

Abstract
Glycoproteins mediate multiple, diverse and critical cellular functions, that are desirable to explore by structural analysis. However, structure determination of these molecules has been hindered by difficulties expressing milligram quantities of stable, homogeneous protein and in determining, which modifications will yield samples amenable to structural studies. We describe a platform proven effective for rapidly screening expression and crystallization of challenging glycoprotein targets produced in mammalian cells. Here, multiple glycoprotein constructs are produced in parallel by transient expression of adherent human embryonic kidney (HEK) 293T cells and subsequently screened in small quantities for crystallization by microfluidic free interface diffusion. As a result, recombinant proteins are produced and processed in a native, mammalian environment and crystallization screening can be accomplished with as little as 65 μg of protein. Moreover, large numbers of constructs can be screened for expression and crystallization and scaled up for structural studies in a matter of five weeks.
Introduction
Glycoproteins are thought to make up about 50% of all human and human-pathogen proteins, but comprise only 3% of structures deposited in the Protein Data Bank (PDB). Structural biology of glycoproteins is hindered by challenges in achieving high-level expression of soluble, stable and homogenous protein capable of forming well-diffracting crystals. Although attachment and processing of glycans is often a prerequisite for correct protein folding and stability, the chemical and conformational heterogeneity of complex-type glycans displayed on proteins produced in mammalian cells generally inhibits formation of a well-ordered crystal lattice1. Multiple rounds of iterative construct re-design or site-directed mutagenesis of N- or O-linked glycosylation sequons, coupled with enzymatic deglycosylation, are usually necessary to identify a protein construct suitable for crystallization. Glycoprotein expression efforts often use Chinese hamster ovary (CHO) cells2, yeast3 and insect cell-based systems4-7. However, these systems are not ideal for rapid construct screening as initial setup of each stable cell line can be time-consuming. Moreover, processing of glycans in yeast, insect and Drosophila cell lines differs from that which occurs in human glycan biosynthetic pathways, possibly influencing protein folding, stability, activity, secretion and immunogenicity8-11. Recent developments using transient transfection protocols of adherent human embryonic kidney (HEK) 293T cells have shown greater promise for higher throughput of mammalian-expressed glycoproteins12,13.
We describe a procedure for screening crystallization using transient expression in adherent HEK293T cells, one-step affinity purification, and microfluidic free interface diffusion. This platform allows cloning, expression and crystallization to be performed in approximately 5 weeks, significantly faster than current protocols. This protocol is now in standard use for all mammalian-expressed glycoproteins in our laboratory14 and by a number of other groups12,13. Its utility is proven by the successful structural determination of a challenging 320 kDa Zaire ebolavirus glycoprotein-antibody complex14.
Considerations for test-scale expression system
The main considerations in the selection of a vector for expression of mammalian glycoproteins are:
the plasmid should have a high copy number in E. coli to produce milligram quantities of DNA required for large-scale transient transfections
the promoter should be strong in mammalian cells
the plasmid should have a high-affinity purification tag to bind low yields of secreted protein from large volumes of conditioned media, and also serve as a high affinity epitope tag for protein detection
the plasmid should have an optimized secretion signal to ensure proper targeting and co- and post-translational processing
the gene of interest should be codon-optimized for mammalian expression
While many commercial vectors are available for expression of human proteins, we have found the pDISPLAY expression system to be of particular utility. The pDISPLAY plasmid has a vector-encoded, C-terminal transmembrane domain and an Ig κ chain leader sequence which targets the protein through the ER and Golgi networks for display on the cell surface. We usually insert a stop codon prior to the vector-encoded transmembrane anchor to allow soluble protein to be released into the cultured media. A strong human cytomegalovirus (CMV) protomer ensures over-expression of the protein of interest, and the presence of a hemagglutinin (HA) tag allows the secreted protein to be selectively purified by immunoaffinity chromatography and detected by Western blot.
Adherent HEK293T cells (Figure 2) are easy to maintain, able to express and properly process recombinant human proteins12,13, and highly suitable for use in small- to medium-sized laboratories. HEK293T cells are available from major cell banks such as ATCC, and are easily cultured using Dulbecco's modified Eagle's medium (DMEM) supplemented with L-glutamine (1× GlutaMAX) and 5% (v/v) fetal bovine serum (FBS) in a 37°C incubator with 5% CO2. Test expressions are performed in 6-well (9.5 cm2 surface area) or in 24-well (1.9 cm2 surface area) cell culture plates. High-level expression in small volumes is best obtained when cells are transfected at 70% confluency and when a commercial reagent such as FuGENE HD (Roche) or GeneJuice (EMD Biosciences) is used for transfection. The detection and analysis of dilute amounts of protein secreted into conditioned media is performed by Western blot analysis using antibodies directed against both linear- and conformational epitopes. The use of an antibody against a linear epitope allows detection of total protein expressed, regardless of quality, while use of an antibody against a conformational epitope (if available) distinguishes properly folded from misfolded protein.
Figure 2. Large-scale expression.

(a) Adherent HEK293T cells grown to 70% confluency at 37°C and 5% CO2. This is the ideal cell density for transient transfection using either commercial or calcium phosphate transfection reagents. Healthy cells should adhere to the surface of the vessel and cluster together in a monolayer. (b) 10-layer Corning CellSTACKS shown in a Labline model 397 37°C CO2 incubator. Although a large incubator (68.5 × 161.3 × 72.5 cm) is shown here, the 33.5 × 20.5 cm footprint of the CellSTACK easily fits in standard CO2 incubators as well. Each CellSTACK layer has 636 cm2 surface area for cell attachment and the 10-layer vessel is capable of holding an average yield of 6.4 × 108 cells.
Considerations for cell culture scale up and purification
A variety of containers, such as T225 cm2 flasks, roller bottles (850 cm2) or CellSTACKS (636 cm2/layer) can be used for 293T culture and expression. We favour the Corning 10-layer CellSTACK system for larger scale expression and 2- or 5-layer CellSTACKS for smaller scale exploratory studies (Figure 2). CellSTACKS provide equivalent or larger surface area for cell attachment and growth than other vessels, yet package cell culture space effectively, thus requiring less incubator space and labour. Although disposable CellSTACKS are more costly than roller bottles, they can be made more cost-effective by washing and recycling the vessel.
