

Abstract
Aquaporin-2 (AQP2), located at the luminal side of the collecting duct principal cells, is a water channel responsible for the final concentration of urine. Lack of function, often occurring through mistargeting of mutated proteins, induces nephrogenic diabetes insipidus (NDI), a condition characterized by large urinary volumes. In the present study, two new mutations (K228E and V24A) identified in NDI-affected individuals from distinct families along with the already reported R187C were analysed in comparison to the wild-type protein (AQP2-wt) using Xenopus laevis oocytes and a mouse collecting duct cell-line (mIMCD-3). Initial data in oocytes showed that all mutations were adequately expressed at reduced levels when compared to AQP2-wt. K228E and V24A were found to be properly targeted at the plasma membrane and exhibited adequate functionality similar to AQP2-wt, as opposed to R187C which was retained in internal stores and was thus inactive. In coexpression studies using oocytes, R187C impeded the functionality of all other AQP2 variants while combinations with K228E, V24A and AQP2-wt only showed additive functionalities. When expressed in mIMCD-3 cells, forskolin treatment efficiently promoted the targeting of AQP2-wt at the plasma membrane (>90%) while K228E only weakly responded to the same treatment (∼20%) and both V24A and R187C remained completely insensitive to the treatment. We concluded that both V24A and K228E are intrinsically functional water channels that lack a proper response to vasopressin, which leads to NDI as found in both compound mutations studied (K228E + R187C and V24A + R187C). The discrepancies in plasma membrane targeting response found in both expression systems stress the need to evaluate such data using mammalian cell systems.
Introduction
Aquaporin-2 (AQP2) is a water channel of homotetrameric structure specifically expressed in principal cells of the kidney collecting duct. Found within intracellular vesicles, AQP2s are shuttled and fused to the luminal membrane in response to basolateral signalling from vasopressin, thus opening water permeability and allowing for water reabsorption from urinary filtrate (Robben et al. 2006). As the final water reabsorption step the activity of AQP2 is paramount to body water homeostasis, and lack of function, as found in individuals bearing incapacitating mutations, induces nephrogenic diabetes insipidus (NDI), a condition where normal water reabsorption is impeded. Affected individuals are unable to concentrate ultrafiltrate, thus producing large urinary volumes.
Extensive biochemical evaluations of NDI-causing mutations (http://www.medicine.mcgill.ca/nephros/aqp2.html) performed using diverse ‘expression’ systems have enabled the elucidation of key aspects of AQP2-related NDI mechanisms. Since most AQP2-related NDI are found to be recessive hereditary traits (Robben et al. 2006), heterozygotes bearing one wild-type form of the protein are usually non-symptomatic as two defective alleles are required to induce the disease. Even though altered forms of the AQP2 protein were often found to be less efficiently synthesized, as shown in studies performed using Xenopus oocytes (Marr et al. 2002a; Guyon et al. 2009b), the lack of function usually originates from their inadequate synthesis and routing (Deen et al. 1995; Lin et al. 2002; Marr et al. 2002a). The mutated protein, believed to be misfolded, is usually retained in intracellular stores (endoplasmic reticulum, ER) and eventually routed for degradation (class II mutations) (Robben et al. 2006). In some cases, mutated forms of AQP2 may also display weakened functionality (Marr et al. 2001, 2002a). In other cases, NDI is inherited as a dominant trait (Robben et al. 2006). These mutations are believed to be restricted to the C-terminal end of the AQP2 protein and usually operate through a dominant negative effect. In this mode of action, the altered protein associates and retains functional AQP2 counterparts within intracellular stores thus preventing normal apical targeting and function (Mulders et al. 1998; Kuwahara et al. 2001).
Past studies using the Xenopus laevis oocyte expression system have been very useful in defining the pathophysiological aspect of AQP2 enabling both functional and biochemical analysis of the protein (Deen et al. 1995; Marr et al. 2001). Water flux measurements are readily accessible through volume evaluations of oocytes challenged with osmotic gradients and most biochemical features including protein synthesis, post-translational modification and targeting can easily be studied in such large cells. Also, the oocyte enables highly controlled coexpression studies (Kamsteeg & Deen, 2000) that allow the unveiling of possible multimeric associations within AQP2 variants (mutants) that may be responsible for the dominant negative effect suspected with some AQP2 mutations. Other expression systems such as yeast (Shinbo et al. 1999) and cell lines (Levin et al. 2001; Marr et al. 2002a; Kamsteeg et al. 2003) have also been used with success, and normally confirm data determined in oocytes. Cell studies were most valuable in deciphering the regulatory means to APQ2 targeting and overall management in its natural mammalian cell environment (Kamsteeg et al. 2003; Umenishi et al. 2005).
In this study, we present the analysis of two new mutations (K228E and V24A) identified in two distinct families along the already reported mutation R187C. In the first family, two individuals are affected, while in the second family, only one individual is affected, all of which were determined to be compound mutations (K228E + R187C and V24A + R187C, respectively). All AQP2-wt-bearing heterozygotes related to these families presented no NDI-related symptoms. Using X. laevis oocytes and the mIMCD-3 cell-line as expression systems, we have performed a complete functional analysis and biochemical characterization for each mutation, probing both pure and coexpression conditions to determine the means by which these mutations induce NDI.
