

Abstract
Protein kinase B (PKB/Akt) plays a critical role in cell survival but the investigation of its involvement has been limited by the lack of specific pharmacological agents. In this study, using novel PKB inhibitors (VIII and XI), we investigated the role of PKB in cardioprotection of the rat and human myocardium, the location of PKB in relation to mitoKATP channels and p38 mitogen-activated protein kinase (p38 MAPK), and whether the manipulation of PKB can overcome the unresponsiveness to protection of the diabetic myocardium. Myocardial slices from rat left ventricle and from the right atrial appendage of patients undergoing elective cardiac surgery were subjected to 90 min ischaemia/120 min reoxygenation at 37°C. Tissue injury was assessed by creatine kinase (CK) released and determination of cell necrosis and apoptosis. The results showed that blockade of PKB activity caused significant reduction of CK release and cell death, a benefit that was as potent as ischaemic preconditioning and could be reproduced by blockade of phosphatidylinositol 3-kinase (PI-3K) with wortmannin and LY 294002. The protection was time dependent with maximal benefit seen when PKB and PI-3K were inhibited before ischaemia or during both ischaemia and reoxygenation. In addition, it was revealed that PKB is located downstream of mitoKATP channels but upstream of p38 MAPK. PKB inhibition induced a similar degree of protection in the human and rat myocardium and, importantly, it reversed the unresponsiveness to protection of the diabetic myocardium. In conclusion, inhibition of PKB plays a critical role in protection of the mammalian myocardium and may represent a clinical target for the reduction of ischaemic injury.
Introduction
Protein kinase B (PKB; also known as Akt) is a serine/threonine kinase, belonging to the AGC superfamily of protein kinases, which plays a prominent role in regulating cell survival, growth, proliferation, angiogenesis, metabolism and migration (Manning & Cantley, 2007). Three mammalian isoforms of PKB/Akt have been identified (named PKBα/Akt1, PKBβ/Akt2 and PKBγ/Akt3) and all are expressed in the myocardium, with PKBα and PKBβ being the most abundant (Matsui & Rosenzweig, 2005). All three PKB isoforms are activated in a phosphatidylinositol 3-kinase (PI-3K)-dependent manner involving either Class 1A or Class 1B PI-3Ks, which in turn are activated by tyrosine kinase and G-protein-coupled receptors, respectively (Duronio, 2008). The phospholipid PIP3 generated following PI-3K activation binds to the pleckstrin homology (PH) domain of PKB and facilitates the translocation of PKB to the plasma membrane. Following recruitment to the cell membrane, PKB is activated via phosphorylation on Thr308 by phosphoinositide-dependent kinase 1 (PDK1; also recruited to the plasma membrane by PIP3) and on Ser473 by a putative PDK2. Several protein kinases have been proposed as the elusive PDK2 including Pak1, which has been suggested as the relevant PDK2 responsible for Ser473 phosphorylation in cardiomyocytes (Mao et al. 2008). Activated PKB phosphorylates a number of downstream targets which have prominent roles in regulating apoptosis including the pro-apoptotic Bcl-2 family member BAD, caspase 9, glycogen synthase kinase 3β (GSK-3β) and the Forkhead family of transcription factors (Manning & Cantley, 2007; Parcellier et al. 2008).
There is considerable evidence indicating a significant role of the PI-3K/PKB pathway in cardioprotection induced by ischaemic preconditioning (IP), ischaemic postconditioning and pharmacological preconditioning (Armstrong, 2004; Matsui & Rosenzweig, 2005; Hausenloy & Yellon, 2007). The vast majority of studies have explored the role of PI-3K/PKB signalling in cardioprotection using the PI-3K inhibitors wortmannin and LY 294002. Until recently no selective pharmacological inhibitors of PKB were available and hence investigating the specific role of PKB (independent of PI-3K) involved both in vitro and in vivo expression of dominant negative or constitutively active PKB mutants (Matsui et al. 2001; Krieg et al. 2004; Uchiyama et al. 2004). The majority of protein kinase inhibitors available to date target the active site and are classified as ATP competitive. Unfortunately, the ATP binding domain is highly conserved amongst the 500 or so protein kinases that have been identified in the human genome and thus the development of selective protein kinase inhibitors is problematic. However, the development of non-ATP competitive inhibitors represents an alternative approach and allosteric PKB inhibitors have been developed which show selectivity over closely related members of the AGC protein kinase family (which includes PKA, PKC and PKG) and in some cases PKB isoform selectivity (Zhao et al. 2005; Barve et al. 2006; Lindsley et al. 2008; Calleja et al. 2009). For example, PKB inhibitor VIII binds to the PH domain of PKB locking the kinase in an inactive state and preventing phosphorylation of Thr308 and Ser473 (Calleja et al. 2009). Similarly, PKB inhibitor XI also interacts with the PH domain of PKB (Barve et al. 2006). The primary aim of this study was to investigate the influence of PKB in the tolerance to ischaemia/reoxygenation (I/R)-induced injury of the mammalian (rat and human) myocardium using novel specific PKB inhibitors binding to the PH domain. A second objective was to define the relationship of PKB with the mitoKATP channel and with p38 MAPK, two identified essential steps in the signal transduction mechanism of cardioprotection by IP.
Methods
Study animals
Male Wistar rats, weighing 250–350 g, were purchased from Charles River Labs (Margate, UK). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and animals received humane care in accordance with the Guidance on the Operation of the Animals (Scientific Procedures) Act 1986 (Her Majesty's Stationery Office, London, UK).
Human study subjects
The right atrial appendage from patients with and without diabetes mellitus undergoing elective coronary artery bypass graft or aortic valve surgery was retrieved at the time of the right atrial cannulation. The patients’ characteristics and the medical treatment recived prior to surgery are described in Table 1. Patients received a standard anaesthetic protocol consisting of temazepam 20 mg and ranitidine 150 mg as pre-medication, 2 h before their scheduled operation. Anaesthesia was then induced with 5–10 mg kg−1 fentanyl, 0.05–0.1 mg kg−1 midazolam and 1 mg kg−1 rocuronium, and maintained with an O2–air mixture and isoflurane to achieve a bispectral index system reading of less than 50. Patients with atrial fibrillation, poor ejection fraction (EF < 30%), and those being treated with opioids, catecholamines or potassium channel openers (nicorandil or diazoxide) were excluded. The investigation conformed to the principles outlined in the Declaration of Helsinki and local ethical approval and patients’ written informed consent were obtained.
Table 1.
Characteristics and medical treatment prior to surgery of right atrial appendage donor patients
| A. Non-diabetics | ||||||
|---|---|---|---|---|---|---|
| Patient no. |
||||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Sex | Male | Male | Female | Male | Female | Male |
| Age (years) | 60 | 37 | 71 | 51 | 85 | 63 |
| Diagnosis | CAD | CAD | AVS | AVS | AVS | AVS |
| Type of surgery | CABG | CABG | AVR | AVR | AVR | AVR |
| Medication | ||||||
| Aspirin | Yes | Yes | Yes | No | No | Yes |
| Clopidogrel | No | No | No | No | No | No |
| Omeprazole | Yes | Yes | No | No | Yes | Yes |
| β-Blocker | Yes | Yes | Yes | No | Yes | No |
| ACE inhibitor | Yes | Yes | Yes | No | Yes | Yes |
| Statin | Yes | Yes | Yes | No | Yes | Yes |
| B. Diabetics | ||||||
| Patient no. | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Sex | Male | Male | Male | Female | Female | Male |
| Age (years) | 38 | 75 | 68 | 79 | 62 | 55 |
| Type of diabetes | I | II | II | II | II | II |
| Diagnosis | CAD | CAD | CAD | AVS | CAD | CAD |
| Type of surgery | CABG | CABG | CABG | AVR | CABG | CABG |
| Medication | ||||||
| Insulin | Yes | No | No | No | No | No |
| Glicazide | No | Yes | Yes | No | No | Yes |
| Metformin | No | No | Yes | Yes | Yes | Yes |
| Aspirin | Yes | Yes | Yes | Yes | Yes | Yes |
| Clopidogrel | Yes | Yes | No | No | Yes | No |
| Omeprazole | Yes | No | No | Yes | No | No |
| β-Blocker | Yes | Yes | No | No | Yes | Yes |
| ACE inhibitor | No | Yes | Yes | No | No | No |
| Statin | Yes | Yes | Yes | No | Yes | Yes |
ACE, angiotensin-converting enzyme; AVR, aortic valve replacement; AVS, aortic valve stenosis; CABG, coronary artery bypass graft; CAD coronary artery disease.
