

Abstract
Monitoring post-control transmission of schistosomes by examining humans becomes less effective as infection rates among humans decrease. Molecular monitoring of prepatent schistosome infection in snails by the polymerase chain reaction (PCR) has been used for studying human-to-snail transmission, and snail prepatent infection rates were found to correspond to infection prevalence and average intensity in human populations contacting the sites studied. We have now developed loop-mediated isothermal amplification (LAMP) assays for identifying Schistosoma mansoni and S. haematobium to facilitate large-scale evaluation of post-intervention transmission potential. LAMP primers were designed based on the Sm1-7 and DraI repeated sequences of the corresponding schistosomes, and amplification by LAMP of these 121-basepair highly abundant sequences provided a detection sensitivity of 0.1 fg of genomic DNA. When these LAMP assays were applied for examining infected laboratory snails, it was possible to identify infection from the first day after exposure to miracidia. The potential advantages of these assays are discussed.
Introduction
The most widely used methods for assessing schistosomiasis endemicity are based on examining the prevalence of Schistosoma infection among humans. The standard screening assays rely on detection of excreted eggs in stool or urine, or the use of questionnaires for self-report of characteristic symptoms.1,2 Such approaches are frequently used for targeting population-based mass drug administration (MDA) in schistosomiasis control campaigns.3 However, despite MDA efforts, Schistosoma transmission usually persists to a greater or lesser extent after delivery of treatment.4 After MDA, human prevalence decreases and the remaining infections are mostly of light intensity, but because of continuing local transmission and exposure to reinfection, risk remains for patients to redevelop schistosomiasis-related morbidities such as anemia, undernutrition, and decreased performance status.5 In addition, after MDA, the number of negative stool or urine test results increases and the performance characteristics of standard diagnostic tests are decreased (especially in terms of sensitivity and negative predictive value), and test-to-test variability is increased.6 Furthermore, where elimination is contemplated, the uncertainty of a zero value poses a programmatic challenge because much larger sample sizes are needed to obtain a high degree of confidence that the observed rate is actually zero.
The detection of schistosome eggs in feces or in urine is particularly insensitive for detection of low-level or light infections,7 and in primate models, egg counts and circulating antigen tests regularly miss detection of infection intensities of up to 10–20 worm pairs per person.8 Infection surrogates, such as microhaematuria in S. haematobium infection, also lose their diagnostic reliability in low-level infections,9 and positive antibody test results (indicative of Schistosoma exposure) may take an indefinite period of time to convert to negativity after elimination of infection.
For more in-depth assessment of post-intervention Schistosoma transmission, various monitoring approaches have been tried to complement or replace human testing. Among these are monitoring of human water use activity,10,11 estimation of egg contamination of water by human excreta,12 detection of cercarial shedding by snails,13 and cercariometry.14,15 Of these approaches, cercarial shedding has been most extensively used (in combination with human water contact data) to provide estimates of snail-to-human transmission. However, cercarial shedding can be highly focal16 and of low frequency, even in areas of significant transmission.17 Where snail shedding rates are low, quite extensive efforts at snail collection and testing are required to accurately assess the impact of intervention on levels of patent snail infection and risk of human re-infection.
To address this issue, we have focused on the development of molecular monitoring of early prepatent (non-shedding) plus patent (shedding) snail infections as a tool for large-scale tracking of residual transmission. We developed polymerase chain reaction (PCR) assays for amplification of the Sm1-7 repeated sequence of S. mansoni,18,19 and of the DraI repeated sequence of S. haematobium.20 These PCRs have proven to be highly sensitive and specific tools for detecting the respective schistosomes in biological materials. By virtue of the fact that snails at all stages of prepatency and patency are detected, we found it possible to detect surprisingly high (30–50%) rates of snail infection persisting at sites where community-based control had reduced human prevalence but not transmission of S. haematobium.21 Cercarial shedding rates did not correspond to infection rates in the human population, whereas rates of snail PCR positivity did.21,22 Molecular detection of snail infection rates appears to be a suitable marker of persisting infection in the human population. This approach resembles those used for assessing transmission of lymphatic filariasis by PCR testing of insect vectors.23 Both methods are examples of xenomonitoring, i.e., monitoring infection in the intermediate host for obtaining evidence on infection in humans.
In rural areas of developing countries, large-scale implementation of molecular transmission monitoring will require tools that are more user-friendly than PCR. The PCR is dependent on complex technology and on specialized training in molecular biology. As an alternative, the loop-mediated isothermal amplification (LAMP) technique may provide the answer to program monitoring needs. The LAMP, initially described in 2000,24 has rapidly gained acceptance for detection of a variety of infectious agents including Plasmodium falciparum and S. japonicum.25,26 This technique does not require complex equipment for DNA amplification or for amplicon detection,27 and is potentially suitable for molecular monitoring in basic laboratory facilities.28 In the present study, we describe the development of two new LAMP assays for detecting S. haematobium and S. mansoni DNA in biological materials and their sensitivity for detecting infected snails.