While FuGENE HD is often used in our laboratory for test transfections, the cost of this reagent is prohibitively expensive for use in large-scale expression. We have found that the use of traditional inexpensive transfection reagents, such as calcium phosphate (reagent of choice in our laboratory) or branched PEI12,15,16, provides excellent transfection efficiencies and cost savings.
In our laboratory, glycoproteins are secreted into litre-scale volumes of conditioned media, and are efficiently purified via a two-step procedure. First, large volumes of media are rapidly concentrated to more manageable amounts using a Centramate tangential flow filtration system. This is followed by capture of the recombinant protein by affinity chromatography on an anti-HA column with high affinity and capacity. Bound proteins are gently eluted from anti-HA columns by competition with a synthetic HA peptide. Afterwards, the anti-HA affinity matrix can be regenerated multiple times without loss of performance. Alternatively, dilute concentrations of secreted protein may be loaded directly onto the affinity column without prior concentration, although this process is slower, or proteins may be fractionated by ammonium sulfate precipitation and reconstituted prior to affinity capture. Many other affinity purification tags such as biotin, glutathione S-transferase (GST), myc-epitope, FLAG, and poly-histidine have also been developed, however we prefer the hemagglutinin tags because of the ease of the antibody-based purification and low cross-reactivity with host cell proteins. In our experience, low-yield proteins purified by metal affinity chromatography retain a number of host cell contaminants and require additional purification steps before crystallization.
Strategies for the removal of N-linked glycans
N-linked glycans attached to proteins expressed in mammalian cells are usually complex, chemically and conformationally heterogeneous in nature, and frequently detrimental to formation of well-ordered crystal lattices. Several strategies have been utilized to overcome the “glycosylation problem”. Two popular methods involve using either a N-acetylglycosaminyltransferase I (GnT I)-deficient HEK293S cell line17 or addition of glycan anabolic inhibitors such as swainsonine or kifunensine13 to cell culture media. Both strategies yield more homogeneous, high mannose-type oligosaccharide chains, which can be removed by EndoH deglycosylation13.
An alternate approach that we favour, involves production of native glycoproteins without inhibitors and enzymatically deglycosylate the resulting protein under native conditions using peptide N-glycosidase F (PNGaseF), followed by Endoglycosidase (Endo)F1, EndoF2 or EndoF3. Each of these enzymes has a unique glycan substrate preference (Figure 3). Of these, PNGaseF has the broadest substrate specificities and cleaves between the asparagine side chain and the innermost N-acetylglucosamine (GlcNAc), thus removing the entire glycan moiety. Occasionally, some glycan attachment sites are buried or masked by the local protein structure and PNGaseF is unable to gain access. However, the EndoF series of glycosidases, which cleave between the two GlcNAc oligosaccharides in the chitobiose core, are less sensitive to protein conformation, and may trim glycans where PNGaseF has failed. In addition, in some cases, removal of terminal sialic acids by neuraminadase, may be beneficial prior to further deglycosylation with EndoF or PNGaseF.
Figure 3. Substrate specificities of selected glycosidases useful for large-scale deglycosylation.

PNGaseF will cleave virtually all N-linked glycans, with the exception of those containing a fucose α(1-3)-linked saccharide to the inner most N-acetylglucosamine (GlcNAc). These fucose α(1-3)-linked glycans are commonly found in glycoproteins from plants, parasitic worms, or insect cells. EndoH primarily cleaves within the chitobiose core in high mannose and some hybrid glycans. EndoF1 cleaves oligomannose and hybrid structures, but not complex structures. EndoF2 and EndoF3 both cleave complex-type glycans. However, EndoF2 prefers biantennary complex-type oligosaccharides and to a lesser extent, oligomannose sugars, while EndoF3 cleaves both biantennary and triantennary glycans. Tetraantennary oligosaccharides are not cleaved by the EndoF series of enzymes and require sequential hydrolysis down to a Man3GlcNAc2 structure. Neuraminadase is an exoglycosidase that removes terminal sialic acid residues from hybrid, biantennary, triantennary and tetraantennary oligosaccharides. It is sometimes necessary to remove these negatively charged saccharides prior to deglycosylation with EndoH, EndoF or PNGaseF.
Considerations for crystallization
The use of robotic liquid handling systems has significantly reduced time and protein quantity required for crystallization screening. Indeed, current robots are able to accurately dispense ∼100 nL of protein or reagent per drop. A recent, additional improvement in protein crystallization is the development of microfluidic circuits to dispense even smaller volumes of protein and reagents for crystallization. For example, the Fluidigm Topaz system allows screening of 96 different crystallization conditions with a mere 1.5 μL of protein, which can be critical for crystallization targets that are difficult to produce. Indeed, an increasing number of mammalian protein structures, including those of glycosylated proteins, have been determined from initial crystallization hits obtained from microfluidic-based diffusion18-25. The system is quite expensive, but in our hands, has been a key determinant in the feasibility of structural analysis of some difficult targets. The Topaz microfluidic screening chips (Figure 4) contain a series of interconnected miniaturized channels and elastomeric rubber valves that control the interface of protein sample and precipitant. Initially, diffusion of protein and precipitant sets a concentration gradient to allow the sampling of a wide area of crystallization space. Subsequently, vapour permeability of the reaction chamber allows dehydration to further concentrate the sample, yielding results in 7 days. Quality hits can then be translated into standard 0.5-2.0 μL size hanging drop, vapour diffusion experiments.
Figure 4. Microfluidic crystallization chip.