Experimental preparation
The in vitro experimental preparation used in these studies has been previously fully characterised (Zhang et al. 2000) and extensively used by our laboratory. Briefly, rats were killed by cervical dislocation and the chest was opened and the heart rapidly removed and placed in ice-cold oxygenated Krebs–Henseleit-Hepes (KHH) buffer at 4°C for 3 min. The atria and the right ventricle were removed and the left ventricle was mounted onto an ice-cooled ground glass plate and then sliced freehand with surgical skin graft blades (Swann-Morton Ltd, Sheffield, UK) to a thickness of between 300 and 500 μm. Myocardial sections, weighing between 30 and 50 mg each, were transferred to conical flasks (25 ml Erlenmeyer flasks, Duran, Astell Scientific, Sidcup, UK) containing 10 ml of KHH-buffered solution and placed in a shaking water bath at 37°C. The myocardial sections were then equilibrated for 40 min in the KHH buffer oxygenated by a continuous flow of 95% O2–5% CO2 gas mixture to obtain a 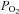 between 25 and 30 kPa and a
between 25 and 30 kPa and a 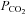 between 6.0 and 6.5 kPa at a temperature of 37°C. At the end of this period, the myocardial slices were subjected to 90 min of simulated ischaemia, induced by bubbling the media with 95% N2 and 5% CO2 in the absence of glucose (pH 6.6–6.9), followed by 120 min of reoxygenation. For the induction of IP, the myocardium was subjected to 5 min ischaemia followed by 5 min reoxygenation prior to the 90 min of ischaemia, a protocol that induces maximal protection in this model (Ghosh et al. 2000).
between 6.0 and 6.5 kPa at a temperature of 37°C. At the end of this period, the myocardial slices were subjected to 90 min of simulated ischaemia, induced by bubbling the media with 95% N2 and 5% CO2 in the absence of glucose (pH 6.6–6.9), followed by 120 min of reoxygenation. For the induction of IP, the myocardium was subjected to 5 min ischaemia followed by 5 min reoxygenation prior to the 90 min of ischaemia, a protocol that induces maximal protection in this model (Ghosh et al. 2000).
The right atrial appendage obtained from cardiac surgery patients was also prepared in an identical manner to that of the rat left ventricle, also described elsewhere (Zhang et al. 2000), and the myocardial sections subjected to an identical protocol of simulated ischaemia and reoxygenation.
Solutions and chemicals
The KHH-buffered solution contained (in mm): NaCl (118), KCl (4.8), NaHCO3 (27.2), MgCl2 (1.2), KH2PO4 (1.0), CaCl2 (1.20), glucose.H2O (10), and Hepes (20) to attain a pH of 7.4 at 37°C.
The agents wortmannin (0.1 μm), LY 294002 (10 μm), chelerythrine (10 μm), 5-hydroxydecanoate (5-HD, 1 mm), SB203580 (10 μm) and diazoxide (100 μm) were purchased from Sigma and their doses were selected following preliminary dose–response studies for each of them (Loubani & Galiñanes, 2002). The new specific PKB inhibitors XI and VIII were purchased from Calbiochem and after performing dose–response studies (0.1, 1, 10 μm for PKB inhibitor XI and 0.05, 0.5 and 5 μm for PKB inhibitor VIII) they were used at the most effective concentration.
Wortmannin, PKB inhibitor VIII, SB203580 and diazoxide were dissolved in DMSO before being added into the KHH buffer. The final concentration of DMSO was <0.1%, a concentration that has been shown to have no effects on the degree of ischaemia/reoxygenation-induced injury (Hassouna et al. 2004). The rest of the agents were dissolved in KHH.
Assessment of tissue injury
Tissue injury was assessed by measurement of creatine kinase (CK) released into the media during the 120 min reoxygenation period. The enzyme activity was measured by a linked-enzyme kinetic assay employing a commercial assay kit (30-3060/R2: Abbott Laboratories, Diagnostic Division, Kent, UK) and using a plate reader (BioTek Instruments, model ELx800uv, Winooski, VT, USA). Results were expressed as IU (mg wet weight)−1 after subtraction of the aerobic control values.
Assessment of cell death
At the end of the experimental protocols, the myocardial slices were incubated for 15 min with 30 μg ml−1 propidium iodide in PBS at pH 7.4. After three washes in PBS, tissues were fixed with 4% paraformaldehyde in PBS and then stored in darkness at 4°C until sectioning. Following this, the tissues were embedded with optical cutting temperature embedding matrix (Tissue-Tek, Agar Scientific Ltd, Stansted, UK). Frozen sections were then cut at 7 μm thickness in a Bright cryotome (model OTF) at ca−22°C, and sections were collected on SuperFrost Plus slides (Menzel-Glaser, Braunschweig, Germany). Before staining, tissues were permeabilised in 0.02 mg ml−1 proteinase-K for 10 min at 37°C in a humidity chamber, and pre-sensitised for 1 min in a microwave oven at 800 W in 0.1% Triton X-100 and 0.1 m tri-sodium citrate at pH 6.0.
A minimum of 100 nuclei per section were counted. To assess apoptosis, the terminal deoxynucleotidyl transferase was used to incorporate fluorescein (FITC)-labelled dUTP oligonucleotides to DNA strand breaks at the 3′-OH termini in a template-dependent manner (TUNEL technique) using a commercially available kit (Roche Diagnostics GmbH, Penzberg, Germany). A fluorescence excitation of 515 nm and FITC fluorescence emission range of 600–630 nm were used. To assess necrosis, propidium iodide-labelled nuclei were excited with helium–neon laser light at 543 nm and fluorescence was detected using 600 nm emission. To count the total number of nuclei, sections were mounted using Antifade mounting medium (Prolong Antifade kit, Invitrogen, Paisley, UK) and stained with 4′,6-diamidino-2-phenylindole (DAPI). For this analysis an excitation at 340 nm and detection at 456 nm were performed.
A fluorescence microscope (Axiovert 200M, Zeiss fluorescence microscope, Göttingen, Germany) at ×40 magnification was used to assess cell necrosis and apoptosis. The images were acquired using the OpenLab v.5 program (Improvision, Coventry, UK). Six to 10 randomised fields from each slide of the triple-labelled high resolution imaging samples were taken as previously described (Hassouna et al. 2004) and analysed using analytical digital photomicroscopy. Briefly, fluorescence signals were evaluated following a colorimetric methodology as described by Basha et al. (1996). The microscope and camera settings were constant and at least three photographs were taken from each field to obtain an average pixel value utilising Adobe Photoshop 6.0 (Adobe Systems Incorporated, San Jose, CA, USA). Necrosis and apoptosis were expressed as a percentage of pixels representing the total nuclei. A pilot study (authors’ unpublished data) performed in our laboratory showed no differences between the colorimetric and manual counting of apoptotic, necrotic or total nuclei.
Western blot analysis
To determine the phosphorylation of PKB (Ser473), tissue samples were homogenised in RIPA buffer containing protease inhibitor, PMSF (1 mm), DDT (0.5 mm), glycerophosphate (25 μm) and sodium orthovanadate (1 mm). The homogenate was centrifuged at 10 000 g for 30 min and the supernatant obtained was analysed for protein concentration using the Bio-Rad DC protein assay kit. Tissue supernatant (20 μg total protein) was electrophoresed on 10% SDS-PAGE and blotted onto nitrocellulose membrane. Following transfer the membranes were washed with Tris-buffered saline (TBS) and blocked for 1 h at room temperature (20–24°C) in blocking buffer (TBS, 5% (w/v) skimmed milk powder, 0.1% (v/v) Tween-20). Blots were then incubated overnight at 4°C with primary rabbit monoclonal antibody against phosphorylated (Ser473) PKB (Cell Signalling Technology) at 1:1000 dilution in blocking buffer. The primary antibody was removed, blots extensively washed three times for 5 min in TBS/0.1% Tween 20 (v/v) and incubated for 1 h at room temperature with goat anti-rabbit secondary antibody coupled to horseradish peroxidase at 1:5000 dilution in blocking buffer. Following removal of the secondary antibody, blots were extensively washed as above and developed using the enhanced chemiluminescence detection system (Amersham, Little Chalfont, UK) and quantified by densitometry using Scion image (Scion, Frederick, MD, USA). The uniform transfer of proteins to the nitrocellulose membrane was routinely monitored by transiently staining the membranes with Ponceau S stain (Sigma Chemical Co.) prior to application of the primary antibody. In addition, replicate samples from each experiment were analysed on separate blots using a rabbit antibody (1:1000) that recognises unphosphorylated (total) PKB (Cell Signalling Technology).
