, we have performed uptake of 100 μM Pi in the presence of various concentrations of NaCl (0−25 mM) in those mutant strains, which show detectable levels of activity (Supporting Information Figure S6). The apparent kinetic parameters for Na+ were determined after subtracting the corresponding control values. The Km value for Na+ of WT ANTR1 (Table 3) is in close agreement with the previously reported value (1.17 ± 0.36 μM (7)). As observed in the case of Pi kinetics, the Vmax for Na+ differs significantly from the previously reported value (99.15 ± 5.17 nmol (mg of protein)−1 h−1), most likely due to changes in the protein expression levels and experimental conditions.
Table 3. Apparent Na+-Dependent Kinetic Parameters for Pi Transport of Wild-Type ANTR1 and Mutantsa.
| mutation | Km (mM) | Vmax (nmol (mg of protein)−1 h−1) | corrected Vmaxb (nmol (mg of protein)−1 h−1) |
|---|---|---|---|
| WT | 0.84 ± 0.38 | 267.0 ± 26.5 | |
| R120K | 1.48 ± 0.90 | 128.0 ± 18.6 | 125.5 |
| S124A | 0.27 ± 0.30 | 67.0 ± 10.9 | 62.0 |
| S124T | 0.88 ± 0.58 | 265.0 ± 38.1 | 222.7 |
| R201K | 2.29 ± 0.83 | 176.5 ± 16.7 | 126.1 |
| R228K | 1.99 ± 0.83 | 365.0 ± 38.6 | 221.2 |
| D382A | 0.49 ± 0.51 | 193.0 ± 37.6 | 119.1 |
| D382E | 0.73 ± 0.53 | 139.0 ± 21.2 | 132.4 |
The uptake of 32Pi (100 μM) was carried out for 3 min in 25 mM Tris−succinate buffer (pH 6.5) in the presence of various concentrations of NaCl. Choline chloride was added to preserve osmolarity. The obtained uptake values for wild-type ANTR1 (WT) and mutant cells were subtracted by those in the control cells. Apparent maximum velocity (Vmax) and Km values were determined by fitting the data to the Michaelis−Menten equation using the nonlinear regression program Prism 5. Data shown are means of two to three experiments ± SE.
Vmax was adjusted to the protein expression levels.
Variations in the Km (Na+) and Vmax values with respect to WT were observed in cells expressing mutants of the Arg-120 and Arg-201 residues, namely, 2−2.5-fold increase in Km and corresponding decrease in corrected Vmax values (Table 3). However, the observed effect on Na+ kinetics in the two mutants most likely reflects the direct effect of mutation on Pi binding and translocation. Notably, substitution of Ser-124 with Ala lowered Vmax by over 4-fold and impaired the Na+ dependency, whereas substitution with Thr did not affect either of the kinetic parameters, emphasizing the importance of the hydroxyl group of this residue for Na+ binding. The mutant R228K displayed over 2-fold increase in Km and only slight effects on Vmax, strengthening that this residue is not directly involved in binding either of the substrate or of the coupling ions. Asp-382 substitutions to Ala or Glu showed only slight effects on Km (Na+) and over 2-fold reduction in Vmax as compared to WT. The kinetic data indicate a participation of Arg-228 and Asp-382 residues in the conformational changes associated with the translocation and required for full activity. Taken together, the results of Na+ kinetic experiments indicate that among the mutated residues, Ser-124 may be involved in Na+ binding and translocation during Pi transport.
Discussion
The structures of four MFS transporters have been solved (12−15), and despite the low amino acid sequence identity, they were found to be highly similar. This led to the hypothesis that all MFS members share an overall similar structure, regardless of the broad substrate specificity. Among the four crystal structures available, the one of GlpT was chosen as a template for homology modeling of thylakoid Pi transporter (ANTR1) from Arabidopsis, as it has been used for modeling of other prokaryotic and eukaryotic MFS transporters (for a recent review, see ref (32)). Moreover, GlpT produced the best alignment scores among the tested structures and is known to function as a glycerol 3-phosphate/Pi antiporter.
To our knowledge, there is no similar study to the present one for direct identification of key residues in Pi transporters from plants or animals. So far, only a homologous structure of a H+-coupled Pi transporter from Saccharomyces cerevisiae (Pho84) has been reported (33), and recently homology modeling has been used to model H+-coupled Pi transporters of type 1 (PHT1) from Medicago truncatula to identify amino acid residues responsible for differences in the affinity between the members of this family (34).