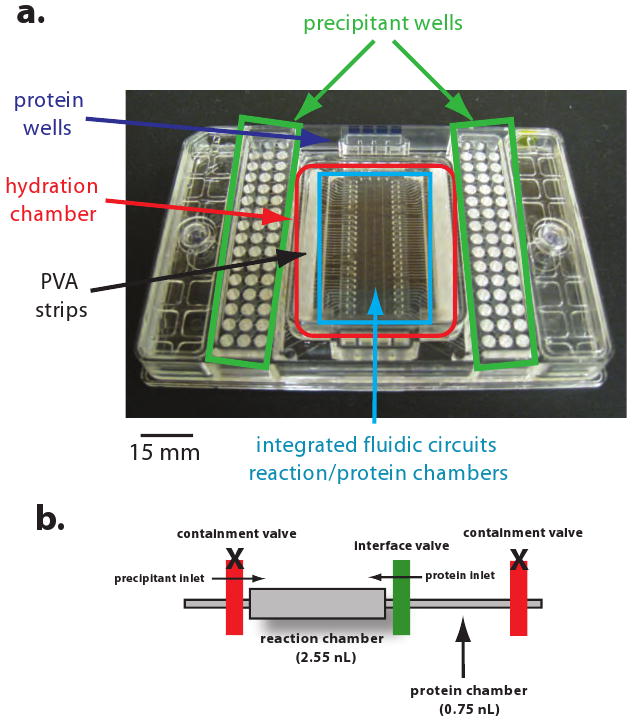
Topaz 4.96 crystallization screening chip shown here contains 96 inlets for the crystallization reagent and inlets for four protein samples. Other chips are available with one or eight protein inlets. Integrated fluidic circuits and reaction chambers are embedded within the hydration chamber.
Materials
Reagents
pDISPLAY expression vector (Invitrogen; cat. #V660-20)
MiniPrep plasmid purification kit (Qiagen; cat. #27106)
HEK293T cells (ATCC; cat. #CRL-11268)
6-well culture plates; surface area: 9.5 cm2 (Corning; cat. #3516)
24-well culture plates; surface area: 1.9 cm2 (Corning; cat. #3526)
Dulbecco's modified Eagle's media (DMEM) (Gibco/Invitrogen; cat. #11965)
100× penicillin/streptomycin (pen/strep) (Gibco/Invitrogen; cat. #15140)
fetal bovine serum (FBS) (Gibco/Invitrogen; cat. #10082)
100× GlutaMAX (Gibco/Invitrogen; cat. #35050)
CO2
FuGENE HD transfection reagent (Roche; cat. #4709691)
MiniProtean 10-15% gradient Tris-HCl polyacrylamide gel (Bio-Rad; cat. #161-1122)
Immobillon-P PVDF membrane (Millipore; cat. #IPVH07850)
Blotting grade blocker non-fat dry milk (Bio-Rad; cat. #170-6404)
Murine IgG anti-HA monoclonal primary antibody (clone 16B12) (Covance Research Products; cat. #MMS-101P)
Goat anti-mouse IgG F(ab')2 alkaline phosphatase conjugated antibody (Pierce; cat. #31324)
Goat anti-human IgG (H+L) alkaline phosphatase conjugated antibody (Pierce; cat. #31310)
SIGMAFAST BCIP/NBT (Sigma; cat. #B5655)
MaxiPrep plasmid purification kit (Qiagen; cat. #12163)
10-layer CellSTACK; surface area: 6360 cm2 (Corning; cat. #3312)
5-layer CellSTACK; surface area: 3180 cm2 (Corning; cat. #3311)
2-layer CellSTACK; surface area: 1272 cm2 (Corning; cat. #3310)
CaCl2 (Sigma; cat. #C5080)
Bleach
Deionized H2O
sterile 1× PBS (Gibco/Invitrogen; cat. #14190)
0.22 μm vacuum filter (Corning; cat. #431097)
anti-HA affinity matrix, clone 3F10 (Roche; cat. #1815016)
Tween-20 (Sigma; cat. #P1379)
Synthetic HA peptide (sequence: YPYDVPDYA)
NaN3 (Sigma; cat. #S8032)
Glycine (Sigma; cat. #G8898)
Amicon Ultra centrifugal concentrators (Millipore)
Pre-Crystallization Test (PCT) (Hampton Research; cat. #HR2-140)
Protein N-glycosidase F (PNGaseF) (500,000 units/mL) (NEB; cat. #P0704L)
Endo-β-N-acetylglucosaminidase F1 (Calbiochem; cat. #324725)
Endo-β-N-acetylglucosaminidase F2 (Calbiochem; cat. #324726)
Endo-β-N-acetylglucosaminidase F3 (Calbiochem; cat. #324727)
Topaz 1.96 or 4.96 screening chips (Fluidigm)
Hydration Fluid (Fluidigm)
OptiMix-1, 2, 3, PEG 96 deep-well block sparse matrix screens (Fluidigm)
Other deep-well crystallization sparse matrix screens (Hampton Research, Qiagen, Molecular Dimensions, Emerald Biosystems, or those developed in-house)
1× Transfer buffer (see REAGENT SETUP)
1× SDS-PAGE running buffer (see REAGENT SETUP)
3× SDS-PAGE non-reducing sample buffer (see REAGENT SETUP)
3× SDS-PAGE reducing sample buffer (see REAGENT SETUP)
2× HBS (see REAGENT SETUP)
1× PBS (see REAGENT SETUP)
1× PBS-Tween-20 (see REAGENT SETUP)
Reagent Setup
1× transfer buffer For 1 L aqueous solution: 3.0 g Tris base, 14.4 g glycine, 200 mL methanol
1× SDS-PAGE running buffer For 1 L aqueous solution: 3.0 g Tris base, 14.1 g glycine, 1.0 g SDS
3× SDS-PAGE non-reducing sample buffer For 10 mL solution: 0.6 g SDS, 3 mL glycerol, 1.8 mL 1.0 Tris-HCl pH 6.8, 1 mg bromophenol blue
3× SDS-PAGE reducing sample buffer For 10 mL solution: 0.6 g SDS, 3 mL glycerol, 1.8 mL 1.0 Tris-HCl pH 6.8, 1 mg bromophenol blue, 1 mL 2-mercaptoethanol
2× HBS For 1 L aqueous solution: 10.0 g HEPES, 16.0 g NaCl, 0.74 g KCl, 0.40 g Na2HPO4.7H2O. Adjust pH of solution to 7.1 and fill to 1.0 L. Sterile filter, aliquot into 50 mL fractions and immediately store at -20°C
1× PBS For 1 L aqueous solution: 8.0 g NaCl, 0.2 g KCl, 1.4 g Na2HPO4 (anhydrous), 0.24 g KH2PO4. Adjust pH of solution to 7.4 and fill to 1.0 L
1× PBS-Tween-20 For 1 L aqueous solution: 8.0 g NaCl, 0.2 g KCl, 1.4 g Na2HPO4 (anhydrous), 0.24 g KH2PO4, 1 mL Tween-20. Adjust pH of solution to 7.4 and fill to 1.0 L
Equipment
Water bath
Laminar flow hood
CO2 incubator
Centrifuge
Microcentrifuge
Electrophoresis system
Western blot transfer apparatus
Centramate tangential flow filtration system (Pall Corporation)
8-channel P20 micropipettor
Topaz Nanoflex IFC Controller (Fluidigm)
AutoInspeX II workstation (Fluidigm)
Vibration-free incubator (for crystallization)
Procedure
Box 1. Test expression.