Study protocols
Myocardial slices (n= 6/group, unless otherwise indicated) were randomly allocated to different groups so that the tissues from each animal or patient donor were not utilised more than once for the same group. Time-matched aerobic controls, values of which are shown in Table 2, were used in each experiment and the rest of the tissues were subjected to 90 min ischaemia/120 min reoxygenation. The following studies were sequentially carried out:
Table 2.
Mean ±s.e.m. values of myocardial slices subjected to time-matched aerobic control for all the studies
| CK leakage (IU (mg wet wt)−1) | Necrosis (% of nuclei) | Apoptosis (% of nuclei) | |
|---|---|---|---|
| Study 1 | |||
| PKB inhibitor VIII | 0.22 ± 0.02 | 10.6 ± 0.9 | 8.6 ± 0.7 |
| PKB inhibitor XI | 0.27 ± 0.07 | 9.3 ± 0.4 | 8.4 ± 1.1 |
| Study 2 | 0.25 ± 0.03 | 16.6 ± 1.5 | 12.8 ± 1.0 |
| Study 3 | 0.24 ± 0.03 | 9.5 ± 1.6 | 10.1 ± 1.5 |
| Study 4 | 0.52 ± 0.07 | 10.5 ± 1.0 | 10.9 ± 0.6 |
| Study 5 | 0.31 ± 0.04 | 8.8 ± 0.6 | 9.9 ± 0.4 |
| Study 6 | 0.33 ± 0.03 | 17.0 ± 3.2 | 14.5 ± 3.2 |
| Study 7 | |||
| Non-diabetics | 0.54 ± 0.08 | 9.1 ± 0.7 | 9.9 ± 0.8 |
| Diabetics | 0.49 ± 0.08 | 9.3 ± 0.8 | 9.0 ± 0.7 |
To investigate the effect of PKB inhibitors on I/R-induced injury and IP (Study 1), rat ventricular muscles were exposed to different concentrations of the PKB inhibitors VIII (0.05, 0.5 and 5 μm) and XI (0.1, 1 and 10 μm) for 20 min prior to I/R as depicted in Fig. 1Aa and b.
To elucidate whether PI-3K inhibitors can reproduce the results obtained with PKB inhibitors on I/R-induced injury and IP (Study 2), rat ventricular muscles were subjected to 20 min exposure with LY 294002 (10 μm) and wortmannin (0.1 μm) before I/R as shown in Fig. 2Aa and b.
To determine the optimal time of administration of PKB and PI-3K inhibitor-induced cardioprotection, slices from rat ventricular myocardium were exposed to the PKB inhibitor XI (1 μm) (Study 3; see Fig. 3A) and wortmannin (0.1 μm) (Study 4; see Fig. 4A) for 20 min before ischaemia; during ischaemia; during reoxygenation; during ischaemia and reoxygenation; and before ischaemia and throughout ischaemia and reoxygenation.
To determine the relationship between PKB inhibition and mitoKATP channels (Study 5; see Fig. 5A), rat ventricular slices were exposed to the PKB inhibitor XI (1 μm) in the presence of the mitoKATP channel blocker 5-HD (10 μm). Similarly, to determine the relationship of PKB with p38 MAPK (Study 6; see Fig. 6A), rat ventricular slices were exposed to the p38 MAPK inhibitor SB203580 (10 μm) in the absence and presence of PKB inhibitor XI (1 μm). For comparison, the effects of 5-HD and SB203580 on IP-induced protection were determined for each study.
To investigate whether PKB inhibition also protects the human myocardium (Study 7; see Fig. 7Aa), myocardial slices from the right atrial appendage of patients undergoing elective cardiac surgery were exposed to the PKB inhibitor XI (1 μm) for 30 min or wortmannin (0.1 μm) for 20 min, both before the 90 min of ischaemia. For comparison, other myocardial slices were subjected to IP. In addition, to investigate whether PKB inhibition can reverse the unresponsiveness to protection of the diabetic myocardium (see Fig. 7Ab), muscle slices from the right atrial appendage of patients with diabetes were also subjected to an identical protocol with the PKB inhibitor XI and wortmannin, with other myocardial slices being treated with IP or the mitoKATP channel opener diazoxide (100 μm) for 10 min before the 90 min of ischaemia.
To determine the effect of PKB inhibition with the inhibitor XI on PKB Ser473 phosphorylation (Study 8), rat ventricular slices (n= 4) were exposed to a 1 μm concentration of this agent for 20 min of aerobic incubation (e.g. no ischaemia). Other myocardial slices (n= 4/group) were exposed to wortmannin (0.1 μm) for 20 min or subjected to IP alone and in combination with the inhibitor XI and wortmannin. At the end of these periods, the slices were frozen in liquid nitrogen and kept at −80°C until analysis.
Figure 1. The effect of PKB inhibitors on I/R-induced injury and IP in rat ventricular myocardium.
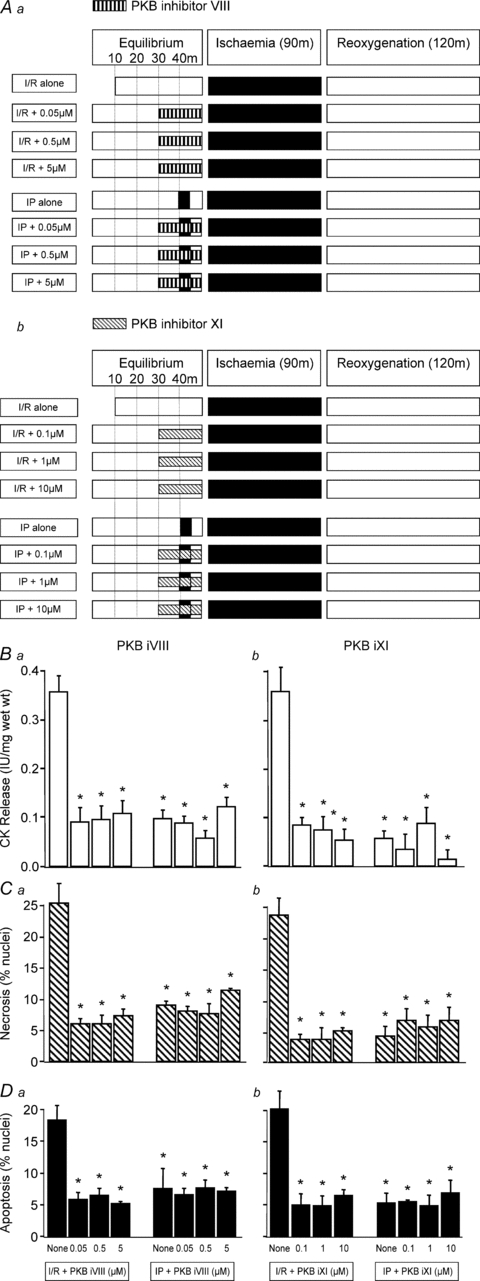
Three different concentrations of PKB inhibitor VIII (0.05, 0.5 and 5 μm) and PKB inhibitor XI (0.1, 1 and 10 μm) were used to investigate their effect on ischaemia/reoxygenation (I/R)-induced injury and ischaemic preconditioning (IP) following the protocols shown in Aa and Ab. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (Ba and Bb), and for necrosis (Ca and Cb) and apoptosis (Da and Db) at the end of 120 min of reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group.
Figure 2. The effect of PI-3K inhibitors on I/R-induced injury and IP in rat ventricular myocardium.