Methods
Ethical approval
All procedures regarding Xenopus laevis treatments and manipulations were performed in accordance with the Canadian guidelines and ethics committee of the Université de Montréal.
Oocyte preparation, injection and maintenance
Ovary nodes were surgically removed from gravid Xenopus laevis frogs anaesthetized with 2-aminobenzoic acid ethyl ester (1.3 g l−1) and bicarbonate (4 g l−1), and oocytes (stage V–VI) were dissected by hand. When required, animals were killed by prolonged anaesthesia. Follicular layers were removed using a collagenase treatment (17.5 mg ml−1, Type 1A, Sigma-Aldrich) for 1 h under mild agitation in a Ca2+ free Barth's solution (in mm: 90 NaCl, 3 KCl, 0.82 MgSO4, and 5 Hepes, pH 7.6) followed by overnight incubation at 18°C in normal Barth's solution (same as above with 0.4 mm CaCl2 and 0.33 Ca(NO3)2) supplemented with horse serum (5%), sodium pyruvate (2.5 mm) and antibiotics (100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin and 0.1 mg ml−1 kanamycin). The oocytes were then injected with water (controls) or the same volume of AQP2 cRNAs in varying (Figs 2, 6 and 7) or fixed quantities for wild-type (1 ng), mutated (5 ng) or a combination of both (as specified in figure legends) and further incubated for 1–3 days before proceeding to experimentation. cRNA preparations were diluted in water and injected with 46 nl per oocyte using a microinjection apparatus (Drummond Scientific, Broomall, PA, USA).
Figure 2. Expression of AQP2 variants in X. laevis oocytes.
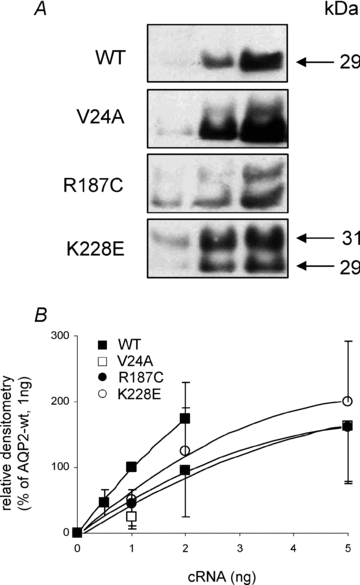
A, Western blot of oocytes injected with increasing amounts of cRNA for wild-type (0.5, 1 and 2 ng) and mutated forms (1, 2 and 5 ng) of AQP2. Each lane represents total membranes from 4 oocytes. B, densitometry evaluation of AQP2 bands (combination of both 29 and 31 kDa bands) extracted from A correlated against cRNA quantities. Data represent mean ±s.d. from triplicates and fitted to monoexponential equation.
Figure 6. Functionality of AQP2-wt in coexpression studies.
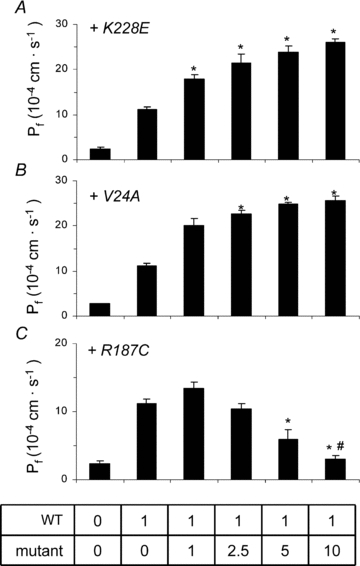
A standard 1 ng AQP2-wt cRNA was injected along with increasing concentrations (0–10 ng) of cRNA for K228E (A), V24A (B) or R187C (C) and water permeability was evaluated using a 20 mosmol l−1 hyposmotic shock. Pf values are given in 10−4 cm s−1 and represent 7–8 determinations for each condition. Asterisks indicate statistical significance (P < 0.05) in comparison to pure 1 ng AQP2-wt value while # indicates non-significance compared to controls (P < 0.05). Data are representative of three individual assays.
Figure 7. Functional analysis of mutant AQP2 in coexpression studies.
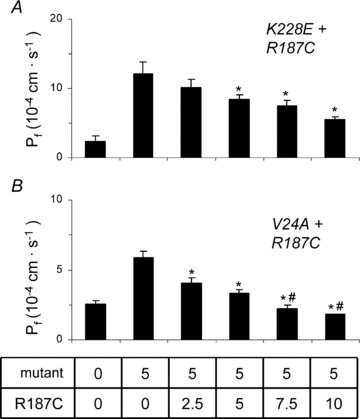
cRNA (5 ng) for either K228E (A) or V24A (B) was injected along with increasing concentrations of R187C cRNA (0–10 ng) and water permeability was evaluated using a 20 mosmol l−1 hyposmotic shock. Pf values are given in 10−4 cm s−1 and represent 7–8 determinations for each condition. Asterisks indicate statistical significance (P < 0.05) in comparison to pure K228E or V24A values while # indicates non-significance compared to controls (P < 0.05). Data are representative of three individual assays.