Materials and Methods
Snails and schistosomes.
Snails of the Puerto Rican strain of Biomphalaria glabrata maintained at Tel Aviv University were exposed individually, when approximately 6 mm in size, to 10 miracidia of an Egyptian strain of Schistosoma mansoni. Biomphalaria glabrata obtained from the Natural History Museum (London, United Kingdom) (NHM no. 3056), were individually exposed to 10 miracidia of S. mansoni, obtained from Besoum, Cameroon. Bulinus wrighti, Oman (NHM No. 3053) were individually exposed, when approximately 3 mm in size, to 5 miracidia of S. haematobium isolated at Ouro Doukoudje, Cameroon. The various snails were preserved in absolute ethanol after survival for 1 day, 6–7 days, or 14 days post-exposure. These specimens were then used for LAMP assay development and standardization.
Adult worms.
Schistosoma haematobium adult worms from Zanzibar were preserved in liquid nitrogen as part of the schistosome collection at the Natural History Museum (London, United Kingdom). They were preserved in absolute ethanol for shipment to Jerusalem and for subsequent storage. Schistosoma mansoni adult worms were harvested from infected mice by perfusion29 and preserved in ethanol.
DNA extraction.
DNA from adult worms was prepared as previously described. Briefly, the worms were kept in lysis buffer (0.1 M EDTA, pH 8.0, 0.1 M Tris-HCl, pH 7.5, 0.2 M NaCl, 1% sodium dodecyl sulfate, 0.2% β-mercaptoethanol, and 100 μg of proteinase K), at 65°C for 1–2 hours. Lysis was followed by phenol-chloroform extraction.
DNA from snails.
Laboratory-infected snails and uninfected snails, which were kept in ethanol, were washed with TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) diluted 1:10, and dried on tissue paper for few seconds to remove excess liquid. This procedure was followed by pulling out the body of the snail from the shell by using a fine needle. Tissue of each snail was kept in separate 1.5-mL tubes and covered with 200 μL of lysis buffer (DNeasy Blood and Tissue Kit; Qiagen, Valencia, CA). DNA extraction was conducted according to the manufacturer's instructions. Extracted DNA was eluted in 200 μL of TE buffer, and a 5-μL aliquot was tested in each LAMP reaction. DNA extraction from snails was also carried by using the High Pure PCR Template Preparation Kit (Roche Diagnostics, Basel, Switzerland). Similar results were obtained.
LAMP primer design.
Sets of forward and backward external primers (F3 and B3) and forward and backward internal primers (FIP and BIP) were designed for S. haematobium and for S. mansoni on the basis of respective tandem repeated sequences DraI20 and Sm1-7,18 each 121 basepairs. Selection of primers used required a number of preliminary LAMP experiments for optimization. Selected LAMP primers are shown in Table 1, and their locations within the respective target repeated sequences are shown in Figure 1.
Table 1.
Loop-mediated isothermal amplification primer sets used for Schistosoma haematobium and S. mansoni DNA amplification*
| Primer set | Primer position | Primers sequence (5′→3′) |
|---|---|---|
| S. haematobium (DraI) | F3 | gat ctc acc tat cag acg |
| B3 | GTC ACC AAT AAT ATG AAAC | |
| FIP: F1c + F2 | CCACCAACAATTTTAAA TTTT ATCAGACGAAAC AAAGAAAAT | |
| BIP: B1c + B2 | GGTCGTATCGTTGTGAAA TTTT CACCAATAATA TGAAACAAT | |
| S. mansoni (Sm1-7) | F3 | GAT CTG AAT CCG ACC AAC CG |
| B3 | AAC GCC CAC GCT CTC GCA | |
| FIP: F1c + F2 | AAATCCGTCCAGTGGTTT TTTT GAAAATCGTTGT ATCTCCG | |
| BIP: B1c + B2 | CCGAAACCACTGGACGGA TTTT TATTTTTAATCT AAAACAAAC ATC |
Figure 1.