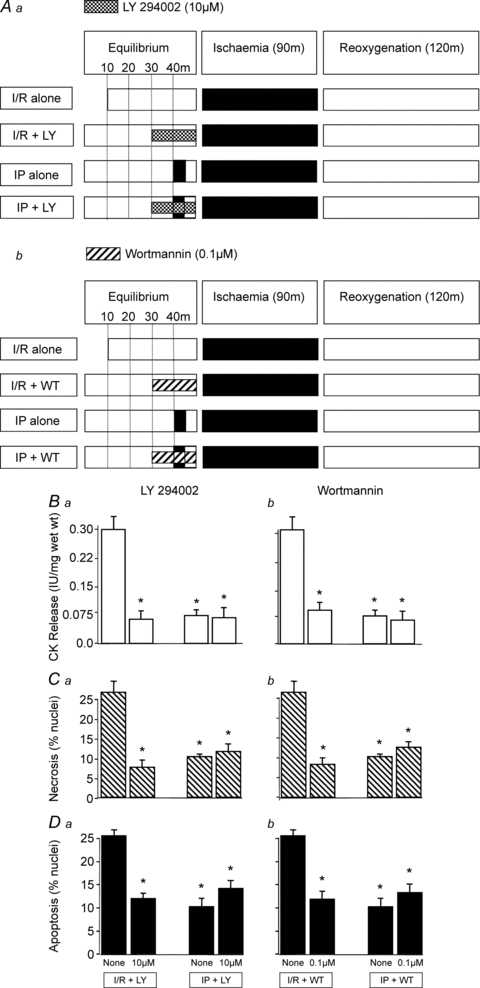
LY 294002 (10 μm) and wortmannin (0.1 μm) were used to investigate the role of PI-3K in ischaemia/reoxygenation (I/R)-induced injury and ischaemic preconditioning (IP) following the protocols shown in Aa and Ab. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (Ba and Bb), and for necrosis (Ca and Cb) and apoptosis (Da and Db) at the end of 120 min of reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group.
Figure 3. The temporal effects of PKB inhibitor XI administration during I/R.
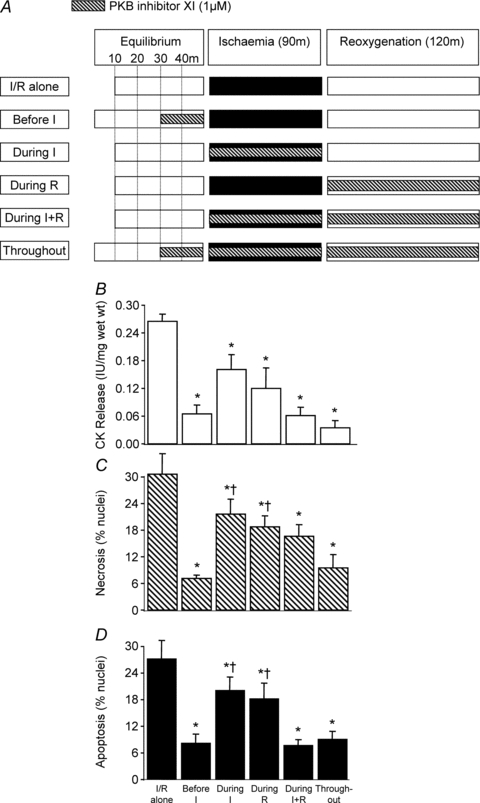
As shown in A, PKB inhibitor XI (1 μm) was administered at 5 different time points during the ischaemia/reoxygenation (I/R) protocol using rat left ventricular myocardial slices: 20 min before ischaemia; during ischaemia; during reoxygenation; during ischaemia and reoxygenation; and before ischaemia and throughout ischaemia and reoxygenation. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (B), and for necrosis (C) and apoptosis (D) at the end of 120 min reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group; †P < 0.05 vs. before I and throughout groups.
Figure 4. The temporal effects of wortmannin administration during I/R.
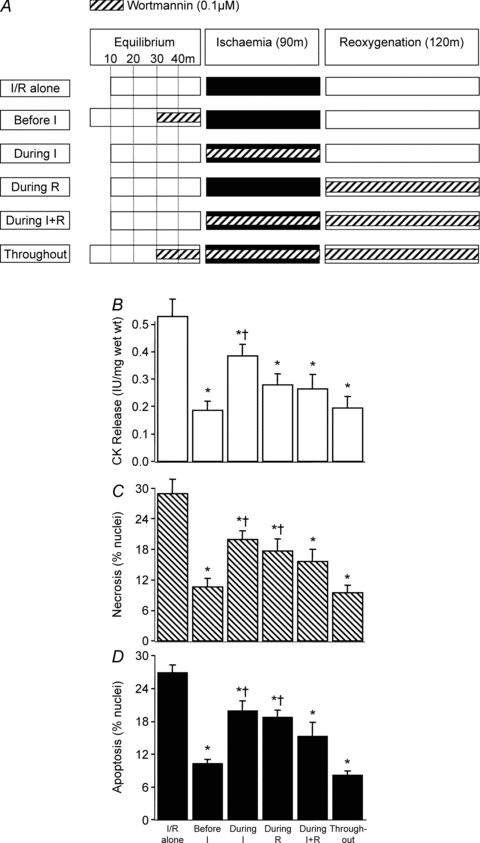
As shown in A, wortmannin (0.1 μm) was administered at 5 different time points during the ischaemia/reoxygenation (I/R) protocol using rat left ventricular myocardial slices: 20 min before ischaemia; during ischaemia; during reoxygenation; during ischaemia and reoxygenation; and before ischaemia and throughout ischaemia and reoxygenation. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (B), and for necrosis (C) and apoptosis (D) at the end of 120 min reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group; †P < 0.05 vs. before I and throughout groups.
Figure 5. The relationship between PKB inhibition and the mitoKATP channel.
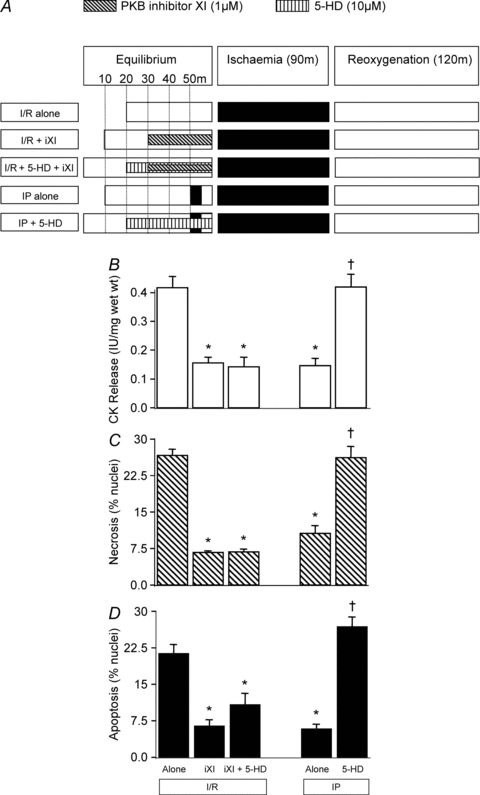
The mitoKATP channel inhibitor 5-HD (10 μm) was used to determine if the mitoKATP channel is upstream or downstream of PKB. For this, PKB inhibitor XI was encapsulated during 5-HD pre-treatment prior to ischaemia/reoxygenation (I/R) of rat left ventricular myocardial slices and compared to PKB inhibitor XI alone, as shown in A. For comparison, ischaemic preconditioning (IP) was also encapsulated during 5-HD administration prior to I/R. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (B), and for necrosis (C) and apoptosis (D) at the end of 120 min reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group; †P < 0.05 vs. IP group.
Figure 6. The relationship between PKB inhibition and p38 MAPK.
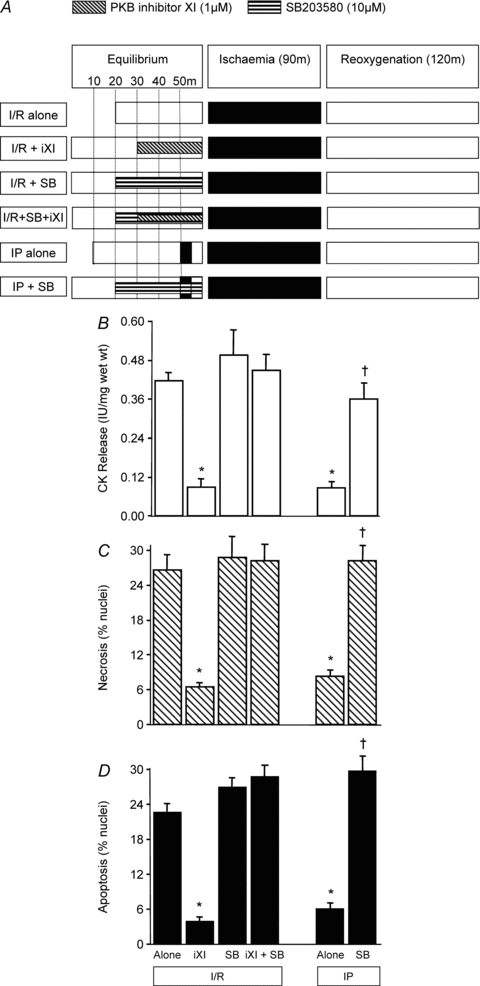
The p38 MAPK inhibitor SB 203580 (10 μm) was used to determine if p38 MAPK is upstream or downstream of PKB. As shown in A, rat left ventricular myocardial slices were pre-treated with SB203580 or PKB inhibitor XI alone or encapsulated with SB203580 prior to ischaemia/reoxygenation (I/R). For comparison, ischaemic preconditioning (IP) was also encapsulated by SB203580 prior to I/R. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (B), and for necrosis (C) and apoptosis (D) at the end of 120 min reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group; †P < 0.05 vs. IP group.