Vectors and cRNA
All constructs used for expression in oocytes were elaborated using the pT7Ts vector. The three point mutations, K228E, V24A and R187C, were inserted using site-directed mutagenesis within the wild-type APQ2 vector (pT7Ts-AQP2-wt) and validity of constructs was confirmed through sequencing. Primers used were 5′ CCGCCAGCCGAGAGCCTGTC 3′ (forward) and 5′ GACAGGCTCTCGGCTGGCGG 3′ (reverse) for K228E, 5′ CTCCTCTTCGCCTTCTTTGGC 3′ (forward) and 5′ GCCAAAGAAGGCGAAGAGGAG 3′ (reverse) for V24A and 5′ AATCCTGCCTGCTCCCTGGC 3′ (forward) and 5′ GCCAGGGAGCAGGCAGGATT 3′ (reverse) for R187C. For coexpression studies, a haemagglutinin (HA) tag was added to pT7Ts-AQP2-wt at the N-terminal position using SpeI and BglII restriction sites. For preparation of cRNAs, all vectors were linearized with SalI and cRNAs synthesized using the mMessage mMachine T7 kit (Ambion, Austin, TX, USA). pcDNA6-AQP2-wt vector used for expression in mIMCD-3 cells was generated by subcloning the full length AQP2 from pT7TS-AQP2-wt using BamH1 and Xba1 unique sites and all three mutations were inserted in pcDNA6-AQP2-wt using the same strategy as described above for pT7Ts-AQP2-wt.
Cell culture maintenance and transfection
mIMCD-3 cells were routinely cultured in DMEM-F12 media supplemented with 10% FBS and antibiotics (100 U ml−1 penicillin and 100 μg ml−1 streptomycin) and maintained in a 95% air, 5% CO2 atmosphere. For transfection with pcDNA6-AQP2 variants, cells were seeded on coverslips and transfection performed using Lipofectamine 2000 (Invitrogen) on 90% confluent monolayers (8 μg per 60 mm Petri dish). After 16–24 h incubation, coverslips were fixed at −20°C for 20 min with formaldehyde (1%) diluted in methanol. For evaluation of plasma membrane targeting, the coverslips were treated with forskolin (50 μm, 45 min) prior to fixation.
Preparation of total and plasma membrane fractions of oocytes
For descriptive methodology, see (Leduc-Nadeau et al. 2006). Briefly, total membranes were prepared by homogenizing 10 oocytes in 1 ml homogenizing buffer (HbA, in mm: 5 MgCl2, 5NaH2PO4, 1 EDTA, 80 sucrose, and 20 Tris pH 7.4) followed by low (250 g for 10 min) and high (16,000 g for 20 min) speed centrifugations. Pellets were resuspended in 20 μl HbA (2 μl solution per oocyte) and frozen until use. For plasma membranes, 40 oocytes were treated for 10 min at room temperature with 0.005% subtilisin A (Sigma-Aldrich) diluted in MES buffered saline for silica (MBSS) (80 mm NaCl, 20 mm Mes, pH 6.0) followed by two 1 h polymerizing steps, first with 1% ludox (Sigma-Aldrich), then with 0.1% polyacrylic acid (Sigma-Aldrich, MW 30,000) again diluted in MBSS and performed at 4°C. The oocytes were then homogenized in cold HbA by hand with a P200 pipettor followed by successive low speed centrifugations (16, 16, 25 and 35 g, all for 30 s at 4°C) keeping the bottom 75 μl and replacing the supernatant with equivalent volumes of fresh HbA. A final centrifugation at 16,000 g for 20 min was performed to pellet the purified plasma membranes which were resuspended in 10 μl HbA and frozen until use.
Western blots
Western blots were performed as described previously (Bissonnette et al. 1999) using total membranes and purified plasma membranes as described above. Samples representing either 2 oocytes (total membranes) or 40 oocytes (purified plasma membranes) were run on a 12% gel and transferred onto a nitrocellulose membrane. The efficiency of the overall procedure was monitored by Ponceau red staining. To prevent non-specific binding of antibodies, the membranes were first blocked with 5% non-fat milk in TBS-T (TBS + Tween 20, 0.1%) and then probed with the specific antibody (α-AQP2 (N-20) 1:100, Santa Cruz Biotech, CA, USA) followed by incubation with secondary antibody (HRP-linked chicken anti-goat 1:25,000, Santa Cruz Biotech). For specific determination of AQP2-wt in coexpression studies using HA-AQP2-wt (Fig. 5), blots were probed using HRP-linked mouse anti-HA antibody (1:500 dilution, Roche, Laval, QC, Canada). Validation of plasma membrane purification was performed by probing Western blots for protein disulphide isomerase (PDI), an endoplasmic reticulum resident enzyme (α-PDI 1:500 followed by HRP-linked donkey anti-rabbit 1:10,000; Santa Cruz Biotech). Blots were revealed using enhanced chemiluminescence detection (Phototope-HRP, New England Biolabs, Pickering, ON, Canada).
Figure 5. Expression of AQP2 in relation to family genotypes.