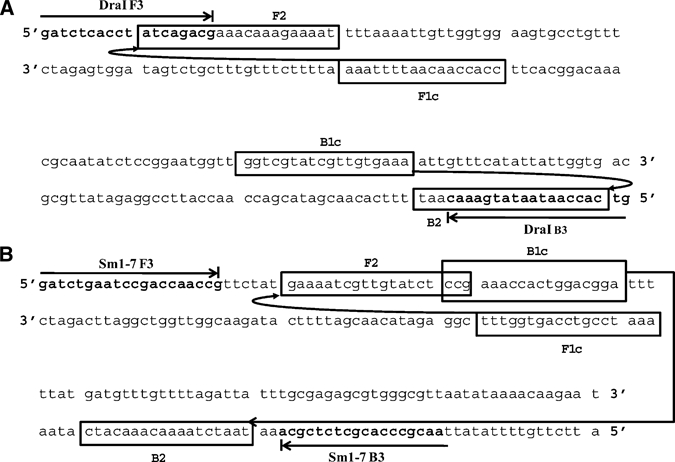
Location of the loop-mediated isothermal amplification forward (F3) and backward (B3) external primers and of internal forward (FIP) and backward (BIP) primers within the respective sequences DraI of Schistosoma haematobium (A) and Sm1-7 of S. mansoni (B).
LAMP assays.
Reaction mixture (25 μL) and amplifications.
The reaction mixture contained primers (40 pmol of FIP and BIP and 5 pmol of F3 and B3 outer primers), DNA polymerase, 8 units of BstI large fragment, 1 mM dNTPs, 0.8 M betaine; 1× reaction buffer (20 mM Tris-HCl, pH 8.8, 10 mM KCl, 10 mM (NH4)2 SO4, 8 mM MgSO4, and 1% Tween 20), and target DNA from S. haematobium or S. mansoni. The reaction was incubated at 63°C in a water bath for 2 hours.
Amplicon detection.
Detection of LAMP-amplified products was performed by using SYBR Green I stain (Invitrogen, Carlsbad, CA). One microliter of 1:50 diluted SYBR Green I was added to the reaction mixture (25 μL). Amplified DNA was seen by exposure to ultraviolet light at 320 nm and subjected to black and white photography. When using SYBR Green at a dilution of 1:10, amplicons could be detected directly by the unaided eye because the color of the reaction solution changed from orange to green in the presence of LAMP amplicon. Amplicon analysis by standard gel electrophoresis was carried out in parallel with SYBR Green staining for quality control.
Results
LAMP amplification of S. haematobium DNA for determining detection sensitivity.
Serial 10-fold dilutions of S. haematobium genomic DNA were amplified by LAMP using DraI LAMP primers (Table 1 and Figure 1A). The results of this amplification experiment are shown in Figure 2A after amplicon examination by agarose gel electrophoresis and staining with SYBR Green I and exposure to ultraviolet light (Figure 2B). Detection sensitivity of this assay was 0.1 fg, which was 10-fold higher than the detection sensitivity achieved by using PCR.19,20
Figure 2.
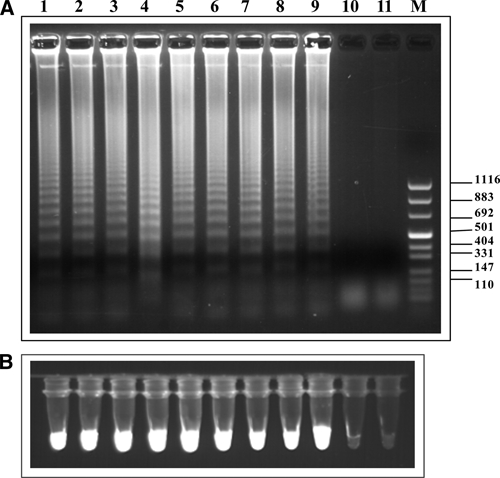
A, Agarose gel electrophoresis of loop isothermal amplified products (LAMP) from different concentrations of Schistosoma haematobium genomic DNA. Ten-fold serial dilutions starting from 10 ng genomic DNA (lane 1) down to 0.001 fg (lane 11) were tested. Lane M, molecular mass marker. B, Fluorescence of LAMP products as indicated in A using SYBR Green I staining followed by ultraviolet illumination at 320 nm. Values on the right are in basepairs
LAMP amplification of S. haematobium-infected snails.
For identification of snails infected with S. haematobium, we applied LAMP by using S. haematobium DraI LAMP primers. The results of amplicon detection in these assays by agarose gel electrophoresis are shown in Figure 3. Infected snails were detected from day 1 after exposure to miracidia. Detection by SYBR Green–generated fluorescence was not possible in the case of examining snails because uninfected snails also showed positive fluorescent signals given the amount of snail DNA present in the reaction mixture. In comparison, differential detection of infected snails by using SYBR Green dye, which is less sensitive than fluorescence, was possible (not shown). Thus, in the presence of amplicon DNA, the DNA level was within the detectable range for SYBR Green, and the color change from orange to green was observed.
Figure 3.