Figure 7. The effect of PKB and PI-3K inhibition on I/R-induced injury in non-diabetic and diabetic human myocardium.
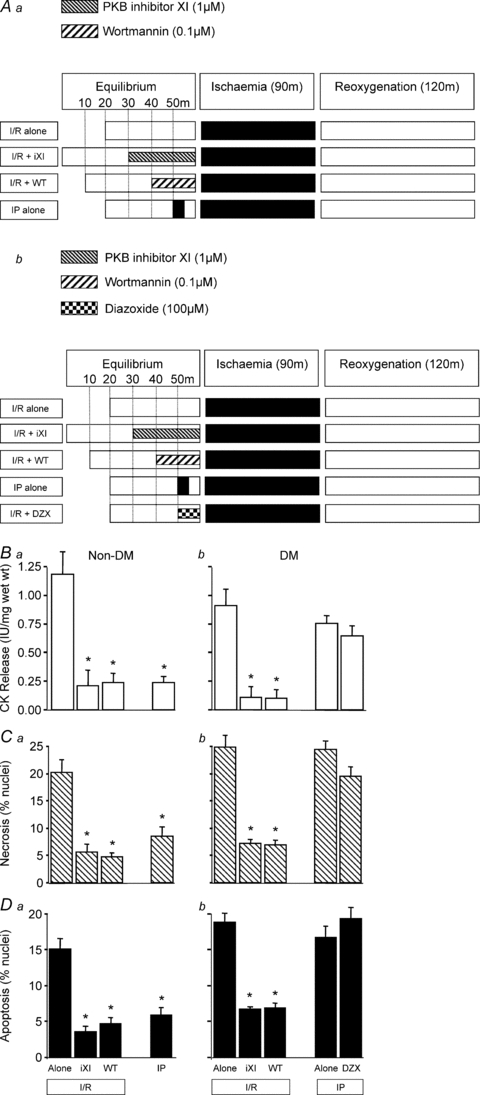
Non-diabetic tissue (Aa) was pre-treated with PKB inhibitor XI (1 μm) or wortmannin (0.1 μm) prior to ischaemia/re-oxygenation (I/R) and compared to ischaemic preconditioning (IP). A similar protocol (Ab) was employed using diabetic human myocardium except with the inclusion of diazoxide (100 μm; mitoKATP channel opener) to induce pharmacological preconditioning. In all cases tissue samples were analysed for CK release during the 120 min reoxygenation (Ba and Bb), and for necrosis (Ca and Cb) and apoptosis (Da and Db) at the end of 120 min reoxygenation. Data represent the mean ±s.e.m. from 6 independent experiments. *P < 0.05 vs. I/R alone group.
Statistical analysis
Data are expressed as mean ±s.e.m. Each reported value was obtained after subtracting the value from the corresponding time-matched aerobic control. One-way ANOVA was used to compare the significance between groups. All the analyses were performed using the SPSS program and differences were considered to be statistically significant if P < 0.05.
Results
Effect of PKB inhibitors on I/R-induced injury and IP in rat ventricular muscle (Study 1)
The results shown in Fig. 1 (panels Ba and b) reveal that the PKB inhibitors VIII and XI significantly reduced ischaemia/reoxygenation (I/R)-induced CK leakage from rat ventricular muscle. It is notable that the degree of protection observed with these novel PKB inhibitors is comparable to that achieved with IP. This figure also shows that both PKB inhibitors VIII and XI significantly reduced the levels of necrotic (panels Ca and Cb) and apoptotic (panels Da and Db) nuclei in rat ventricular muscle subjected to I/R. The cardioprotection obtained with the PKB inhibitors was similar to that seen with classical IP, and, interestingly, the combination of PKB inhibition and IP did not result in additional benefit.
Effect of PI-3K inhibitors on I/R-induced injury and IP in rat ventricular muscle (Study 2)
Having observed marked cardioprotection with the PKB inhibitors VIII and XI, we investigated whether inhibition of PI-3K would also induce similar protection. As shown in Fig. 2 both LY 294002 (10 μm) and wortmannin (0.1 μm) significantly reduced CK leakage and the levels of apoptotic and necrotic nuclei in rat ventricular muscle subjected to I/R but had no effect on IP. Overall, the degree of protection induced by PI-3K inhibition with LY 294002 and wortmannin was comparable to that induced by IP.
Temporal effect of PKB and PI-3K inhibitor-induced cardioprotection (Studies 3 and 4)
Having established that PI-3K and PKB inhibitors induce cardioprotection when applied 20 min prior to I/R we performed a series of experiments to determine if this is the optimal time for administration. As shown in Fig. 3B–D, the PKB inhibitor XI when administered during ischaemia or reoxygenation was less effective than when given during both periods. It is worth noting that the administration of PKB inhibitor XI during ischaemia and reoxygenation was as cardioprotective as the administration before ischaemia or when given throughout the entire experimental period (e.g. prior to ischaemia, during ischaemia and during reoxygenation). Figure 4B–D shows that similar results to those seen with the PKB inhibitor XI were observed with the PI-3K inhibitor wortmannin.
Relationship between PKB inhibition and mitoKATP channels (Study 5)
To determine the relationship of PKB with the mitoKATP channel we explored the effect of PKB inhibitor XI in the presence of the mitoKATP channel blocker 5-HD (10 μm). For comparison, we determined the effect of 5-HD on IP-induced protection. As shown in Fig. 5, encapsulating the PKB inhibitor XI with 5-HD did not significantly reduce the cardioprotection induced by PKB inhibition. In contrast, and in agreement with previous studies, 5-HD abolished IP-induced cardioprotection. These observations suggest that PKB is downstream of the mitoKATP channel since protection from I/R injury triggered by PKB inhibitor XI is still evident in the presence of the mitoKATP channel blocker 5-HD.
Relationship between PKB inhibition and p38 MAPK (Study 6)
These experiments were designed to determine the relationship of PKB with p38 MAPK, which we have previously reported to be downstream of the mitoKATP channel (Loubani & Galiñanes, 2002). To achieve this, we explored the effect of the p38 MAPK inhibitor SB203580 (10 μm), in the absence and presence of PKB inhibitor XI, on I/R-induced CK release and apoptosis/necrosis. For comparison we determined the effect of SB203580 on IP since previous studies have shown that inhibiting p38 MAPK reverses protection (Loubani & Galiñanes, 2002). As shown in Fig. 6B–D, PKB inhibitor XI was not cardioprotective when encapsulated with SB203580. As expected, SB203580 alone had no effect on I/R-induced CK release but blocked IP. Similar profiles were observed when monitoring the levels of apoptotic and necrotic nuclei under the above conditions. Overall these observations suggest that PKB is upstream of p38 MAPK since protection from I/R injury by the inhibition of PKB is abolished by p38 MAPK inhibition.
Effect of PKB inhibition on I/R-induced injury in human myocardium (Study 7)
Having shown that PKB inhibitors VIII and XI induce cardioprotection in rat ventricular muscle we investigated whether a similar phenomenon occurs in human myocardium. Right atrial muscles were obtained from non-diabetic and diabetic patients undergoing elective cardiac surgery and subjected to the same I/R protocol employed for rat myocardium. The results shown in Fig. 7 demonstrate that in non-diabetic human myocardium PKB inhibitor XI, wortmannin and IP reverse I/R-induced CK leakage (panel Ba). Similarly, all three modes of cardioprotection significantly reduced the levels of necrotic and apoptotic nuclei (panels Ca and Da). These observations clearly indicate that, as seen in the rat ventricular myocardium, inhibiting PKB and PI-3K prior to I/R induces cardioprotection in human atrial myocardium similar to that achieved by classical IP.
Next we investigated whether PKB inhibitor XI induces cardioprotection in diabetic human myocardium, which we have previously shown to be resistant to both IP and pharmacological preconditioning with the mitoKATP channel opener diazoxide (Ghosh et al. 2001; Hassouna et al. 2006). As shown in Fig. 7Bb, Cb and Db, PKB inhibitor XI and wortmannin induced cardioprotection in diabetic human myocardium, whereas IP and diazoxide were ineffective. Overall these results reveal that inhibition of PKB is cardioprotective for human myocardial tissue, and its effect is independent of functional mitoKATP channels.