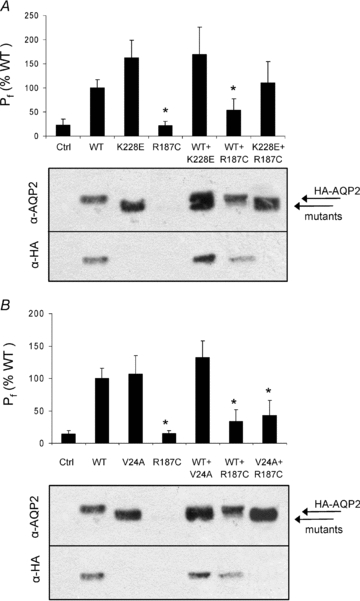
Oocytes were injected with AQP2 cRNA combinations in accordance with Fig. 1 reproducing genotypes presented for family 1 (A) and family 2 (B). With regard to translation efficiency, cRNAs were injected at 1 ng (HA-AQP2-wt) or 5 ng (mutant) with the exception of double mutant conditions (K228E + R187C and V24A + R187C) where both mutants were injected at 5 ng each. Functionality was evaluated using a mild hyposmotic shock (−20 mosmol l−1). Pf values are expressed as percentage activity of pure AQP2-wt and represent 7–8 determinations for each condition. Western blots of purified plasma membrane were first probed using α-AQP2 for total AQP2 staining then stripped and probed again using α-HA to specifically identify wt-AQP2. Data are representative of three individual assays.
Immunofluorescence
Oocytes
Immunofluorescence detection in oocytes was performed according to the protocol described earlier for fixed oocytes (Bissonnette et al. 1999). Control, wild-type (1 ng) and mutant (10 ng) injected oocytes were incubated for 72 h before the immunofluorescence assay. Oocytes were rinsed 3 times with serum-free Barth's solution and fixed for 15 min in ice-cold methanol solution containing 1% formaldehyde. Fixed oocytes were rinsed again three times and incubated overnight in a 30% sucrose Barth's solution. Oocytes were embedded in Tissue-tek medium diluted 1:7 in water (Sakura Finetek, USA), frozen, sliced (10 μm thickness) on a cryostat and mounted on slides. Slices were blocked for 30 min at room temperature using a 2% BSA solution in PBS. Incubation with anti-AQP2 antibody at a dilution of 1:20 in PBS + BSA was performed in a wet chamber at room temperature for one hour, followed by three rinses in PBS. Incubation with secondary antibody (Alexa Fluor 488-conjugated anti-goat antibody, 1:1000 in blocking solution, Molecular Probes, Eugene, OR, USA) was performed as for the first antibody and rinsed accordingly. Slides were mounted using an anti-quenching agent (Prolong antifade, Molecular Probes) prior to observation.
Cells
Immunofluorescence of cell monolayers was performed on confluent mIMCD-3 cells grown and transfected on coverslips (see above) as described previously (Bissonnette et al. 1999). AQP2 proteins (green) were visualized using the same protocol as for oocytes. Prior to fixation, some coverslips were treated with forskolin (50 μm, 45 min, de Mattia et al. 2005) to evaluate the cAMP-dependent plasma membrane insertion of all AQP2 variants through positive identification (fluorescence) of membrane ruffles at the plasma membrane. PDI was used as ER marker and visualized (red) using a specific antibody (mouse α-PDI, 1:50 dilution, Santa Cruz Biotech) with corresponding secondary antibody (Alexa 568-labelled α-mouse, 1:1000 dilution, Molecular Probes). Nuclei were stained by incubating slides in 4′,6-diamidino-2-phenylindole (DAPI) for 30 min (Sigma, 1 μg ml−1 in PBS). As for the oocytes, slides were mounted using an anti-quenching agent (Prolong antifade) prior to visualization (×60) and micrograph treatments were performed using an Olympus IX-81 microscope and Image-Pro Plus v.5.0 software.
Volume measurements
Functionality of AQP2 was assessed by water flux measurements in water-injected and AQP2-injected oocytes. Briefly, the oocytes were placed in a 0.07 ml bath on the stage of an inverted, low power microscope equipped with a camera and a recording system for analysis of the oocyte cross-section. The oocytes were challenged to a mild hyposmotic solution performed by removing mannitol from the isotonic media (−20 mosmol l−1), and the osmotically induced swelling monitored. The variations in volume were used to determine water permeability values (Pf) which are given in 10−4 cm s−1. A more detailed description of this set-up and procedure is presented elsewhere (Duquette et al. 2001).
Data analysis
All experiments were performed at least three times using different oocyte batches or cell cultures and values are presented as mean ±s.e.m. One-way ANOVA + Tukey's statistical analysis was performed on all Pf determinations. Molecular mass and densitometry analyses were performed using software from Alpha Imager 2000 (Alpha Innotech Corp, San Leandro, CA, USA).
Results
Family description
This report presents two new mutations for AQP2, K228E and V24A, respectively, identified in individuals from families 1 and 2 (Fig. 1) along with the already described mutation R187C identified here in both families. Also in both families, only the compound heterozygote individuals presenting both mutations (K228E + R187C in family 1 and V24A + R187C in family 2) are affected with severe NDI while individuals bearing only one mutation (heterozygotes) are all healthy. The V24A mutation was identified in a 39-year-old woman presenting with polyuria and polydipsia as well as dehydration periods. Mean urinary volumes were 14.2 l (24 h)−1 with corresponding osmolality of 106 mmol (kg H2O)−1. K228E was identified in two siblings (4 months) presenting with mild to severe phenotype including fever with dehydration periods and inability to concentrate urine in the dehydration test (283 mmol (kg H2O)−1).