Integrating information from a combination of structural modeling, MSAs, and substrate docking, we have identified and selected for mutagenesis five charged or hydrophilic residues, which are highly conserved in plant ANTRs, mammalian SLC17 members, and even MFS members (GlpT), namely, Arg-120, Ser-124, Arg-201, Arg-228, and Asp-382. The mutated variants of ANTR1 were thereafter expressed and functionally characterized in E. coli. Immunoblot analysis confirmed that each of the mutant proteins was expressed although in variable amounts (Figure 4 inset). Data from Figure 4 clearly show that substitution of the three Args (120, 201, and 228) for Glu residue and of Asp-382 for Asn residue resulted in an inactive ANTR1 transporter. Notably, these mutations may affect the general physiology of the bacterial cells via an unknown mechanism, since they result in transport activities even lower than the control cells. All other mutants had sufficient activity to allow measurement of kinetic parameters, attesting that the mutated proteins were functional.
The kinetic characterization of the mutants revealed interesting insights into the roles of each of the five residues in the function of ANTR1 in Pi transport, which have been schematically illustrated in Figure 5. Based on Pi-dependent kinetics, Arg-201 was found to be the most critical residue for substrate binding and translocation. For the Na+ dependency of the transport, Ser-124 was found to be a key residue, which may function as a transient binding site for Na+ ions. Residue Arg-120 was found to play an important role in Pi binding and associated conformational changes. Finally, Arg-228 and Asp-382 may participate in interactions allowing conformational changes to occur at the cytoplasmic surface of the transporter. On the basis of the conservation analysis of these residues (Table 1), our findings may have relevance for the mechanism of transport by plant and mammalian ANTR1 homologues. Particularly, residue Arg-201 is fully conserved among all of these homologues, whereas Ser-124 is fully conserved only among plant homologues at the ARAMEMNON database. As compared to those two residues, Arg-120, Arg-22, and Asp-382 are fully conserved even in GlpT (Figure 1), and therefore our findings may have implications for the general MFS type of transport mechanism.
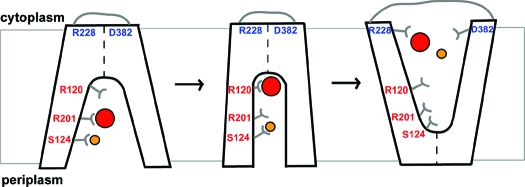
Figure 5.

Schematic diagram of substrate binding and alternating-access mechanism of ANTR1. Like MFS transporters, the conformational cycle of ANTR1 may feature a state open to the periplasmic side, an intermediate state with the substrate and coupling ion bound, and one state open to the cytoplasmic side of the membrane. The diagram shows the proposed roles for five residues analyzed in this study: Arg-120, Ser-124, Arg-201, Arg-228, and Asp-382. The Pi substrate is represented by a red disk and Na+ ion by a yellow disk.
In previous studies, Arg-45 in GlpT, corresponding to Arg-120 in ANTR1, has been demonstrated using mutagenesis as a key residue for binding of either Pi or anionic phosphoryl group of glyceraldehyde 3-phosphate (35). Nevertheless, the corresponding residue in the SLC17 member VGLUT2 (Arg-88) was not found to be critical for glutamate transport (36). The same report has found instead the residue corresponding to Arg-201 in ANTR1 to be essential for glutamate transport, whereas neither of the two Arg residues were found to be important for Pi transport by VGLUT2. The results obtained in the present study clearly demonstrate the importance of Arg-120 and Arg-201 for Pi transport by ANTR1. The fact that Arg-201 is fully conserved in the SLC17 family implies that this residue may be important for binding of both organic and inorganic anionic substrates. For comparison, the His-128 residue shown to be essential for glutamate transport by VGLUT2 (36) is not conserved in ANTR1 and also neither in human NTPs nor in sialin and may therefore be specific for glutamate binding/transport. The docking predictions performed for ANTR1 indicated a distinct binding position for Pi and glutamate in the transportation cavity, at the level of Arg-120 and Arg-201, respectively. The Arg-201 residue may only be a transient binding site for Pi in ANTR1, to attract it inside the cavity (Figure 5), since docking at this position was found to be less favorable than for Arg-120. In our previous biochemical characterization of ANTR1, glutamate could compete for the Pi transport (by 50%); however, no glutamate transport activity could be detected (7). Those data could now be explained based on the findings of the docking predictions and biochemical analysis performed in the present study, indicating that Arg-201, which is close to the best docking position of glutamate, may at the same time be important for Pi transient binding and transport. For comparison, the two positions corresponding to Arg-120 and Arg-201 were found to be energetically similar when glutamate substrate was docked into the VGLUT1 model (27). Those predictions and previous biochemical studies (36) may support the hypothesis that both residues are stable binding sites for glutamate in VGLUTs.