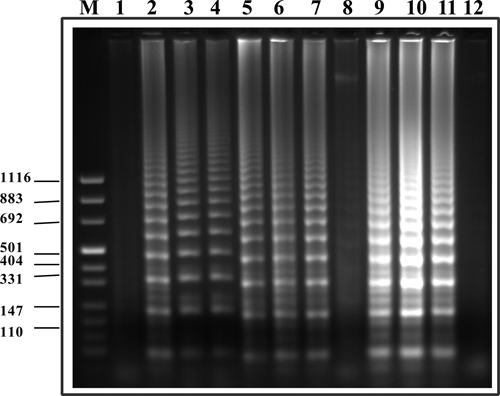
Agarose gel electrophoresis of loop-mediated isothermal amplification products after targeting DraI S. haematobium repetitive sequence within infected snails. Lanes 1 and 8, uninfected snails; lanes 2–4, snails after one day of infection; lanes 5–7, snails after seven days of infection; lanes 9–11, snails after 14 days of infection; lane 12, negative control (no DNA); lane M, molecular mass marker. Values on the right are in basepairs.
LAMP amplification of S. mansoni DNA for determining detection sensitivity.
As described earlier regarding determination of the sensitivity of LAMP for detecting S. haematobium DNA, the same process was used for amplifying serial 10-fold dilutions of S. mansoni genomic DNA by using a LAMP assay and corresponding Sm1-7 LAMP primers (Table 1 and Figure 1B). The results of this amplification are shown in Figure 4, which shows results of agarose gel electrophoresis (Figure 4A) and parallel results of staining with SYBR Green I and exposure to ultraviolet light at 320 nm (Figure 4B). Detection sensitivity of this LAMP assay was 0.1 fg, which is 10-fold more sensitive than that of the corresponding PCR assay.18,20
Figure 4.
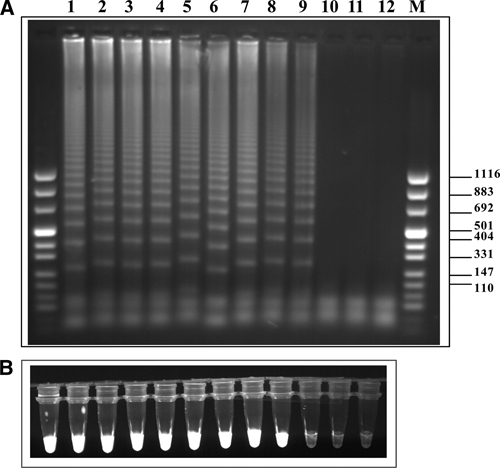
A, Agarose gel electrophoresis of loop-mediated isothermal amplification (LAMP) products from different concentrations of Schistosoma mansoni genomic DNA. Ten-fold serial dilutions starting with 10 ng of genomic DNA (lane 1) down to 0.001 fg (lane 11) were tested. Lane 12, negative control (no DNA); lane M molecular mass marker. B, Fluorescence of LAMP products as indicated in A using SYBR Green I staining followed by ultraviolet illumination at 320 nm. Values on the right are in basepairs.
LAMP amplification of S. mansoni-infected snails.
For identification of snails infected with S. mansoni, we applied the LAMP assay and Sm1-7 primers. The results of amplicon detection by agarose gel electrophoresis in these assays are shown in Figure 5. Detection by SYBR Green–generated fluorescence was not possible in this case, but detection by SYBR Green color change was possible (not shown). Infected snails were detected from day 1 after exposure to miracidia.
Figure 5.
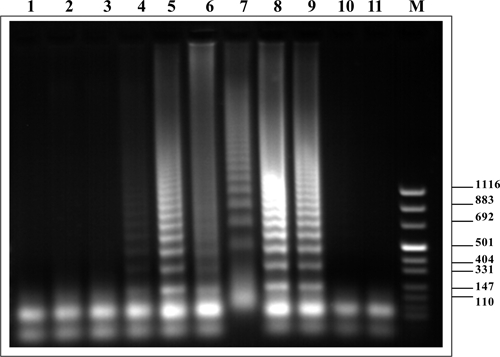
Agarose gel electrophoresis of loop-mediated isothermal amplification products after targeting Sm1-7 Schistosoma mansoni repetitive sequence from infected snails. Lanes 1–3, uninfected snails; lanes 4–6, snails after one day of infection; lanes 7–9, snails after seven days of infection; lanes 10 and 11, negative control (no DNA); lane M: molecular mass marker. Values on the right are in basepairs.