Effect of PKB inhibitor XI on PKB Ser473 phosphorylation (Study 8)
Western blot analysis of PKB Ser473 phosphorylation was performed using rat ventricular muscle in order to confirm PKB inhibition by PKB inhibitor XI. As shown in Fig. 8, PKB Ser473 phosphorylation was detectable in aerobic and ischaemic tissue. As expected, both PKB inhibitor XI and wortmannin reduced levels of PKB Ser473 phosphorylation in aerobic tissue following 20 min pre-treatment with the inhibitors. IP also induced a significant reduction in Ser473 phosphorylation. The reduction in PKB Ser473 phosphorylation following IP is in contrast to previous studies which have reported IP-induced increases in Ser473 phosphorylation (Tong et al. 2000; Mocanu et al. 2002). Treatment with inhibitor XI during the equilibrium period prior to IP (with inhibitor present) also reduced levels of PKB Ser473 phosphorylation. Overall these observations suggest that IP induces a marked reduction in Ser473 PKB phosphorylation which is in line with the protection observed using the PKB inhibitors.
Figure 8. Western blot analysis of PKB Ser473 phosphorylation in rat ventricular myocardium.
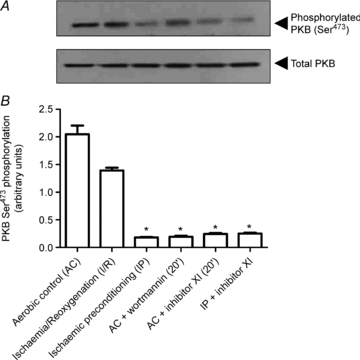
Representative immunoblot (A) and quantitative analysis (B) of PKB Ser473 phosphorylation in samples obtained from the following protocols: aerobic control (AC); ischaemia/reoxygenation (I/R); ischaemic preconditioning (IP); 20 min pre-treatment with wortmannin (0.1 μm) or PKB inhibitor XI (1 μm) prior to AC and inhibitor XI treatment during the equilibration and ischaemic preconditioning (IP + inhibitor XI). Insulin-induced PKB Ser473 phosphorylation from DDT1MF-2 cells was used as a positive control. Data represent the mean ±s.e.m. from 4 independent experiments and are expressed as a percentage of the aerobic control values. *P < 0.05 vs. aerobic control.
Discussion
The present studies have demonstrated that: (i) inhibition of PKB activity with novel specific agents results in a potent cardioprotection of the rat left ventricular myocardium and of the human atrial myocardium, an effect that can be reproduced by the blockade of PI-3K; (ii) maximal protection is obtained when both PKB and PI-3K inhibition are applied before ischaemia and when given during ischaemia and reoxygenation; (iii) PKB is located downstream of the mitoKATP channels and upstream of p38 MAPK; (iv) the unresponsiveness of the diabetic myocardium to cardioprotective interventions such as IP can be overcome by inhibition of PKB and PI-3K. These findings advance our knowledge of the mechanism of cardioprotection of the mammalian heart, are of clinical relevance and warrant further discussion.
Selectivity and action of PKB inhibitors
Previously, and because of the lack of specific PKB inhibitors, the role of this kinase in ischaemia/reperfusion-induced injury and cardioprotection has been investigated by using blockers of the upstream kinase PI-3K such as wortmannin and LY 294002 (Tong et al. 2000). Recently, a novel family of specific PKB inhibitors has been developed (Zhao et al. 2005; Barve et al. 2006; Calleja et al. 2009) and used extensively in oncology studies (Lindsley et al. 2008). However, to the best of our knowledge, these new inhibitors have not been used in studies related to ischaemic injury and cardioprotection.
For these studies, two PKB inhibitors, VIII and XI, both cell permeable, were selected. Inhibitor VIII is a quinoxaline compound that potently and selectively inhibits PKB1/PKB2 activity (IC50= 58 nm, 210 nm and 2.12 μm for PKB1, PKB2 and PKB3, respectively, in in vitro kinase assays) by interacting with the PH domain (Zhao et al. 2005). It does not exhibit inhibitory effects against other non-PKB PH domains, or other closely related AGC family kinases such as PKA, PKC and GSK, even at concentrations as high as 50 μm (Zhao et al. 2005). Inhibitor XI is a copper complex that interacts with both the PH and the kinase domains of PKB and potently inhibits its kinase activity (IC50= 100 nm) (Barve et al. 2006). As shown in this study inhibitor XI significantly reduced PKB Ser473 phosphorylation in rat left ventricle tissue. The dose–response studies performed for each inhibitor were constructed on base to the IC50 values and clearly demonstrated that both inhibitors VIII and XI greatly reduced ischaemic injury (CK values) and cell necrosis and apoptosis. It is worth noting that the degree of protection afforded by the two PKB inhibitors was similar to that of IP and that the use in combination of each of the inhibitors with IP did not result in additional benefit, suggesting that both interventions use an identical cellular signalling cascade. Furthermore, the results obtained with the PI-3K blockers wortmannin and LY 294002 convincingly showed that inhibition of PKB protects the human and rat myocardium against ischaemia/reoxygenation-induced injury. These results, that were similar in the rat and in the human myocardium, are in apparent contraposition to the more accepted view that activation of PKB is protective (Armstrong, 2004; Matsui & Rosenzweig, 2005; Wang et al. 2009). In trying to understand the differing results, it is necessary to clarify once more that previously the role of PKB could not be appropriately investigated because of the lack of specific blockers. However, it has been reported that inhibition of PI-3K blocks the protective effect of preconditioning (Tong et al. 2000) but at the same time it has been shown that the genetic suppression of PI-3Kα isoform activity (upstream of PKB) induces resistance against prolonged ischaemia (Ban et al. 2008). The latest findings contrast with the observation that mice cardiomyocytes lacking 3′-phosphoinositide-dependent kinase-1 (PDK1), a kinase phosphorylating PKB, are more sensitive to hypoxic injury and animals with this deficit develop heart failure and die suddenly at an early stage of adult life (Mora et al. 2003). Also, the reduced expression of PDK1 in hypomorphic mutant mice abolishes the cardioprotection induced by IP (Budas et al. 2006). By using the PI-3K inhibitor LY 294002, other investigators have concluded that activation of PKB is mediating the protection afforded by preconditioning adult rat left ventricular myocytes (Uchiyama et al. 2004); however, the dose used in this study was 5 times greater than the one used in our studies (50 μmversus 10 μm, respectively). Adding to the controversy is the finding that rapamycin, a blocker of mTOR (which is phosphorylated by PKB on Ser2448), can abrogate opioid-mediated cardioprotection when added before ischaemia (Gross et al. 2004) and also the beneficial effect of preconditioning when added at the start of reperfusion (Hausenloy et al. 2004) whilst other investigators have reported that acute treatment with rapamycin is in fact cardioprotective (Khan et al. 2006). Therefore it could be hypothesised that the final effect on protection, or the absence of it, is due to activation or blockade of specific isoforms of the various protein kinases involved. This is an area of critical importance for therapeutic purposes and, certainly, a full elucidation would require further investigations.
Another possible explanation for the reported apparent contradictory results could be the variable outcome measures used in each of the studies. Thus, for example, apart from the species differences, some studies examined the recovery of the left ventricular function (Tong et al. 2000; Ban et al. 2008; Wang et al. 2009) whereas others measured reduction in infarct size in isolated in vitro perfused hearts (Hausenloy et al. 2004; Budas et al. 2006; Khan et al. 2006) or in in vivo animals (Gross et al. 2004). In the present studies, the release of CK and the rate of necrosis and apoptosis in strips of cardiac muscle were assessed to evaluate tissue injury and protection. Yet other investigators using isolated cardiomyocytes analysed intracellular Ca2+ overload and hypercontracture (Mora et al. 2003) or cell viability, lactate dehydrogenase release and apoptosis (Uchiyama et al. 2004). Hence, the variety of experimental models used, along with the different tissue manipulations and degrees of injury inflicted, might have contributed to the confounding results.