Figure 1. Segregation of AQP2 alleles in two families.
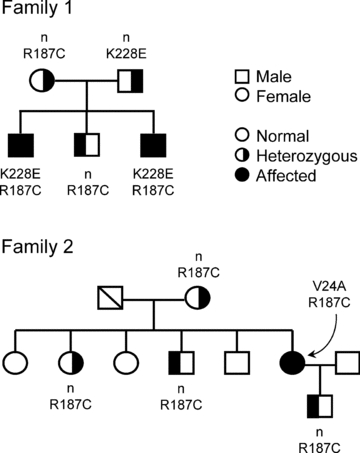
Genotypes presenting segregation of both wild-type (open symbols) and mutated (filled symbols) forms of AQP2 in the two families tested. Clinical evaluation indicates that only individuals with compound mutations are affected with NDI. n indicates normal alleles.
Expression of mutated AQP2 in oocytes
Since mutated forms of AQP2 are often ill-expressed in oocytes, the relative abundance efficiencies of all three AQP2 mutations along with wild-type were initially evaluated through densitometry analysis of Western blots. All AQP2 variants were thus expressed using increasing amounts of cRNA (0.5–2 ng for AQP2-wt and 1–5 ng for mutant forms) and densitometry values plotted against nanograms of injected cRNA. Figure 2A presents a typical Western blot with increasing AQP2 labelling from which mean densitometry values of five individual experiments were used to evaluate their relative expression efficiencies (Fig. 2B). The Western blot profile found for wild-type protein essentially depicts a major 29 kDa band along a very faint band at 31 kDa. In comparison, mutated forms of AQP2 usually display a more intense 31 kDa band of varying intensity which may even be equivalent to that of 29 kDa (R187C). This heavier form of AQP2 corresponds to an N-glycosylation intermediate whose fate remains unclear. As shown, the wild-type form of AQP2 was expressed more efficiently so that 1 or 2 ng of AQP2-wt cRNA globally compares to 5 ng cRNA of any mutant. Since the 31 kDa form is not expected to actually participate in functional channels, a ratio of 1:5 was thus chosen in coexpression studies to evaluate interactions between wild-type and mutant forms of AQP2. In the following set of experiments, all three mutants were compared to AQP2-wt looking at both functionality (Fig. 3A) and plasma membrane targeting (Fig. 3C and D). For this analysis, oocytes were injected with either 1 ng AQP2-wt cRNA or 5 ng cRNA coding for each mutation. As seen in Fig. 3B, where total AQP2 proteins are evaluated (total membrane preparations), abundance levels of all APQ2 variants tested were found to be equivalent thus validating the 1:5 cRNA ratio determined previously in Fig. 2. In all assays performed on mutants, the 29 kDa band was always found to be predominant with the exception of R187C which usually displays equivalent 29 and 31 kDa bands. When purified plasma membranes from the same oocytes were probed for AQP2 (Fig. 3C), all variants except R187C were found to be adequately targeted at the plasma membrane. It is also noteworthy that the 31 kDa bands are absent from the plasma membrane fractions. The absence of ER-resident protein PDI staining in the lower blot of Fig. 3C, as opposed to Fig. 3B, demonstrates the absence of intracellular membrane contaminants in the purified plasma membrane fractions tested, thus validating the purification procedure (Leduc-Nadeau et al. 2006). Similarly, immunofluorescence labelling of AQP2 performed on oocyte slices (Fig. 3D) also demonstrated the plasma membrane localization for all AQP2 variants under study with the exception of R187C which is retained in intracellular stores, most probably the ER. These plasma membrane targeting results also correlate with functionality assays (Fig. 3A) as all variants adequately targeted also display appropriate water permeability values (in this experiment, Pf values are (14.4 ± 5.5), (16.3 ± 3.5) and (20 ± 7) × 10−4 cm s−1, for AQP2-wt, V24A and K228E, respectively). As expected, the only mistargeted mutant, R187C, displayed no specific activity (Pf= (2.6 ± 0.6) × 10−4 cm s−1) similar to water-injected oocytes (Pf= (2.8 ± 1.0) × 10−4 cm s−1). Overall statistical analysis of functionality for all AQP2 variants (7 independent assays) comparing expressions of 1 ng wild-type against 5 ng for mutant forms gave Pf values of (14.2 ± 4.5) × 10−4 cm s−1 for AQP2-wt (n= 64), (16 ± 6) × 10−4 cm s−1 for V24A (n= 24), (22 ± 5) × 10−4 cm s−1 for K228E (n= 24) and (2.4 ± 0.7) × 10−4 cm s−1 for R187C (n= 24), which is similar to controls ((2.8 ± 1.1) × 10−4 cm s−1, n= 64).
Figure 3. Expression of AQP2 variants in X. laevis oocytes.