Day 1- Cloning and Sequencing TIMING ∼1 ½ weeks
1) Clone gene of interest into pDISPLAY or another appropriate expression vector. Amplify and purify vector (Qiagen MiniPrep kit is helpful for purification).
Day 11- Small-scale expression TIMING 4 days
-
2) Add 2 × 106 HEK293T cells to a 6-well culture plate with 2 mL DMEM, 1× pen/strep, 1× GlutaMAX and 5% (v/v) FBS. Incubate at 37°C with 5% CO2 for 4 hours to allow cell attachment. CO2 is necessary to activate the bicarbonate buffer in DMEM. For higher-throughput, add 4.0 × 105 HEK293T cells to a 24-well cell-culture plates with 0.5 mL DMEM, 1× pen/strep, 1× GlutaMAX and 5% (v/v) FBS.
CAUTION: All cell cultures are considered biohazardous because of their potential to be infectious. Experimenters should wear proper protective clothing and work should be performed in an approved laminar flow hood using aseptic techniques. After completion of experiments, all surfaces, waste media, glassware, consumables and equipment should be disinfected according to institutional and governmental guidelines.
-
3) Pipet 3 μL of FuGENE HD directly into 97 μL of serum-free DMEM.
CRITICAL STEP: Always dilute FuGENE HD into serum-free medium and do not allow undiluted reagent to come into contact with plastics.
-
4) Add 1 μg of Qiagen MiniPrep-purified DNA to the FuGENE-DMEM mixture, gently mix and incubate 30 minutes.
CRITICAL STEP: Always use a greater μL volume of FuGENE HD reagent than μg mass of DNA (we use a 3:1 ratio). To prevent DNA shearing, gently mix.
5) Pipet the FuGENE-DNA mixture drop-wise onto 70% confluent HEK293T cells. Ensure even dispersal.
6) Incubate the cells at 37°C with 5% CO2 for 4 days.
Day 15- Western blot analysis TIMING 1 day
-
7) Decant the supernatant and microcentrifuge at 16,000g for 20 minutes. To detect expression, mix 10 μL aliquots of cell culture supernatant with 3× SDS-PAGE non-reducing sample buffer and separate at 200 V for 45 minutes on a non-denaturing 10-15% SDS-Tris-HCl polyacrylamide gel or another suitable polyacrylamide gel.
CRITICAL STEP: Do not denature sample by heating or adding reducing agent, as this will interfere with recognition by antibodies directed against conformational epitopes, preventing analysis of proper protein fold.
8) Activate the Immobillon-P membrane by soaking 2 minutes in 100% methanol followed by a 2 minute soak in 1× transfer buffer.
9) Assemble Western blot transfer apparatus, fill reservoir with 1× transfer buffer and transfer at 100V for 1 hour.
10) Block the transferred membrane for at least 1 hour in 5% (w/v) non-fat milk in 1× PBS-Tween-20 or overnight with 3% (w/v) non-fat milk in 1× PBS-Tween-20.
11) Incubate the membrane with primary antibody (1 μg/mL) with 3% (w/v) non-fat milk in 1× PBS-Tween-20 for 1 hour.
12) Wash by rocking immunoblot in 25 mL of 1× PBS-Tween-20 for 10 minutes and decant. Repeat three times.
13) Incubate with appropriate secondary antibody for 1 hour (we use 1:1000 dilution of alkaline phosphatase-conjugated antibody).
14) Wash with 25 mL of 1× PBS-Tween-20 for 10 minutes and decant. Repeat three times.
15) Dissolve 1 tablet of SIGMAFAST BCIP/NBT in 10 mL of deionized H2O. Develop blot to desired intensity, rinse with deionized H2O and let dry. Colour development should occur quickly.
Box 2. Large-scale expression and purification.
Preparation- HEK293T cell scale-up and MaxiPrep DNA TIMING 1 week
1) Scale up HEK293T to ∼2.0 × 108 cells. Three T225 cm2 flasks of HEK293T cells grown to 100% confluency contains ∼2.0 × 108 cells.
2) Purify 1 mg of DNA for transfection using the Qiagen MaxiPrep plasmid purification kit. A 1 L overnight culture of E. coli should produce 2-3 mg of pDISPLAY vector.
Day 1- 10-stack preparation TIMING 1 hour
3) Pre-warm all media to 37°C prior to addition of cells. Add ∼2.0 × 108 HEK293T cells into a 10-layer CellSTACK containing 1.2 L DMEM, 1× pen/strep, 1× GlutaMAX and 5% (v/v) FBS and incubate overnight at 37°C with 5% CO2 to allow for cell attachment and growth.
Day 2- Large-scale expression TIMING 4 days
4) Prepare the calcium phosphate-DNA mixture in laminar flow hood. Mix 1 mg DNA, 6.8 mL of 2 M CaCl2 and 60 mL of deionized H2O on ice. All solutions should be sterile filtered. If DNA is dilute (<0.5 mg/mL), appropriately decrease the amount of deionized water added.
5) Pipet this solution dropwise onto 67 mL of pre-chilled 2× HBS on ice. Vortex or mix well. 2× HBS should be made freshly or stored frozen to prevent pH drift.
6) Incubate on ice for 30 minutes. Solution should become cloudy from formation of a fine white precipitate.
7) Add calcium phosphate-DNA mixture to the media in the 10-layer CellSTACK. For optimal transfection, cells should be at ∼70% confluency. Thoroughly mix with media, distribute evenly over cells and incubate in CO2 incubator (5% CO2) at 37°C for 4 days.