It has been proposed that either Na+ or H+ ions are required for transport of Pi by ANTR1 (7,8). For LacY, a His residue inside the cavity has been proposed to bind the H+ ions involved in cotransport (16). There are only three His residues on the inside of the ANTR1 protein, located opposite to Arg-120 and Arg-201, namely, His-330, His-333, and His-444. None of them is fully conserved among plant ANTRs. Moreover, they do not have direct access to the cavity, even though the side chains are pointing in the right direction. If any of the ANTRs would be a H+-coupled transporter, dramatic changes must occur to expose any of these histidines, or there must be some other types of residues involved in H+ binding/transport. Nevertheless, all six members of the ANTR family have been characterized as H+-dependent Pi transporters when expressed in yeast (8). This implies that there must be some other types of residues involved in H+ cotransport, which may be present among the ones listed in Table 1.
Na+/solute cotransporters require mainly OH-containing groups (Ser, Thr) but also acidic residues (Glu, Asp) for binding and translocation of the coupling ion, as revealed in the recent crystal structure of LeuTA (37). The target for Na+ binding in this study has been Ser-124. This residue is located at the bottom of the cavity in the cytoplasmic-open conformation (Figure 5). It may be a fast operating gateway by short-lived binding of Na+ ions. Its closest partner in coordinating the Na+ ions may be Thr-342 residue from the other domain, at a distance of around 4 Å between OH groups, in the modeled ANTR1 structure.
Arg-228 and Asp-382 are actually the most conserved residues among the five studied ones (Table 1), even though they both may reside close to the cytoplasmic surface, and are far away from the substrate-docking site (Figure 5). The R228E and D382N mutants completely lost the transport activity, whereas a partial loss was obtained in R228K, D382E, and D382A mutants. Although it is not clear at the present stage which type of interactions the two residues make, we suggest that their presence at positions 228 and 382, respectively, is required for full activity. An attractive possibility could be that, due to its basic properties, Arg-228 may participate in transient binding of anionic substrates, such as Pi, on the way out from the cavity into the cytoplasm. Notably, ANTR1 amino acid sequence was shown to contain a seven-residue consensus pattern for the MFS anion:cation symporter family, which includes Arg-228 (6,7,38). Conserved cationic residues such as Arg are also part of other MFS family specific consensus sequences (38) and could be important for both structural and functional aspects. A relevant question is why among all studied mutants R228E accumulates in the largest amounts as compared to WT (200%) and is inactive (Figure 4). We tempt to speculate that this mutant is partially folded/misfolded for two possible reasons: When close to the cytoplasmic surface, the positively charged side chains may interact with negatively charged phosphate groups at the lipid bilayer surface or may promote packing of TMs by H-bond formation with neighboring residues, resulting in proper folding and/or insertion of the protein (39,40).
It is important to consider the fact that the crystal structure of GlpT was obtained in the absence of a substrate, revealing a cytoplasmic-open conformation and with a tightly closed periplasmic side (12). In the case of LacY, the same conformation was obtained in the presence of a lactose homologue, β-d-galactopyranosyl 1-thio-β-d-galactopyranoside (13). Recently, it has been shown that, in the presence of the native substrate, LacY adopts the periplasmic-open conformation, indicating that the structure of the binding pocket undergoes a rocker-switch conformational change (41). Experimental evidence for as much as 15−17 Å large periplasmic-open cavity has been reported for LacY (42), which implies that the cavity can be closed at the cytoplasmic side. In order for transformed E. coli cells to take up Pi via ANTR1, a hydrophilic pathway must open at the periplasmic side to allow access of Pi to the binding site. Since Pi transport shows strong dependency on Na+ concentration (Supporting Information Figure S6), initial or simultaneous Na+ binding may be required. To explain the effects of various substitutions on Pi and Na+ kinetics (Tables 2 and 3), we propose that binding of Na+ ions by Ser-124 may allow opening at the periplasmic side and access of Pi inside the cavity (Figure 5). The positively charged Arg-201 may attract Pi in the cavity before reaching its binding position, close to Arg-120, in the intermediate conformation. This results in the structural change leading to the cytoplasmic-open conformation and subsequent release of the substrate and coupling ion. We suggest that Arg-228 and Asp-382 participate in interactions at the cytoplasmic side, possibly by attracting the substrate out from the cavity, preparing the structure for the periplasmic-open conformation and a new transport cycle. Arg-228 may also be important for proper insertion/folding of the protein in the membrane.