Discussion
With the advent of large-scale mass treatment campaigns for schistosomiasis in Africa, the ecology of S. mansoni and S. haematobium transmission in disease-endemic areas is likely to change. An essential factor in determining the overall effectiveness of population-based treatment is whether such broad treatment coverage to humans can significantly reduce local transmission and reduce the risk for reinfection among high risk subgroups, such as school age children. Consistent with the need for new tools for monitoring schistosomiasis transmission, we previously developed two PCR assays for sensitive detection identification of infected snails,18–20 and provided evidence that detection of snail infection by PCR enables identification of a large proportion of infected snails even where cercarial shedding (patency) is rare.21,22 Furthermore, in Coastal Kenya, snail prepatent infection rates (as measured by PCR) were found to correspond to prevalence and average intensity of infection in human communities that had specific transmission sites.21,22 This xenomonitoring, which is based on determining snail infection rates, provided a measure of transmission potential (from the human population) that persisted after community-based drug therapy. With the potential advantages of this molecular monitoring in mind, we have developed new, simpler DNA amplification assays for S. haematobium and for S. mansoni based on the LAMP assay.24 This assay appears to provide technology that can facilitate large-scale monitoring of prepatent snail infection that will, in turn, enable better assessment of intervention impact on parasite transmission.
The LAMP assay has already been used for amplification of DNA of other microorganisms including parasites, notably S. japonicum.26 Unlike PCR, LAMP does not require amplification cycles by thermocycling or amplicon detection by electrophoresis. Given these features, LAMP is potentially useful for work in the field and has already used in rural laboratories in developing areas for the diagnosis of tuberculosis.28
In the present study, we have tested laboratory-infected snails, and some points still need to be addressed in considering LAMP implementation in the field. First, a simple and inexpensive DNA preparation method is required. In the present study, as in most other studies of LAMP development, DNA extraction kits were used for pre-isolation of target nucleic acid material. For achieving field applicability, the expensive DNA extraction kits should be replaced by user-friendly and affordable DNA preparation tools, particularly where large numbers of samples are to be examined. Also, to be applicable in field laboratories, LAMP reaction mixtures will need to be premixed, ready for use, and storable under field-laboratory conditions. Reaction mixtures having these features are now available for PCR30 and can likely be developed for LAMP. For now, practical use of LAMP in field laboratories for diagnosis of schistosomiasis and for monitoring schistosomiasis transmission will require further system development and validation. This development and validation can be performed in association with ongoing intervention projects and in other areas where schistosomiasis prevalence is decreasing or approaching elimination.
Diagnosis of schistosomiasis mansoni infection in humans by using Sm1-7 sequence amplification for copro-PCR,31 or plasma-PCR32 has already been accomplished and used as a tool for detection of low-grade infections, with high detection sensitivity. Using LAMP assays for this purpose should add the inbuilt operational facility of LAMP and another order of magnitude of detection sensitivity, namely 0.1 fg (Figures 2 and 4) compared with 1 fg detection sensitivity of the corresponding PCR assays.18,20 The high detection sensitivity of the LAMP assays should facilitate examination of pooled snails for monitoring larger areas or in areas where transmission has reached low values, which requires testing of many snails. Such a pooling strategy has facilitated detection of infected filarial vectors.33–35
The specificity of the LAMP assay depends largely on the primers used. We have used primers designed from the Sm1-7 repeated sequence of S. mansoni and the DraI sequence of S. haematobium. These repeated sequences are 121 basepairs and are shorter than sequences typically targeted for LAMP, (e.g., the LAMP primer design site [http://primerexplorer.jp/v4_manual/02.html], which provides primer design details only for sequences ≥ 200 basepairs). The independent primer design of our relatively short sequences, as shown in Figure 1, required multiple trials for optimization. The high abundance of these sequences, their tandem arrangement in large arrays, and their well-studied specificity provided the reasoning for developing corresponding LAMP assays based on these relatively short sequences. We have found that avoiding excessive overlap between sequences of the internal and external primers was important for successful amplification. It should be mentioned that the S. japonicum retrotransposon SjR2 has been recently targeted for diagnosis by LAMP,26 and retrotransposons of other schistosomes may also prove suitable for this purpose if providing suitable detection sensitivity and specificity.
Regarding specificity, detection of S. haematobium DraI sequence, which is group specific,20 can provide a basis for monitoring S. haematobium transmission in areas where animal schistosomes, that share this sequence, are rare. Other groups have used other sequences for schistosome species differentiation, such as cyclooxygenase I36 and internal transcribed spacer 1.37 We have used the Sh110/SmSL inter-repeat sequence of S. haematobium for designing primers capable of differentiating S. haematobium from related animal schistosomes by using PCR38 with a detection sensitivity of 10 pg, which is higher than that described for other tests designed for this purpose.36,37 Preliminary results obtained by a LAMP assay based on this inter-repeat sequence indicate that its detection sensitivity can reach an even higher range. Considering that S. bovis and S. mattheei, the major animal schistosomes related to S. haematobium, are not detected by Sh110/SmSL-PCR,38 a corresponding LAMP assay is not expected to produce amplicons and should therefore enable a clear differentiation of S. haematobium DNA from DNA of these abundant animal schistosomes.