Day 6- Harvest and concentration TIMING 4 hours
8) Harvest supernatant 4 days post-transfection. Centrifuge the supernatant at 10,000g for 20 minutes and filter using a 0.22 μm vacuum filter apparatus. TIP: The 10-layer CellSTACKS can be re-used. Disinfect the CellSTACK by soaking overnight in 1.5 L 10% (v/v) bleach. HEK293T cells should detach. Rinse the CellSTACK copiously with sterile filtered deionized H2O, followed by 1× sterile PBS. Store the CellSTACK in 1.0 L sterile 1× PBS supplemented with 10× pen/strep until use. The CellSTACK layers should appear transparent after cleaning and, in our hand, can be re-used effectively two additional times.
9) Concentrate the supernatant to 150 mL using the Centramate tangential flow filtration system. Buffer-exchange the concentrated sample into PBS by adding 500 mL of 1× PBS and concentrating down to 150 mL, repeating this process five times. Recirculate the sample through the system for 30 minutes to recover additional sample from the membrane.
Day 6- Purification TIMING 1 ½ days
10) Pre-equilibrate a 1 mL anti-HA affinity column with 1× PBS and load sample by gravity at a flow rate <1 mL/min.
11) Wash the column with 10 mL of 1× PBS-Tween-20.
12) Wash the column with 30 mL of 1× PBS.
13) Dissolve the synthetic HA peptide in 1× PBS (1 mg/mL) and pre-warm to 37°C.
14) Pipette 1 mL of HA peptide to the anti-HA column and allow peptide to enter the resin. Collect flow-through. Turn off column flow once peptide reaches the bed height and incubate entire column at 37°C for 15 minutes.
15) Repeat step 13 two additional times.
16) Pipette 1 mL of 1× PBS onto the column and flow into resin until the meniscus reaches the bed height. Collect the flow-through.
17) Pool fractions according to SDS-PAGE analysis.
18) Steps 9-16 can be repeated to capture additional protein.
19) Regenerate anti-HA column with 10 column volumes of 0.1 M glycine pH 2.2, and store at 4°C in 1× PBS with 0.02% (w/v) NaN3.
Box 3. Deglycosylation TIMING 2 days.
-
Perform a time-course test deglycosylation reaction. Set up the following series of digestions at room temperature and at 37°C:
Glycosidase Units of glycosidase Protein (μg) Buffer Final volume (μL) PNGaseF 500 40 8 μL 10× PBS pH 7.4 80 EndoF1 0.04 40 8 μL 1M sodium citrate pH 5.5 80 EndoF2 0.01 40 8 μL 1M sodium acetate pH 4.5 80 EndoF3 0.01 40 8 μL 1M sodium citrate pH 5.5 80 Take 10 μL aliquots at time points of 0, 2, 4, 6, 8, 24, and 48 hours. To stop the reaction, immediately add 3× SDS-PAGE reducing sample buffer to each aliquot and heat sample at 95°C for 5 minutes. Note: In some cases, removal of the terminal sialic acid is required prior to deglycosylation by PNGaseF, EndoH, or the EndoF series. Moreover, sequential or simultaneous deglycosylation by a cocktail of enzymes may be necessary.
Analyze the deglycosylation reaction by Coomassie-stained SDS-PAGE or anti-HA-probed immunoblot analysis.
Select deglycosylation enzyme and reaction condition yielding the most homogeneous sample. Scale up reaction for preparative use.
Separate deglycosylated glycoprotein from enzyme and cleaved glycans using size exclusion chromatography prior to crystallization.
Box 4. Microfluidic crystallization screening TIMING setup: 3 hours; crystal growth: 7 days.
The following is a summary of the Topaz microfluidic crystallization process; consult the manufacturer's instructions for more detailed information.
Concentrate purified protein to 10-20 mg/mL using an Amicon Ultra-0.5 microcentrifugal concentrator. It may be helpful to determine ideal protein concentration for crystallization using the Pre-Crystallization Test (PCT) kit from Hampton Research or Qiagen. In general, initial concentration of protein needs to be higher than that typically used for vapour diffusion experiments, due to the direct mixing of both precipitant and protein in the reaction chambers in the free interface diffusion process.
Pipette 750 μL of appropriately diluted Fluidigm Hydration Fluid onto each PVA strip inside the hydration chamber and close the chamber lid. For most screens, a 2:1 ratio of hydration fluid concentrate to water is sufficient, while for other screens, such as OptiMix-3, a 1:1 dilution is better.
Run the Prep script on the Topaz IFC Controller to activate control valves and charge the hydrated chip.
-
Using an 8-channel micropipettor, add 15 μL of crystallization sparse matrix screen (OptiMix-1,2,3, PEG or other commercial/in-house screens) into the appropriate precipitant well.
CRITICAL STEP: Perform this within 20 hours of steps 2-3 to avoid dehydration of the crystallization reagent in the precipitant well. Examine the chip under a light microscope to ensure the absence of bubbles at the bottom of reagent wells, which would interfere with proper dispensing of reagent into the microfluidic circuitry.
Pipet 1.5 μL of protein into the protein inlet [marked on the chip with blue square(s)].
Run the Load script. Check under a light microscope that all chambers are filled.
Record a time=0 image scan (negative control) using the AutoInspeX II workstation.
Initiate diffusion using the FID Control RT script.
Store screening chip in a vibration-free incubator and monitor for crystal hits and growth over a 7 day period using the AutoInspeX II.
Score the crystallization results (we use a class I-IV scale, Figure 6). Select best crystallization hits (class I or II) for translation to traditional, 0.5-2.0 μL size hanging drop vapour diffusion. Class III or IV crystals may require additional screening in Fluidigm 1.96 screening chips to improve crystal quality. Chances of successful translation increase when crystals of high quality (class I or II) are chosen. See manufacturer's translation guide for more detailed instructions.
Troubleshooting
Exhaustive protocols on tissue culture and expression are available in sources such as Cold Spring Harbor Protocols. In Table 1, we present some of the more common problems and their solutions.