The similar findings seen in the present studies with the use of the specific PKB inhibitors VIII and XI and with the PI-3K inhibitors wortmannin and LY 294002 is restricting a greater mechanistic insight. Despite it, the intriguing observations reported in our studies may reflect ‘cross-talk’ of protein kinase cascades associated with cardioprotection. There is evidence that the PI-3K/PKB pathway interacts with ERK1/2 and p38 MAPK/JNK signalling. For example, PKB phosphorylates and inhibits Raf-1 leading to attenuation of ERK1/2 signalling (Zimmermann & Moelling, 1999). Hence, inhibition of PKB activity in our experimental model of ischaemic injury may enhance ERK1/2 signalling and thus promote protection. It is notable that in isolated rat hearts reperfusion-induced ERK1/2 activation is enhanced in the presence of the PI-3K inhibitor LY 294002 (Hausenloy et al. 2004). However, this apparent ‘cross-talk’ of protein kinase cascades did not result in enhanced cardioprotection (Hausenloy et al. 2004). PKB also phosphorylates and activates three protein kinases (ASK1, MLK3 and SEK1/MKKK4) involved in the upstream activation of p38 MAPK/JNK (Song et al. 2005). Therefore inhibition of PKB could trigger the blockade of p38 MAPK/JNK signalling but such a possibility would not explain our findings since blockade of p38 MAPK by SB203580 abrogated the protection induced by the PKB inhibitor XI. Clearly further detailed studies are required to explore the kinetics of PKB, ERK1/2, p38 MAPK and JNK activation during our experimental regime in the absence and presence of PKB inhibitors VIII and XI. The effect of PKB on GSK-3β activity, another important kinase in the signal transduction pathway of cardioprotection, is also unclear and needs to be elucidated, particularly in view of the reported conflicting results on the role of GSK-3β (Juhaszova et al. 2004; Gomez et al. 2008; Murphy & Steenbergen, 2008; Nishino et al. 2008).
Importance of the time of administration
Our studies are also the first to demonstrate that blockade of the PKB activity confers different degrees of myocardial protection depending on the time of application (e.g. before ischaemia, during ischaemia or during reoxygenation). Thus, although protection was obtained in each instance, maximal benefit was only seen when PKB was blocked before ischaemia or throughout both ischaemia and reoxygenation periods. These results, which were reproduced by blocking PI-3K with wortmannin, suggest that the PKB activity state during ischaemia and reoxygenation is critical in determining the tolerance of the tissue to an ischaemic insult. However, the demonstration by other laboratories that activation of PKB is the mechanism of protection by ischaemic post-conditioning (Zhu et al. 2006) raises again the issue of the importance of the kinase's activity status before, during and after ischaemia. This apparent contradiction may also lead to the hypothesis that both blockade and activation of PKB can be either beneficial or not depending on the time that it occurs.
Location of PKB in the cell signalling cascade for cardioprotection
MitokATP channels are central to the signalling of cardioprotection, although they are not the end-effectors of protection (Pain et al. 2000). It has been demonstrated that the opening of these channels generates free radicals leading to the activation of kinases and preconditioning (Pain et al. 2000), an action mediated by the inner mitochondrial membrane connexin43 (Heinzel et al. 2005). Although mitoKATP channels have been implicated in preconditioning by many studies, the evidence for their structure and function, and even their existence, remains inconclusive (for review see Hanley & Daut, 2005). Furthermore, the pharmacological agents used, such as the opener diazoxide and the inhibitor 5-HD, exert other well-documented actions that may question their utility as selective tools to dissect the underlying signalling mechanism of preconditioning (for review see Hanley & Daut, 2005). Despite these concerns, using the selective mitoKATP channel blocker 5-HD, our results indicate that PKB is located downstream of mitoKATP channels, since the protection induced by the PKB blocker inhibitor XI was unaffected by 5-HD. Other investigators have shown that mitoKATP channel openers BMS-191095 and diazoxide induce PKB phosphorylation (Wang et al. 2004; Ahmad et al. 2006), suggesting location of the kinase downstream of the channel. However, it is notable that the effect of diazoxide on PKB-phosphorylation reported by Wang et al. (2004) was not blocked by 5-HD. In contrast, the work of Garlid's group suggests that PKB is located upstream of the mitoKATP channel (Garlid et al. 2009).
Here we have also demonstrated that the protection induced by the PKB inhibitor XI can be completely abrogated by the p38 MAPK blocker (blocking the α and β isoforms) SB203580, suggesting that PKB is upstream of p38 MAPK. Previously, using an identical experimental model, we have shown that p38 MAPK is downstream of PKC and mitoKATP channels in the human myocardium (Loubani & Galiñanes, 2002).
The role of the p38 MAPK pathway in ischaemic preconditioning is also controversial since either activation or inhibition of this protein kinase reportedly promotes cardioprotection (Schulz et al. 2002; Steenbergen, 2002). This may reflect distinct roles for p38 MAPK isoforms since studies have shown that the p38α isoform contributes to cell death whereas the p38β isoform mediates cell hypertrophy (Wang et al. 1998). Both p38 MAPKα and p38 MAPKβ isoform activities are increased by the induction of ischaemia, although with prolonged ischaemia p38β activity decreases towards baseline levels in non-preconditioned hearts, and remains elevated in preconditioned hearts (Schulz et al. 2003). However, the apparent controversy surrounding p38 MAPK and indeed many of the other protein kinases implicated in cardioprotection strengthens our observations reported in this study concerning the apparent detrimental role of PKB activation during ischaemic injury. The reasons for these differences could include: (i) the use of varying in vivo and in vitro model systems; (ii) the timing of kinase activation/inhibition during the experimental protocol; (iii) the selectivity and/or concentration of the protein kinase inhibitors used, and (iv) ‘cross-talk’ between different protein kinase cascades.
The molecular interactions between PKB, mitoKATP channels and p38 MAPK are not clear. As mentioned above, there is evidence that mitoKATP channel opening increases reactive oxygen species production (Pain et al. 2000) leading to the activation of protein kinases such as PKCɛ (Garlid et al. 2009). However, at present it is not known if PKB activity is directly or indirectly regulated via mitoKATP channel opening, despite reports describing activation of PKB by diazoxide (Wang et al. 2004; Ahmad et al. 2006). As described by Heusch et al. (2008), PKB can also interact with a number of different mediators, depending on the stimuli. PKB plays a critical role in cardioprotection (Heusch, 2009) but its effect may depend on the species and experimental model used (Skyschally et al. 2009). Clearly further studies are required to explore the complex interplay between PKB, PKC, p38 MAPK and mitoKATP channels.
PKB inhibition overcomes the deficit of diabetes for cardioprotection
Previously we have demonstrated that mitochondrial dysfunction is the cause of the unresponsiveness of the diabetic myocardium to protection by IP (Ghosh et al. 2001) and the present finding that PKB blockade overcomes this deficit confirms the location of PKB being downstream of the mitoKATP channels. Similar results were obtained in our laboratory when activators of PKC and p38 MAPK were used (Loubani & Galiñanes, 2002), further supporting the location of these kinases beyond the mitochondria. These results may have clinical relevance since the manipulation of the activity of these protein kinases may reduce the ischaemic injury in subjects with diabetes and suffering an acute coronary syndrome or undergoing cardiac surgery.
Study limitations
The present studies were performed in an in vitro model and therefore care must be taken when extrapolating the present results to clinical conditions. The use of such an in vitro model has the important advantage of achieving better control of the experimental conditions than in more complex biological systems; however, the use of in vitro preparations may also be associated with inadvertent phosphorylation of survival kinases (Stensløkken et al. 2009) that could influence the results and be responsible for some of the conflicting findings reported in the literature. It is worth noting that despite the existing differences between the atrial and ventricular tissues, the myocardium from the rat left ventricle and from the human right atrial appendage responded in an identical manner. Finally, although the PKB inhibitors used in this study reportedly exhibit high specificity and selectivity for PKB (Zhao et al. 2005; Barve et al. 2006), it cannot be entirely ruled out that the results obtained could reflect modulation of alternative cell survival pathways. Studies using transgenic technology also report opposing results and, therefore, in the future it may be necessary that a combination of approaches, pharmacological and transgenic modifications, and also an expanded and comprehensive number of functional, biochemical and genetic markers are used in a complementary manner.
Conclusions
In conclusion, blockade of PKB activity protects the mammalian myocardium against ischaemic injury and its location beyond the mitoKATP channels make it a target for clinical use, particularly in the presence of diabetes where dysfunctional mitochondria are responsible for the unresponsiveness to cardioprotection.