Given that major differences can exist in snail-schistosome compatibility in different areas,39 snail prepatency rates (as determined by snail molecular monitoring) cannot be expected to serve as a direct indicator of snail-to-human transmission potential. However, changes in snail prepatency rates over time after an intervention can be expected to reflect changes in the force of transmission from humans to snails in the study area. This finding, in turn, is likely the outcome of changes in prevalence and mean intensity of infection among humans in the same area, as we have previously found in Coastal Kenya.22 However, this concept has to be tested in various areas of defined levels of endemicity. Examination of this monitoring approach will be greatly facilitated if the tests can be carried out in field laboratories close to the transmission sites, rather than employing PCR–based testing in remote central laboratories. Using LAMP-based assays will thus enable making use of field laboratories often located in areas endemic for schistosomiasis, without requiring expensive instruments and without having to train special cadres of molecular technologists. The relevance of snail molecular-monitoring by LAMP-based assays will be even more pronounced once area-wide validation will evolve into large-scale application alongside control campaigns.
Acknowledgments
We thank Dr. David Rollinson and Dr. Aidan Emery (Department of Zoology, Natural History Museum, London United Kingdom) and Professor Daniel Gold (Tel-Aviv University, Tel-Aviv, Israel) for providing the snails used in this study.
Footnotes
Financial support: This study was supported by National Institutes of Health research grants R01TW008067 and R21AI076672 funded by the Fogarty International Center and the National Institute of Allergy and Infectious Diseases.
Authors' addresses: Ibrahim Abbasi and Joseph Hamburger, Department of Parasitology, Kuvin Center for the Study of Infectious and Tropical Diseases, Hebrew University, Hadassah Medical School, Jerusalem, Israel, E-mails: ibrahima@ekmd.huji.ac.il and hambu@cc.huji.ac.il. Charles H. King, Center for Global Health and Diseases, Case Western Reserve University School of Medicine, Cleveland, OH, E-mail: chk@cwru.edu. Eric M. Muchiri, Division of Vector Borne and Neglected Tropical Diseases, Ministry of Public Health and Sanitation, Nairobi, Kenya, E-mail: ericmmuchiri@gmail.com.
References
- 1.Rabarijaona LP, Boisier P, Ravaoalimalala VE, Jeanne I, Roux JF, Jutand MA, Salamon R. Lot quality assurance sampling for screening communities hyperendemic for Schistosoma mansoni. Trop Med Int Health. 2003;8:322–328. doi: 10.1046/j.1365-3156.2003.01019.x. [DOI] [PubMed] [Google Scholar]
- 2.Lengeler C, Utzinger J, Tanner M. Screening of schistosomiasis with questionnaires. Trends Parasitol. 2002;18:375–377. doi: 10.1016/s1471-4922(02)02318-8. [DOI] [PubMed] [Google Scholar]
- 3.Fenwick A. New initiatives against Africa's worms. Trans R Soc Trop Med Hyg. 2006;100:200–207. doi: 10.1016/j.trstmh.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Satayathum SA, Muchiri EM, Ouma JH, Whalen CC, King CH. Factors affecting infection and reinfection with Schistosoma haematobium in coastal Kenya: survival analysis during nine-year, school-based treatment program. Am J Trop Med Hyg. 2006;75:83–92. [PMC free article] [PubMed] [Google Scholar]
- 5.King C, Dickman K, Tisch DJ. Reassessment of the cost of chronic helminthic infection: a meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet. 2005;365:1561–1569. doi: 10.1016/S0140-6736(05)66457-4. [DOI] [PubMed] [Google Scholar]
- 6.Carabin H, Marshall CM, Joseph L, Riley S, Olveda R, McGarvey ST. Estimating the intensity of infection with Schistosoma japonicum in villagers of Leyte, Phillipines. Part I: a Bayesian cumulative logit model. The Schistosomiasis Transmission and Ecology Project (STEP) Am J Trop Med Hyg. 2005;72:745–753. [PubMed] [Google Scholar]
- 7.Noya BA, Guevara RR, Colmenares C, Losada S, Noya O. Low transmission areas of schistosomiasis in Venezuela: consequences on the diagnosis treatment, and control. Mem Inst Oswaldo Cruz. 2006;101((Suppl 1)):29–35. doi: 10.1590/s0074-02762006000900006. [DOI] [PubMed] [Google Scholar]