Table 1. Troubleshooting.
| Problem | Possible Reason | Solution |
|---|---|---|
| Test expression | ||
| Cells don't attach | Cells are sick or dead | Check cell viability using trypan blue staining |
| Contamination with bacteria, yeast or fungi | Check cells for signs of contamination under the Microscope | |
| Decontaminate all surfaces, media and equipment. Thaw new cells and start over. | ||
| Media without divalent ions, or wrong plate type used | Check media and tissue-culture appropriate plates | |
| Cloudy medium | Contamination with bacteria, yeast or fungi | Check cells for contamination under the Microscope; Decontaminate |
| No/low test expression levels | Poor transient transfection | Try a higher ratio of transfection reagent to DNA |
| Check that cells are ∼70% confluent before transfection | ||
| Try different transfection reagent | ||
| Cells are dead/not viable | Check cell viability using trypan blue staining | |
| Check for contamination | ||
| Protein is not folded correctly | Check the cell pellet for insoluble, unsecreted protein | |
| Test expression of other construct variants | ||
| Codon usage | Check the DNA sequence for the use of rare Codons; if necessary, synthesize a version of the gene codon-optimize for expression in mammalian cells | |
| Large-scale expression | ||
| Media does not maintain a constant colour/No buffering | Bicarbonate-based buffer is not activated | Ensure a constant level of 5% CO2 |
| Use gas-permeable CellSTACK caps | ||
| Low protein yield | Poor transient transfection | Make sure DNA is precipitated with calcium phosphate. Calcium phosphate-DNA solution should turn cloudy with a fine white precipitate |
| Ensure pH of 2× HBS is 7.1 | ||
| Optimize the amount of DNA | ||
| Check DNA purity; the A260:A280 ratio should be 1.8 or greater | ||
| Contamination with bacteria, yeast or fungi | Check cells under microscope for signs of contamination | |
| Suboptimal growth of cells | Transfection efficiencies may decrease if cells have been passaged for many generations. Start a new culture. | |
| Purification | ||
| BSA contamination after anti-HA purification | Insufficient wash | Increase volume of PBS-Tween-20 wash to 60 mL |
| Increase Tween-20 in wash to 0.2% (v/v) | ||
| Low protein yield allowing excess non-specific binding of BSA | Use a smaller anti-HA column | |
| Optimize construct or codon usage for higher protein yields | ||
| Protein does not bind to purification matrix | Acidic pH/poor buffering | Check pH of sample and buffer exchange into 1× PBS if necessary |
| Loading of sample too rapid | Slow the flow rate (keep under 1 mL/min) | |
| Inaccessible purification tag | Check that purification tag is intact by performing a reduced and denatured Western blot analysis; if accessibility is a problem, sub-clone tag to the opposite terminus or add a linker between the protein and tag | |
| Column was not properly regenerated prior to use | Regenerate column using 0.1 M glycine pH 2.0 | |
| Little or no protein is eluted | Protein is degraded | Add protease inhibitors and perform purification at 4°C |
| Protein retained on column | Elute any column bound protein using 0.1 M glycine pH 2.0, collect eluant, and detect the presence of protein by Western blot; if protein is retained on column, optimize HA-peptide elution protocol by incubating HA peptide for longer periods of time at 37°C | |
| Protein has precipitated on column due to aggregation or instability | Test expression of other construct variants | |
| Deglycosylation | ||
| No or incomplete deglycosylation | Glycan site is sterically hindered | Sequential or simultaneous deglycosylation using a cocktail of glycosidases; use neuraminadase to remove terminal sialic acid, followed by the EndoF series, EndoH, or PNGaseF |
| Point mutations to delete restricted glycan sites | ||
| Degradation of protein | Glycosidase contains contaminating proteases | Use recombinant glycosidases, rather than those purified from natural sources |
| Add protease inhibitors to deglycosylation reaction | ||
| Crystallization | ||
| Some chambers are not filled | Air bubble in channels or in the reagent well | Dispense more reagent/protein in well and reload |
| Crystals disappear/deteriorate over time | Evaporation of the reaction chamber may lead to mechanical stresses on the crystal | Document images of each reaction chamber on a daily basis |
| No crystallization hits | Insufficient sampling of crystallization space | Use different sparse matrix screens to sample alternate regions of crystallization space |
| Protein quality and conformational flexibility | Characterize the protein using DLS, native gels. Strive for homogeneity and monodispersity | |
| Create new protein variants using deuteriumexchange mass spectrometry (DXMS) to identify and eliminate flexible regions26,27. | ||
| Protein solution is undersaturated (especially if all wells are clear) | Increase protein concentration | |
| Abundance of salt crystal hits | Dehydration of reaction chamber | It is normal for reaction chamber to dry out, allowing salt crystals to form over time; be skeptical of crystal hits that appear on Day 7 |
| Ensure the PVA strips are evenly coated with hydration fluid and hydration chamber is tightly closed | ||
| Crystal screens have high salt content as a precipitant | Use sparse-matrix screens that have lower salt concentrations or are primarily PEG-based | |
| Distinguishing salt from protein crystals | Look for salt crystal formation in the precipitant inlet side of reaction chamber or for identical crystals formed by different proteins in same precipitant conditions | |
| Set up control reaction with buffer (no protein) | ||
| In situ diffraction of Topaz crystallization chip crystals at the synchrotron (prototype setup on BL8.3.1 at the Advanced Light Source, Berkeley, CA USA) | ||
| Trouble translating hits to vapour diffusion | Free interface diffusion and vapour diffusion feature different crystallization kinetics | Try different well to protein ratios |
| Try different PEG to salt/additive ratios | ||
| Optimize crystal hits in 1.96 chip to obtain higher quality hits prior to translation | ||
| Try capillary counterdiffusion crystallization | ||
Anticipated Results
The expression and crystallization platform described here was instrumental in determination of the structure of the trimeric, prefusion Zaire ebolavirus glycoprotein (EBOV GP)14. Crystal structures of viral glycoproteins are difficult to achieve given the challenges to production, purification, crystallization and diffraction inherent in the heavily glycosylated, oligomeric, metastable, and flexible nature of these proteins. In the case of EBOV GP, the strategy described here allowed rapid expression screening for over 100 constructs containing various truncations and multiple combinations of mutations to N-x-S/T glycosylation sequons. Moreover, the HEK293T cell productions could be scaled up to quantities which allowed growth and screening of 50,000 crystals. It is anticipated that this platform will be a useful tool not just for glycosylated proteins, but also for mammalian proteins in general.