Acknowledgments
We are grateful to the Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Spain (BA06/90037) and the British Heart Foundation (FS/06/072) for the funding of this work and to Mrs Nicola Harris for secretarial assistance. The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- CK
creatine kinase
- 5-HD
5-hydroxydecanoate
- IP
ischaemic preconditioning
- I/R
ischaemia/reoxygenation
- KHH
Krebs–Henseleit-Hepes
- PDK
phosphoinositide-dependent kinase
- PH
pleckstrin homology
- PI-3K
phosphatidylinositol 3-kinase
- PKB
protein kinase B
- p38 MAPK
p38 mitogen-activated protein kinase
Author contributions
J.L.-P., M.A.H. and V.K.L. performed the studies and contributed to the interpretation of the results and the writing of the manuscript. J.M.D. and M.G. designed the studies and contributed to the interpretation of the results and the writing of the manuscript. Experiments were performed at the University of Leicester and the Nottingham Trent University. All authors approved the final version of the manuscript.
References
- Ahmad N, Wang Y, Haider KH, Wang B, Pasha Z, Uzun O, Ashraf M. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am J Physiol Heart Circ Physiol. 2006;290:H2402–H2408. doi: 10.1152/ajpheart.00737.2005. [DOI] [PubMed] [Google Scholar]
- Armstrong SC. Protein kinase activation and myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2004;61:427–436. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Ban K, Cooper AJ, Samuel S, Bhatti A, Patel M, Izumo S, Penninger JM, Backx PH, Oudit GY, Tsushima RG. Phosphatidylinositol 3-kinase γ is a critical mediator of myocardial ischaemic and adenosine-mediated preconditioning. Circ Res. 2008;103:643–653. doi: 10.1161/CIRCRESAHA.108.175018. [DOI] [PubMed] [Google Scholar]
- Barve V, Ahmed F, Adsule S, Banerjee S, Kulkarni S, Katiyar P, Anson CE, Powell AK, Padhye S, Sarkar FH. Synthesis, molecular characterization, and biological activity of novel synthetic derivatives of chromen-4-one in human cancer cells. J Med Chem. 2006;49:3800–3808. doi: 10.1021/jm051068y. [DOI] [PubMed] [Google Scholar]
- Basha G, Yap P, Penninckx F. Comparative study of classical, colorimetric and immunologic staining methods for the assessment of tumor cell viability. Tumour Biol. 1996;17:354–361. doi: 10.1159/000218000. [DOI] [PubMed] [Google Scholar]
- Budas GR, Sukhodub A, Alessi DR, Jovanovic A. 3′Phosphoinositide-dependent kinase-1 is essential for ischemic preconditioning of the myocardium. FASEB J. 2006;20:2556–2558. doi: 10.1096/fj.06-6252fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e1000017. doi: 10.1371/journal.pbio.1000017. doi 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Costa ADT, Quinlan CL, Pierre SV, Dos Santos P. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol. 2009;46:858–866. doi: 10.1016/j.yjmcc.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galiñanes M. Preconditioning the human myocardium by simulated ischemia: studies on the early and delayed protection. Cardiovasc Res. 2000;45:339–350. doi: 10.1016/s0008-6363(99)00353-3. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galiñanes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;38:711–718. doi: 10.1016/s0735-1097(00)01161-x. [DOI] [PubMed] [Google Scholar]
- Gomez J, Paillard M, Thibault H, Derumeauz G, Ovize M. Inhibition of GSK3-β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–966. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Daut J. KATP channels and preconditioning: a re-examination of the role of mitochondrial KATP channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Hassouna A, Loubani M, Matata BM, Fowler A, Standen NB, Galiñanes M. Mitochondrial dysfunction as the cause of the failure to precondition the diabetic human myocardium. Cardiovasc Res. 2006;69:450–458. doi: 10.1016/j.cardiores.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Hassouna A, Matata BM, Galiñanes M. PKC-ɛ is upstream and PKC-α is downstream of mitoKATP channels in the signal transduction pathway of ischaemic preconditioning of human myocardium. Am J Physiol Cell Physiol. 2004;287:C1418–C1425. doi: 10.1152/ajpcell.00144.2004. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Mocanu MM, Yellon DM. Cross-talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning. Cardiovasc Res. 2004;63:305–312. doi: 10.1016/j.cardiores.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;114:208–221. doi: 10.1016/j.pharmthera.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin43 deficient mice. Circ Res. 2005;97:583–586. doi: 10.1161/01.RES.0000181171.65293.65. [DOI] [PubMed] [Google Scholar]
- Heusch G. No RISK no…..cardioprotection? A critical perspective. Cardiovasc Res. 2009;84:173–175. doi: 10.1093/cvr/cvp298. [DOI] [PubMed] [Google Scholar]
- Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Zimin BD, Wang S, Ytrehus K, Antos CL, Olsen EN, Sollot SJ. Glycogen synthase kinase 3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1534–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischaemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Krieg T, Qin Q, Philipp S, Alexeyev MF, Cohen MV, Downey JM. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am J Physiol Heart Circ Physiol. 2004;287:H2606–H2611. doi: 10.1152/ajpheart.00600.2004. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Barnett SF, Layton ME, Bilodeau MT. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8:7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- Loubani M, Galiñanes M. Pharmacological and ischemic preconditioning of the human myocardium: mitoKATP channels are upstream and p38MAPK is downstream of PKC. BMC Physiol. 2002;2:10. doi: 10.1186/1472-6793-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao KM, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J, Liang Q. Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol. 2008;44:429–434. doi: 10.1016/j.yjmcc.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Matsui T, Tao J, del Monte F, Lee K-H, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Bell RM, Yellon DM. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J Mol Cell Cardiol. 2002;34:661–668. doi: 10.1006/jmcc.2002.2006. [DOI] [PubMed] [Google Scholar]
- Mora A, Davies AM, Bertland L, Sharif I, Budas GR, Jovanovic S, Mouton V, Kahn CR, Lucocq JM, Gray GA, Jovanovic A, Alessi DR. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity by hypoxia. EMBO J. 2003;22:4666–4676. doi: 10.1093/emboj/cdg469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Does inhibition of glycogen synthase kinase protect in mice? Circ Res. 2008;103:226. doi: 10.1161/CIRCRESAHA.108.181602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino Y, Webb JG, Davidson SM, Ahmed AJ, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber M. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–314. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang X-M, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2008;20:21–30. doi: 10.1016/j.cellsig.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Schulz R, Belosjorow S, Gres P, Jansen J, Michel MC, Heusch G. P38 MAP kinase is a mediator of ischemic preconditioning in pigs. Cardiovasc Res. 2002;55:690–700. doi: 10.1016/s0008-6363(02)00319-x. [DOI] [PubMed] [Google Scholar]
- Schulz R, Gres P, Skyschally A, Duschin A, Belosorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexion43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J. 2003;17:1355–1367. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, Schulz R, Heusch G. Ischemic postconditioning in pigs: no casual role for RISK activation. Circ Res. 2009;104:15–18. doi: 10.1161/CIRCRESAHA.108.186429. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenbergen C. The role of p38 mitogen-activated protein kinase in myocardial ischemia/reperfusion injury; relationship to ischemic preconditioning. Basic Res Cardiol. 2002;97:276–285. doi: 10.1007/s00395-002-0364-9. [DOI] [PubMed] [Google Scholar]
- Stensløkken KO, Rutkovskiy A, Kaljusto ML, Hafstad AD, Larsen TS, Vaag J. Inadvertent phosphorylation of survival kinases in isolated perfused hearts: a word of caution. Basic Res Cardiol. 2009;104:412–423. doi: 10.1007/s00395-009-0780-1. [DOI] [PubMed] [Google Scholar]
- Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res. 2000;87:309–315. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–3049. doi: 10.1161/01.CIR.0000130647.29030.90. [DOI] [PubMed] [Google Scholar]
- Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R972–R978. doi: 10.1152/ajpregu.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ahmad N, Kudo M, Ashraf M. Contribution of Akt and endothelial nitric oxide synthase to diazoxide-induced late preconditioning. Am J Physiol Heart Circ Physiol. 2004;287:H1125–H1131. doi: 10.1152/ajpheart.00183.2004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Ghosh S, Ockleford CD, Galiñanes M. Characterization of an in vitro model for the study of the short and prolonged effects of myocardial ischaemia and reperfusion in man. Clin Sci (Lond) 2000;99:443–453. [PubMed] [Google Scholar]
- Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME, Lindsley CW. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:905–909. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
- Zhu M, Feng J, Lucchinetti E, Fischer G, Xu L, Pedrazzini T, Schaub MC, Zaugg M. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res. 2006;72:152–162. doi: 10.1016/j.cardiores.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]