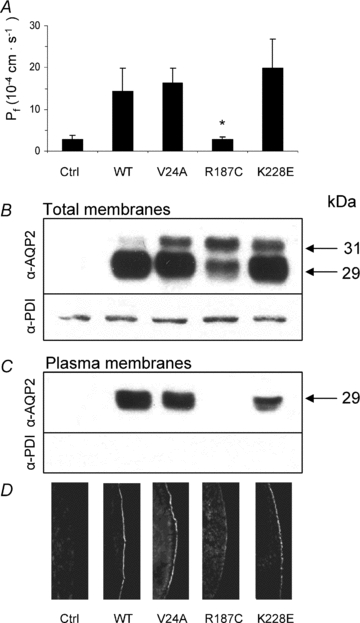
A, functional evaluation of all AQP2 variants. Oocytes were injected with cRNA coding for either wild-type (WT, 1 ng) or mutated (V24A, R187C and K228E, 5 ng) AQP2 along with controls (Ctrl) and incubated for 72 h prior to testing for water permeability capacities (see Methods). Pf values are in 10−4 cm s−1 and represent 7–8 determinations per condition. Data are representative of 5–7 individual assays. The same oocytes were tested in Western blot using either total membranes (B, 1 oocyte per lane) or purified plasma membranes (C, 40 oocytes per lane) fractions. PDI detection was performed on the same material to confirm the quality of the purification procedure for plasma membrane in C. D, immunofluorescence labelling of all AQP2 in the same samples showing retention of R187C within intracellular stores as opposed to AQP2-wt, V24A and K228E.
Studies were also performed to determine the relative plasma membrane targeting efficacy for all AQP2 species under study. To do so, Western blots from both total and plasma membranes samples were probed for AQP2 and relative densities of specific bands for each mutant were evaluated and compared to AQP2-wt, which served as reference, i.e. plasma over total membrane densities ratio for AQP2-wt = 1. Results averaging five assays indicated that both V24A (1.2 ± 0.3) and K228E (1.2 ± 0.4) have similar plasma membrane targeting efficacies in comparison to AQP2-wt. Also, relative single channel activity levels were assessed by comparing Western blot densitometry values of each AQP2 variant, as found at the plasma membrane, against their respective activity levels (Pf values). Once again taking AQP2-wt as reference (Pf over blot density = 1), both V24A (ratio = 1.0 ± 0.2) and K228E (ratio = 1.3 ± 0.2) displayed single channel functionalities similar to AQP2-wt.
More relevant targeting studies were performed using the renal mIMCD-3 cell line. To do so, cells transfected with all three mutant AQP2 forms were compared to AQP2-wt for their ability to reach the plasma membrane in response to forskolin treatment. As seen in the top panel of Fig. 4, AQP2-wt responded adequately and showed positive plasma membrane localization (identification of plasma membrane ruffles) of the protein after forskolin treatment (arrow) in over 90% of observations (n > 50) while R187C remained sequestered within intracellular stores with the same treatment (n > 30). These two results are in agreement with those previously found in oocytes. For the two remaining mutants, plasma membrane targeting induced by forskolin was either of low occurrence for K228E (20%, n > 50) or completely absent for V24A (n > 30).
Figure 4. Immunofluorescence analysis of AQP2 in transfected IMCD-3 cells.
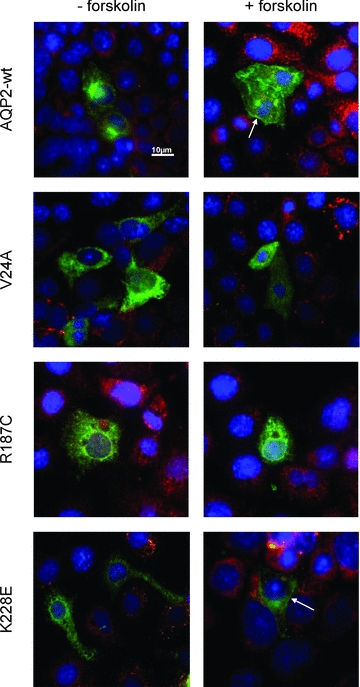
mIMCD-3 cells were transfected with pcDNA6-AQP2-wt, -V24A, -R187C or -K228E, incubated for 16–24 h and treated (+forskolin) or not (–forskolin) with forskolin (50 μm, 45 min) prior to fixation. PDI was used as an ER marker (red) and DAPI was used as a nuclear stain (blue). Plasma membrane staining is found for both AQP2-wt and -K228E (arrows) but not for R187C or V24A. Magnification ×60. Scale bar, 10 μm.