Constructs selected for scale-up should be characterized by a single strong band at the proper molecular weight on both linear anti-HA and conformation-dependent antibody immunoblots, indicating adequate expression level, monodispersity and proper folding (Figure 5a, for one example). In our laboratory, large-scale expression in HEK293T cells routinely results in the production of ∼2-10 mg of protein/L of media. It is expected that glycans produced in HEK293T cells can be efficiently removed by either PNGaseF or the EndoF series of glycosidases (Figure 5b). However, deglycosylation is often incomplete due to poor steric access of the attachment site by the local conformation of the protein. In the case of EBOV GP, resistant glycan sites were removed by site-directed mutagenesis of the N-x-S/T glycosylation sequons14.
Figure 5. Test expression and deglycosylation of glycoproteins.
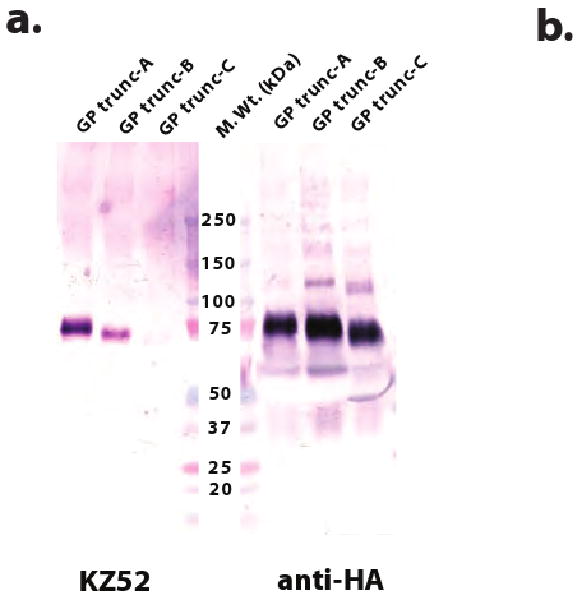
(a) Small-scale expression screening of selected Zaire ebolavirus GP truncation variants. Conditioned media containing EBOV GP variants was harvested 4 days post-transfection, and 10 μL of each culture supernatant was separated on a non-denaturing 10-15% gradient SDS-Tris-HCl polyacrylamide gel and probed with mAb-anti-HA or mAb-KZ52 (conformational). The KZ52 antibody28 was isolated from a human survivor of a 1995 outbreak in Kikwit, Democratic Republic of the Congo (formerly Zaire) and recognizes an epitope bridging both the attachment and fusion subunits of Zaire ebolavirus GP14. All EBOV GP variants were overexpressed and secreted, as illustrated by the anti-HA probed immunoblot. However, only wild-type GP and GP trunc-A were recognized by KZ52, suggesting that this particular truncation has not significantly altered the native fold. (b) Control of N-linked glycosylation in HEK293T-expressed EBOV GP. Coomassie-stained, 10-15% gradient, SDS-Tris-HCl polyacrylamide gel analysis of EBOV WT GP trunc-A (lane 1), PNGaseF- (lane 3) or EndoF3- (lane 4) treated GP trunc-A. As a positive control for complete deglycosylation, EBOV GP trunc-A was denatured and subsequently treated with PNGaseF (lane 2). Note that two sites are resistant to deglycosylation; analysis of the crystal structure confirms that local protein structure at these sites would limit enzyme access.
Fluidigm microfluidic free interface diffusion crystallization uses only 6 μL of protein (10 mg/mL) to screen 384 sparse matrix conditions and yields results within 7 days. Protein crystals usually appear closer to the right-hand side of the chamber, which is the region of highest protein concentration. However, the concentration gradient formed in the horizontal axis of the chamber may sometimes permit crystallization in the middle of the chamber. Crystals closest to or contained within the precipitant inlet side (far left) are usually salt. Crystallization hits can be scored into four classes (I-IV; Figure 6). Class I crystals appear perfect, with sharp edges and thickness in three dimensions. Class II crystals are similar to class I, but have minor defects, such as irregular, jagged or rounded edges or satellites. Class III crystals are small needle or crystalline clusters, while class IV crystals are showers of microcrystals or spherulites. In the case of EBOV GP and other projects in our group, the success of translation is highly dependent on the quality of the crystal hit and number of unique hits obtained. Class I and II crystal hits have high translation percentages and can often be translated directly into similar conditions of the crystallization screen. Translation of class III and IV crystals tends to be difficult and likely requires additional optimization of pH, salts, PEG or additives using a 1.96 chip to yield more crystalline class I and II-type crystals prior to translation.
Figure 6. Crystallization using microfluidic free interface diffusion.
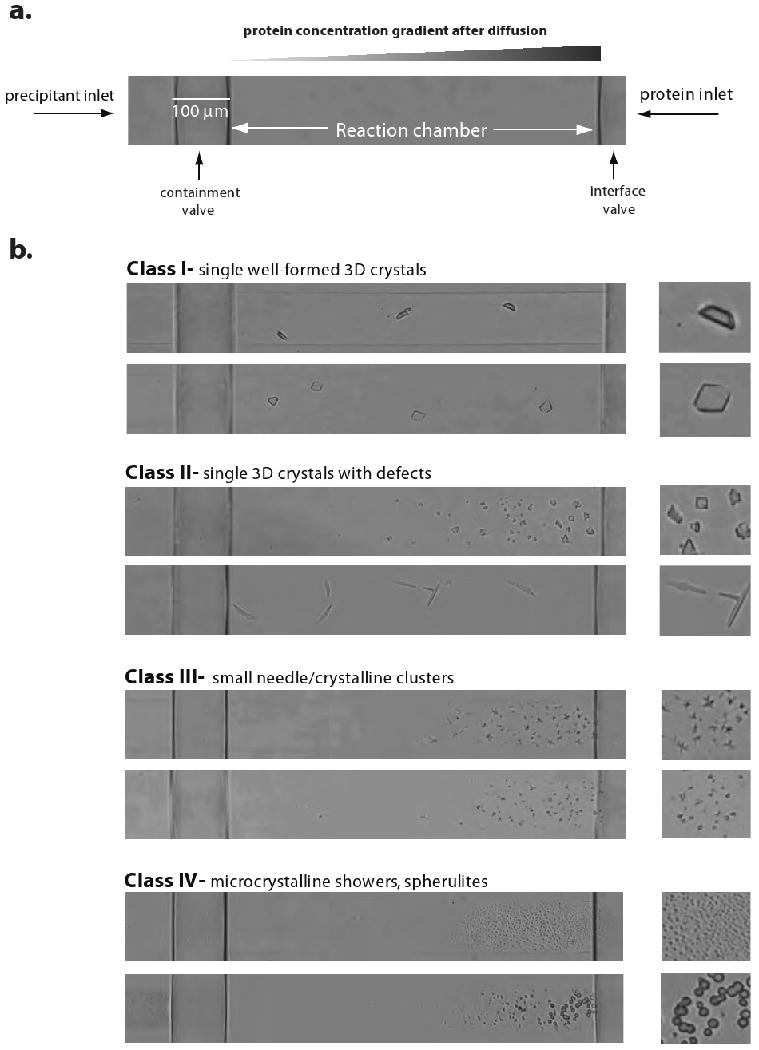
(a) In each reaction chamber of a Topaz crystallization chip, the release of the containment and interface valves allows protein and precipitant to enter from the right and left sides of the chamber, respectively. This creates a protein and precipitant concentration gradient in the chamber, which allows the sampling of a wide area of crystallization space. (b) Selected examples of EBOV GP-KZ52 crystals belonging to the four classes of hits obtained by free interface diffusion. Classes I and II are characterized by single three-dimensional crystals with no or minor visible defects, and are immediately suitable for translation to vapour diffusion experiments. Class III and IV hits are typically poor quality clusters or microcrystals and probably require further optimization prior to translation.