- 8.Wilson A, van Dam GJ, Kariuki TM, Farah IO, Deelder AM, Coulson PS. The detection limits for estimates of infection intensity in schistosomiasis mansoni established by a study in non-human primates. Int J Parasitol. 2006;36:1241–1244. doi: 10.1016/j.ijpara.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Savioli L, Hatz C, Dixon H, Kisumku UM, Mott KE. Control of morbidity due to Schistosoma haematobium on Pemba Island: egg excretion and hematuria as indicators of infection. Am J Trop Med Hyg. 1990;43:289–295. doi: 10.4269/ajtmh.1990.43.289. [DOI] [PubMed] [Google Scholar]
- 10.Chandiwana SK, Woolhouse ME, Bradley M. Factors affecting the intensity of reinfection with Schistosoma haematobium following treatment with praziquantel. Parasitology. 1991;102:73–83. doi: 10.1017/s0031182000060364. [DOI] [PubMed] [Google Scholar]
- 11.Fulford AJ, Ouma JH, Kariuki HC, Thiongo FW, Klumpp R, Kloos H, Sturrock RF, Butterworth AE. Water contact observations in Kenyan communities endemic for schistosomiasis: methodology and patterns of behaviour. Parasitology. 1996;113:223–241. doi: 10.1017/s0031182000082007. [DOI] [PubMed] [Google Scholar]
- 12.Vercruysse J, Shaw DJ, Bont J. Index potential contamination for schistosomiasis. Trends Parasitol. 2001;17:256–261. doi: 10.1016/s1471-4922(01)01937-7. [DOI] [PubMed] [Google Scholar]
- 13.Jordan P. Schistosomiasis: The St. Lucia Project. Cambridge, United Kingdom: Cambridge University Press; 1985. [Google Scholar]
- 14.Ouma JH, Sturrock RF, Klumpp PK, Kariuki HC. A comparative evaluation of snail sampling and cercariometry to detect Schistosoma mansoni transmission in a large-scale, longitudinal field-study in Machakos, Kenya. Parasitology. 1989;99:349–355. doi: 10.1017/s0031182000059060. [DOI] [PubMed] [Google Scholar]
- 15.Aoki Y, Sato K, Muhoho ND, Noda S, Kimura E. Cercariometry for detection of transmission sites for schistosomiasis. Parasitol Int. 2003;52:403–408. doi: 10.1016/s1383-5769(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 16.Woolhouse ME, Chandiwana SK. Spatial and temporal heterogeneity in the population dynamics of Bulinus globosus and Biomphalaria pfeifferi and the epidemiology of their infection with schistosomes. Parasitology. 1989;98:21–34. doi: 10.1017/s0031182000059655. [DOI] [PubMed] [Google Scholar]
- 17.Kariuki HC, Clenon JA, Brady MS, Kitron U, Sturrock RF, Ouma JH, Ndzovu ST, Mungai P, Hoffman O, Hamburger J, Pellegrini C, Muchiri EM, King CH. Distribution patterns and cercarial shedding of Bulinus nasutus and other snails in the Msambweni area, Coast Province, Kenya. Am J Trop Med Hyg. 2004;70:449–546. [PubMed] [Google Scholar]
- 18.Hamburger J, Turetsky T, Kapeller I, Deresievicz R. Highly repeated short DNA sequences in the genome of Schistosoma mansoni recognized by a species-specific probe. Mol Biochem Parasitol. 1991;44:73–80. doi: 10.1016/0166-6851(91)90222-r. [DOI] [PubMed] [Google Scholar]
- 19.Hamburger J, He N, Xin XY, Ramzy RM, Jourdane J, Ruppel A. A polymerase chain reaction assay for detecting snails infected with bilharzia parasites (Schistosoma mansoni) from very early prepatency. Am J Trop Med Hyg. 1998;59:872–876. doi: 10.4269/ajtmh.1998.59.872. [DOI] [PubMed] [Google Scholar]
- 20.Hamburger J, He N, Abassi I, Ramzy RM, Jourdane J, Rupple A. Polymerase chain reaction assay based on a highly repeated sequence of Schistosoma haematobium: a potential tool for monitoring schistosome-infested water. Am J Trop Med Hyg. 2001;65:907–911. doi: 10.4269/ajtmh.2001.65.907. [DOI] [PubMed] [Google Scholar]
- 21.Hamburger J, Hoffman O, Kariuki CH, Muchiri EM, Ouma JH, Koech DK, Sturrock RF, King CH. Large-scale polymerase chain reaction-based surveillance of Schistosoma haematobium DNA in snails from transmission sites in coastal Kenya: a new tool for studying the dynamics of snail infection. Am J Trop Med Hyg. 2004;71:765–773. [PubMed] [Google Scholar]
- 22.King CH, Sturrock RF, Kariuki CH, Hamburger J. Transmission control for schistosomiasis. Why it matters now. Trends Parasitol. 2006;22:575–582. doi: 10.1016/j.pt.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Farid HA, Morsy ZS, Helmy H, Ramzy RM, El Setouhy M, Weil GJ. A critical appraisal of molecular xenomonitoring as a tool for assessing progress toward elimination of lymphatic filariasis. Am J Trop Med Hyg. 2007;77:593–600. [PMC free article] [PubMed] [Google Scholar]