Coexpression studies
As mentioned above, only the two compound mutation individuals (K228E + R187C in family 1 and V24A + R187C in family 2) are affected. In that context, a study was performed where oocytes were injected with APQ2 variants in accordance with the reported family genotypes presented in Fig. 1. The goal of this study was to correlate functionality results, as determined by water permeability evaluations, with their respective plasma membrane expression levels, as determined by Western blot. Oocytes were thus injected either with pure AQP2 variants or with a mixture of cRNAs to allow an equivalent coexpression of AQP2 products (1 ng HA-tagged AQP2-wt + 5 ng mutant cRNAs or 5 ng of each mutant). The functionality results presented in Fig. 5A and B are mean ±s.d. (as % of AQP2-wt alone) of 22–30 determinations per condition collected within six separate experiments. As seen in Fig. 5A representing family 1, K228E displays a functionality superior to that of the wild-type (162 ± 37%) while R187C is without activity, similar to water-injected oocytes (22 ± 9 and 22 ± 13%, respectively). Co-injecting mutant forms along with AQP2-wt either increased (+K228E, 169 ± 58%) or partially inhibited (+R187C, 52 ± 24%) the level of activity in comparison to AQP2-wt alone. Lastly, co-injecting K228E along with R187C also generated intermediate activity levels (107 ± 45%). Asterisks indicate a statistical difference from pure AQP2-wt (P < 0.05). In addition, Western blots were also performed to evaluate the impact of these expression conditions on the presence of both total and wild-type forms of AQP2 at the plasma membrane. For that purpose, an HA-tagged version of AQP2-wt was used to allow its discrimination against mutant forms of the protein within a same sample. Care was taken to confirm that the presence of the HA-tag at the C-terminus did not impede the normal functionality of the protein (data not shown). Western blots were thus performed on purified plasma membranes using either an anti-AQP2 antibody to evaluate total AQP2 (upper blot) or an anti-HA antibody to specifically evaluate the wild-type form of AQP2 (lower blot). As seen in the blots presented in Fig. 5A, all AQP2-injected oocytes with the exception of R187C displayed appropriate labelling at plasma membranes, which correspond to the functionality data shown in the graph above. The lower blot specifically presenting HA-AQP2-wt indicates that the wild-type protein was present in plasma membranes of all conditions, including when coexpressed with K228E or R187C. The higher molecular mass found with HA-AQP2-wt (∼2 kDa) is due to the addition of the HA-tag. Also of interest, we find that staining for HA-AQP2-wt is more intense in the presence of K228E and reduced in the presence of R187C in comparison to HA-AQP2-wt alone (α-HA blot).
For the second family case study (Fig. 5B), similar results were found; functionality of V24A was found to be equivalent to AQP2-wt (109 ± 29%) while R187C remained completely inactive, similar to control oocytes (15 ± 5 and 14 ± 5%, respectively). Also, co-injecting AQP2-wt was additive with V24A (136 ± 26%) and inhibitory with R187C (36 ± 17%), similar to when both mutations were co-injected together (43 ± 24%). Once again, the corresponding Western blot presents AQP2 labelling at the plasma membrane that is compatible with activity, i.e. adequate labelling for AQP2-wt and V24A but not R187C (α-AQP2 blot). The presence of AQP2-wt at the plasma membrane, as detected using anti-HA antibody (α-HA blot), was also found in both coexpression conditions and again seemed to be increased in the presence of V24A and reduced in presence of R187C.
A distinctive feature of some AQP2 mutations, known as the dominant negative effect, is found when a mutated form of the protein combines with and sequesters its wild-type counterpart within intracellular stores. This association between wild-type and mutant forms of AQP2 results in a reduction of properly targeted AQP2 channels with concomitant lack of function. In order to further investigate interrelations within AQP2 variants, we have measured the water permeability of oocytes injected with a fixed 1 ng cRNA coding for AQP2-wt with increasing cRNA concentrations (0–10 ng) coding for each mutation in the study (Fig. 6). The profiles found for both K228E (panel A) and V24A (panel B) are similar; AQP2-wt generates an increase in water permeability over control oocytes which is gradually enhanced by the addition of increasing cRNA coding for either mutation. Addition of 10 ng mutant cRNA increased the wild-type-induced permeability by (14.9 ± 2.9) × 10−4 cm s−1 and (14.4 ± 1.0) × 10−4 cm s−1 for K228E and V24A, respectively. These inductions are compatible with Pf values determined previously with independent expressions of the same mutants (Fig. 3A). On the other hand, results found when co-injecting R187C (Fig. 3C) are completely opposite to AQP2-wt-dependent Pf gradually decreasing to control levels (water-injected oocytes) within 10 ng R187C cRNA ((−8.1 ± 1.1) × 10−4 cm s−1). While coexpressions of wild-type and mutant AQP2 reflect the prevailing conditions found for AQP2-wt-bearing heterozygote members in both families studied, Fig. 7 presents analyses of coexpressions representing the two affected conditions. In both assays, a standard 5 ng cRNA for either K228E (panel A) or V24A (panel B) is challenged against increasing concentrations of R187C (0–10 ng). Once again, R187C drastically diminishes the Pf values of both K228E ((−9.0 ± 1.8) × 10−4 cm s−1), and V24A ((−4.1 ± 0.4) × 10−4 cm s−1). Although the effect of R187C on the three forms of AQP2 tested is similar, its inhibitory action against both mutants is more pronounced with Pf reductions evidenced at a 1:1 ratio for both K228E and V24A (Fig. 7A and B) while a 1:5 ratio is necessary to reach a similar effect with AQP2-wt (Fig. 6C). These functional assays were also tested through Western blot evaluations, probing key conditions found in this study (HA-AQP2-wt alone (1 ng) or in combination with untagged mutants (5 ng)), in order to evaluate the impact of all mutations on the expression and targeting of the AQP2-wt counterpart. To do so, total membranes were probed with α-AQP2 to identify all AQP2 forms (evaluation of AQP2 abundances) while plasma membrane fractions purified from the same samples were probed using α-HA (evaluation of HA-AQP2-wt targeting at the plasma membrane; Fig. 8). As shown in Fig. 8A, AQP2-wt is equally expressed in all conditions (AQP2-wt) while its presence at the plasma membrane (Fig. 8B, α-HA) seems to be modulated by expression conditions with both V24A and K228E promoting the presence of AQP2-wt at the plasma membrane while R187C reduces it. These results are in agreement with those presented in Fig. 5A and B (α-HA blots).