The present study based on Arabidopsis ANTR1 provides the first data on the 3-D molecular interactions of a Pi transporter with its substrate and the coupling ion. The homology model can be used to target other key amino acids responsible for Pi binding, membrane interaction, and interactions with other components. The functionally important residues in ANTR1 may help to understand the mechanism of solute transport by SLC17 homologues and could even be expanded to other MFS members. In vivo inhibitors should offer possibility to mimic the phenotype of plant ANTR1 mutants.
Acknowledgments
The full-length RAFL09-06-K07 cDNA clone was provided by RIKEN BioResource Center (Japan).
Supporting Information Available
Table S1 and Figures S1−S6 as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by grants from the Swedish Research Council (to C.S. and B.L.P.), the Swedish Research Council for Environment, Agriculture, and Space Planning (Formas) (to C.S.), Linköping University (to C.S. and B.P.), and the Helge Ax:son Johnson Foundation (to P.M.K.).
Footnotes
Abbreviations: ANTR1, anion transporter 1; GlpT, glycerol 3-phosphate/phosphate antiporter; IPTG, isopropyl 1-thio-β-d-galactopyranoside; MFS, major facilitator superfamily; MSA, multiple sequence alignment; Pi, inorganic phosphate; SLC, solute carrier; TM, transmembrane segment; VGLUT, vesicular glutamate transporter; WT, wild-type ANTR1.
Supplementary Material
References
- Block M. A.; Douce R.; Joyard J; Rolland N. (2007) Chloroplast envelope membranes: a dynamic interface between plastids and the cytosol. Photosynth. Res. 92, 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linka M.; Weber A.P. M. (2010) Intracellular metabolite transporters in plants. Mol. Plant 3, 21–53. [DOI] [PubMed] [Google Scholar]
- Spetea C.; Schoefs B. (2010) Solute transporters in plant thylakoid membranes—key players during photosynthesis and stress. Comm. Integr. Biol. 3, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flugge U. I. (1999) Phosphate translocators in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 27–45. [DOI] [PubMed] [Google Scholar]
- Weber A. P.; Schwacke R.; Flügge U. I. (2005) Solute transporters of the plastid envelope membrane. Annu. Rev. Plant Biol. 56, 133–164. [DOI] [PubMed] [Google Scholar]
- Roth C.; Menzel G.; Petétot J. M.; Rochat-Hacker S.; Poirier Y. (2004) Characterization of a protein of the plastid inner envelope having homology to animal inorganic phosphate, chloride and organic-anion transporters. Planta 218, 406–416. [DOI] [PubMed] [Google Scholar]
- Pavón L. R.; Lundh F.; Lundin B.; Mishra A.; Persson B. L.; Spetea C. (2008) Arabidopsis ANTR1 is a thylakoid Na+-dependent phosphate transporter: functional characterization in Escherichia coli. J. Biol. Chem. 283, 13520–13527. [DOI] [PubMed] [Google Scholar]
- Guo B.; Jin Y.; Wussler C.; Blancaflor E. B.; Motes C. M.; Versaw W. K. (2008) Functional analysis of the Arabidopsis PHT4 family of intracellular phosphate transporters. New Phytol. 177, 889–898. [DOI] [PubMed] [Google Scholar]
- Guo B.; Irigoyen S.; Fowler T. B.; Versaw W. K. (2008) Differential expression and phylogenetic analysis suggest specialization of plastid-localized members of the PHT4 phosphate transporter family for photosynthetic and heterotrophic tissues. Plant Signal. Behavior 3, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spetea C.; Hundal T.; Lundin B.; Heddad M.; Adamska I.; Andersson B. (2004) Multiple evidence for nucleotide metabolism in the chloroplast thylakoid lumen. Proc. Natl. Acad. Sci. U.S.A. 101, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer R. J.; Edwards R. H. (2004) Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pfluegers Arch. 447, 629–635. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Lemieux M. J.; Song J.; Auer M.; Wang D. N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–20. [DOI] [PubMed] [Google Scholar]
- Abramson J.; Smirnova I.; Kasho V.; Verner G.; Kaback H. R.; Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615. [DOI] [PubMed] [Google Scholar]
- Yin Y.; He X.; Szewczyk P.; Nguyen T.; Chang G. (2006) Structure of the multidrug transporter EmrD from Escherichia coli. Science 312, 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T.; Heymann J. A.; Shi D.; Sarker R.; Maloney P. C.; Subramaniam S. (2002) Three-dimensional structure of a bacterial oxalate transporter. Nat. Struct. Biol. 9, 597–600. [DOI] [PubMed] [Google Scholar]