Figure 1. Flowchart of the HEK293T-microfluidic crystallization screening platform.

Using this system, constructs can be screened for expression and crystallization and scaled-up for structural studies in a matter of five weeks.
Acknowledgments
The authors would like to thank Dr. Robyn Stanfield at The Scripps Research Institute for assistance with the Fluidigm microfluidic system and members of the Ollmann Saphire laboratory for advice and help. This work is supported by NIH NIAID operating grants (AI053423 and AI067927) and a Career Award by the Burroughs Wellcome Fund to E.O.S. and a fellowship to J.E.L. from the Canadian Institutes of Health Research. This is manuscript #XXXXX from The Scripps Research Institute.
References
- 1.Kwong PD, et al. Probability analysis of variational crystallization and its application to gp120, the exterior envelope glycoprotein of type 1 human immunodeficiency virus (HIV-1) J Biol Chem. 1999;274:4115–4123. doi: 10.1074/jbc.274.7.4115. [DOI] [PubMed] [Google Scholar]
- 2.Cockett MI, Bebbington CR, Yarranton GT. High level expression of tissue inhibitor of metalloproteinases in Chinese hamster ovary cells using glutamine synthetase gene amplification. Biotechnol. 1990;8:662–667. doi: 10.1038/nbt0790-662. [DOI] [PubMed] [Google Scholar]
- 3.Cereghino JL, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 4.Bernard A, Payton M, Radford KR. Protein expression in the baculovirus system. Curr Protoc Protein Sci. 2001;Chapter 5(Unit5):5. doi: 10.1002/0471140864.ps0505s00. [DOI] [PubMed] [Google Scholar]
- 5.Shrestha B, Smee C, Gileadi O. Baculovirus expression vector system: an emerging host for high-throughput eukaryotic protein expression. Methods Mol Biol. 2008;439:269–289. doi: 10.1007/978-1-59745-188-8_19. [DOI] [PubMed] [Google Scholar]
- 6.Kost TA, Condreay JP, Jarvis DL. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat Biotechnol. 2005;23:567–575. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith GE, Summers MD, Fraser MJ. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol Cell Biol. 1983;3:2156–2165. doi: 10.1128/mcb.3.12.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatia PK, Mukhopadhyay A. Protein glycosylation: implications for in vivo functions and therapeutic applications. Adv Biochem Eng Biotechnol. 1999;64:155–201. doi: 10.1007/3-540-49811-7_5. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins N, Curling EM. Glycosylation of recombinant proteins: problems and prospects. Enzyme Microb Technol. 1994;16:354–364. doi: 10.1016/0141-0229(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 10.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiol. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins N, Parekh RB, James DC. Getting the glycosylation right: implications for the biotechnology industry. Nat Biotechnol. 1996;14:975–981. doi: 10.1038/nbt0896-975. [DOI] [PubMed] [Google Scholar]
- 12.Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. 2006;D62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- 13.Chang VT, et al. Glycoprotein structural genomics: solving the glycosylation problem. Structure. 2007;15:267–273. doi: 10.1016/j.str.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JE, et al. Structure of the Ebola virus glycoprotein bound to a human survivor antibody. Nature. doi: 10.1038/nature07082. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demeneix B, Behr JP. Polyethylenimine (PEI) Adv Genet. 2005;53:217–230. [PubMed] [Google Scholar]
- 16.Kichler A. Gene transfer with modified polyethylenimines. J Gene Med. 2004;6 1:S3–10. doi: 10.1002/jgm.507. [DOI] [PubMed] [Google Scholar]
- 17.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chirifu M, et al. Crystal structure of the IL-15-IL-15Ralpha complex, a cytokine-receptor unit presented in trans. Nat Immunol. 2007;8:1001–1007. doi: 10.1038/ni1492. [DOI] [PubMed] [Google Scholar]
- 19.English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. Structural basis for the histone chaperone activity of Asf1. Cell. 2006;127:495–508. doi: 10.1016/j.cell.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. 2008;105:1820–1825. doi: 10.1073/pnas.0712064105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevens J, et al. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 22.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu SH, Latham CF, Gee CL, James DE, Martin JL. Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc Natl Acad Sci USA. 2007;104:8773–8778. doi: 10.1073/pnas.0701124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kothe M, et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry. 2007;46:5960–5971. doi: 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- 25.Yasui N, et al. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc Natl Acad Sci USA. 2007;104:9988–9993. doi: 10.1073/pnas.0700438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantazatos D, et al. Rapid refinement of crystallographic protein construct definition employing enhanced hydrogen/deuterium exchange MS. Proc Natl Acad Sci USA. 2004;101:751–756. doi: 10.1073/pnas.0307204101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spraggon G, et al. On the use of DXMS to produce more crystallizable proteins: structures of the T. maritima proteins TM0160 and TM1171. Protein Sci. 2004;13:3187–3199. doi: 10.1110/ps.04939904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maruyama T, et al. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol. 1999;73:6024–6030. doi: 10.1128/jvi.73.7.6024-6030.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]