- 24.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:e63i–vii. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poon LL, Wong BW, Ma EH, Chan KH, Chow LM, Abeyewickreme W, Tangpukdee N, Yuen KY, Guan Y, Looareesuwan S, Peiris JS. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin Chem. 2006;52:303–306. doi: 10.1373/clinchem.2005.057901. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Rong R, Zhang HQ, Shi CJ, Zhu XQ, Xia CM. Sensitive and rapid detection of Schistosoma japonicum DNA by loop-mediated isothermal amplification (LAMP) Int J Parasitol. 2010;40:327–331. doi: 10.1016/j.ijpara.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Mori Y, Nagamine K, Tomita N, Notomi T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun. 2001;289:150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- 28.Boehme CC, Nabeta P, Henostroza G, Raqib R, Rahim Z, Gerhardt M, Sanga E, Hoelscher M, Notomi T, Hase T, Perkins MD. Operational feasibility of using loop-mediated isothermal amplification for diagnosis of pulmonary tuberculosis in microscopy centers in developing countries. J Clin Microbiol. 2007;45:1936–1940. doi: 10.1128/JCM.02352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smithers SR, Terry RJ. The infection of laboratory hosts with cercariae of Schistosoma mansoni and the recovery of adult worms. Parasitology. 1965;55:695–700. doi: 10.1017/s0031182000086248. [DOI] [PubMed] [Google Scholar]
- 30.Sohni Y, Kanjilal S, Kapur V. performance of five commercial real-time PCR reagent systems using TaqMan assays for B. antracis detection. Clin Biochem. 2008;41:640–644. doi: 10.1016/j.clinbiochem.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Pontes LA, Oliveira MC, Katz N, Dias-Neto E, Rabello A. Comparison of a polymerase chain reaction and the Kato Katz technique for diagnosing infection with Schistosoma mansoni. Am J Trop Med Hyg. 2003;68:652–656. [PubMed] [Google Scholar]
- 32.Wickman D, Panning M, Quack T, Kramme S, Burchard GD, Grevelding C, Drosten C. Diagnosing schistosomiasis by detection of cell-free parasite DNA in human plasma. PLoS Negl Trop Dis. 2009;3:e422. doi: 10.1371/journal.pntd.0000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katholi CR, Toe L, Merriweathe A, Unnasch TR. Determining the prevalence of Onchocerca volvulus infection in vector populations by PCR screening of pools of black flies. J Infect Dis. 1995;172:1414–1417. doi: 10.1093/infdis/172.5.1414. [DOI] [PubMed] [Google Scholar]
- 34.Yameogo L, Toe L, Hougard JM, Boatin BA, Unnasch TR. Pool screen polymerase chain reaction for estimating the prevalence of Onchocerca volvulus infection in Simulium damnosum sensu lato: results of a field trial in an area subject to successful vector control. Am J Trop Med Hyg. 1999;60:124–128. doi: 10.4269/ajtmh.1999.60.124. [DOI] [PubMed] [Google Scholar]
- 35.Helmy H, Fischer P, Farid HA, Bradley MH, Ramzy RM. Test strip detection of Wuchereria bancrofti amplified DNA in wild-caught Culex pipiens and estimation of infection rate by the PoolScreen algorithm. Trop Med Int Health. 2004;9:158–163. doi: 10.1046/j.1365-3156.2003.01155.x. [DOI] [PubMed] [Google Scholar]
- 36.Webster BL, Rollinson D, Stothard JR, Huyse T. Rapid diagnostic multiplex PCR (RD-PCR) to discriminate Schistosoma haematobium and S. bovis. J Helminthol. 2010;84:107–114. doi: 10.1017/S0022149X09990447. [DOI] [PubMed] [Google Scholar]
- 37.Weyher A, Phillips-Conroy J, Fischer K, Weil G, Chansa W, Fischer P. Molecular identification of Schistosoma mattheei from feces of Kinda (Papio cynocephalus kindae) and greyfoot baboons (Papio cynocephalus gryseipes) in Zambia. J Parasitol. 2009 doi: 10.1645/GE-2186.1. August 22. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 38.Abbasi I, King CH, Sturrock RF, Kariuki C, Muchiri E, Hamburger J. Differentiation of Schistosoma haematobium from related schistosomes by PCR amplifying an inter-repeat sequence. Am J Trop Med Hyg. 2007;76:950–955. [PMC free article] [PubMed] [Google Scholar]
- 39.Webster JP, Davies CM. Coevolution and compatibility in the snail-schistosome system. Parasitology. 2001;123:S41–S56. doi: 10.1017/s0031182001008071. [DOI] [PubMed] [Google Scholar]