- Guan L.; Kaback H. R. (2006) Lessons from lactose permease. Annu. Rev. Biophys. Biomol. Struct. 35, 67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C. J.; Maloney P. C.; Wang D. N. (2008) Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 62, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S. K.; Morimoto R.; Otsuka M.; Omote H. (2006) Transmembrane topology of vesicular glutamate transporter 2. Biol. Pharm. Bull. 29, 547–549. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Graaff E.; Fischer K.; Catoni E.; Desimone M.; Frommer W. B.; Flügge U. I.; Kunze R. (2003) ARAMEMNON, a novel database for Arabidopsis integral membrane proteins. Plant Physiol. 131, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády G. E.; Simon I. (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850. [DOI] [PubMed] [Google Scholar]
- Abagyan R.; Totrov M. (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W; Moss D. S; Thornton J. M (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Glaser F.; Pupko T.; Paz I.; Bell R. E.; Bechor D.; Martz E.; Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164. [DOI] [PubMed] [Google Scholar]
- Landau M.; Mayrose I.; Rosenberg Y.; Glaser F.; Martz E.; Pupko T.; Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrignanò T.; De Meo P. D.; Cozzetto D.; Talamo I. G.; Tramontano A. (2006) The PMDB protein model database. Nucleic Acids Res. 34, D306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almqvist J.; Huang Y.; Laaksonen A.; Wang D. N.; Hovmöller S. (2007) Docking and homology modeling explain inhibition of the human vesicular glutamate transporters. Protein Sci. 16, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni B.; Rosteck P. R.; Nadi N. S.; Paul S. M. (1994) Cloning and expression of a cDNA encoding a brain-specific Na(+)-dependent inorganic phosphate cotransporter. Proc. Natl. Acad. Sci. U.S.A. 91, 5607–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K.; Tatsumi S.; Sonoda T.; Yamamoto H.; Minami H.; Taketani Y.; Takeda E. (1995) Cloning and functional expression of a Na(+)-dependent phosphate co-transporter from human kidney: cDNA cloning and functional expression. Biochem. J. 1305, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aihara Y.; Mashima H.; Onda H.; Hisano S.; Kasuya H.; Hori T.; Yamada S.; Tomura H.; Yamada Y.; Inoue I.; Kojima I.; Takeda J. (2000) Molecular cloning of a novel brain-type Na(+)-dependent inorganic phosphate cotransporter. J. Neurochem. 74, 2622–2625. [DOI] [PubMed] [Google Scholar]
- Harris R. M.; Webb D. C.; Howitt S. M.; Cox G. B. (2001) Characterization of PitA and PitB from Escherichia coli. J. Bacteriol. 183, 5008–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M. J. (2007) Eukaryotic major facilitator superfamily transporter modeling based on the prokaryotic GlpT crystal structure. Mol. Membr. Bio.l 24, 333–341. [DOI] [PubMed] [Google Scholar]
- Lagerstedt J. O.; Voss J. C.; Wieslander A.; Persson B. L. (2004) Structural modeling of dual-affinity purified Pho84 phosphate transporter. FEBS Lett. 578, 262–268. [DOI] [PubMed] [Google Scholar]
- Liu J.; Versaw W. K.; Pumplin N.; Gomez S. K.; Blaylock L. A.; Harrison M. J. (2008) Closely related members of the Medicago truncatula PHT1 phosphate transporter gene family encode phosphate transporters with distinct biochemical activities. J. Biol. Chem. 283, 24673–24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C. J.; Enkavi G.; Wang D. N.; Tajkhorshid E. (2009) Structural basis of substrate selectivity in the glycerol-3-phosphate: phosphate antiporter GlpT. Biophys. J. 97, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge N.; Yoshida Y.; Yatsushiro S.; Omote H.; Moriyama Y. (2006) Vesicular glutamate transporter contains two independent transport machineries. J. Biol. Chem. 281, 39499–39506. [DOI] [PubMed] [Google Scholar]
- Yamashita A.; Singh S. K.; Kawate T.; Jin Y.; Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223. [DOI] [PubMed] [Google Scholar]
- Pao S. S.; Paulsen I. T.; Saier M. H. Jr (1998) Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Sansom M. S. (2008) Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 18, 425–431. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2006) Insertion of an arginine residue into the transmembrane segments corrects protein misfolding. J. Biol. Chem. 281, 29436–29440. [DOI] [PubMed] [Google Scholar]
- Guan L.; Mirza O; Verner G.; Iwata S.; Kaback H. R. (2007) Structural determination of wild-type lactose permease. Proc. Natl. Acad. Sci. U.S.A. 104, 15294–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Guan L.; Freites J. A.; Kaback H. R. (2008) Opening and closing of the periplasmic gate in lactose permease. Proc. Natl. Acad. Sci. U.S.A. 105, 3774–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.