Figure 8. Effects of mutant's coexpression on AQP2-wt abundance and plasma membrane targeting.
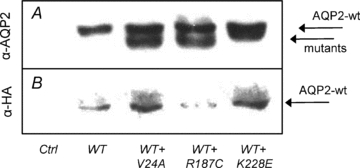
Oocytes injected with water (Ctrl), HA-AQP2-wt alone (WT, 1 ng) or with untagged mutants (+, 5 ng) were probed for overall AQP2 abundances in total membranes using α-AQP2 (A) or specifically for AQP2-wt in purified plasma membranes using α-HA (B).
Discussion
Many mutations responsible for NDI have been identified so far and expression in Xenopus oocytes has proven to be a tool of choice for both biochemical and functional studies. Key pathophysiological data related to NDI, such as misrouting or inadequate processing of altered AQP2 proteins, were identified using this approach (Tamarappoo & Verkman, 1998; Marr et al. 2002a). Most frequently, NDI is transmitted to lineage as a recessive trait, which implies that two mutated alleles are required to induce the disease. Although expressing AQP2 mutations in oocytes is not expected to reproduce actual phenotypes reported by affected individuals, coexpression studies have permitted some important features related to this condition to be highlighted. One important trait sometimes found in AQP2-related NDI is the dominant negative behaviour in which a defective subunit associates with its functional counterpart(s) and impedes the normal processing and/or targeting of the oligomer, thus sequestering the complex in internal stores and preventing the normal activity of the whole (Robben et al. 2006). These mutations, essentially located at the C-terminal end of the protein, are responsible for the autosomal dominant form of transmission of NDI. A few reports in the past have presented such interactions between wild-type and mutated forms of AQP2, usually through co-immunoprecipitation assays (Kamsteeg et al. 1999; Kamsteeg & Deen, 2001; Marr et al. 2002b).
The present study focuses on two new mutations identified from distinct families, K228E and V24A, as well as R187C found in both families and already reported in the literature (Mulders et al. 1998; Tamarappoo & Verkman, 1998; Shinbo et al. 1999). As indicated in clinical data, only the individuals presenting compound mutations (K228E + R187C and V24A + R187C) are affected with NDI and since heterozygotes are not affected, no dominant negative behaviour of pathological impact is suspected for these mutations. The schematic representation of the AQP2 protein in Fig. 9 shows the positions of all three mutations under study. V24A, situated within the first transmembrane domain, represents a modest modification with the replacement of valine with a similar non-polar amino acid of smaller size. Functionality for that mutant form is thus not surprising. On the other hand, the R187C mutation is located immediately after the second NPA motif which is implicated in the binding and transfer of water molecules. The arginine in position 187 is most probably a constituent of the water channel structure, similar to R181 in the closely related AQP1 channel (Wu et al. 2009). Moreover, replacing a large basic amino acid (arginine) by a small neutral one (cysteine) at such a sensitive location represents a profound modification most inclined to alter the protein's functionality. The last mutant, K228E, is located at the C-terminal end of the protein, near the last transmembrane domain. Even though this mutation implies changing a bulkier basic lysine for a smaller glutamic acid, and thus changing the charge of the residue, this amino acid is found in a region of low secondary structure which may be more compliant to such modifications. We thus set to characterize these new mutations, looking at functionality, maturation and targeting properties through a series of AQP2 expressions in oocytes and mammalian cells with the goal of identifying the means by which each mutation operates.
Figure 9. Schematic representation of AQP2 protein.
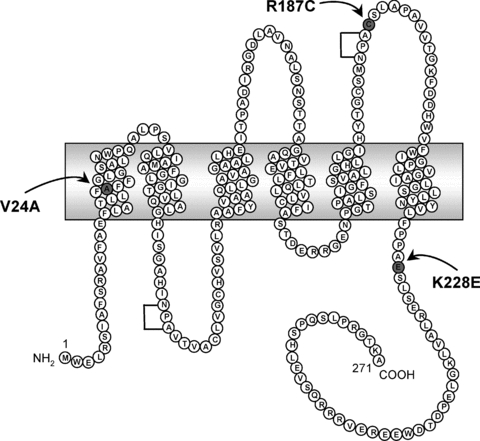
Depiction of the human AQP2 protein showing the positions of V24A, R187C and K228E mutations. Brackets indicate conserved NPA motifs.
Processing, functionality and targeting of mutated AQP2
Figure 3 presents an overall characterization of AQP2 expression in oocytes and describes both the abundance and targeting of the protein as well as its functionality as a water channel. As seen in Figure 3B, all AQP2 variants are adequately synthesized, presenting a typical 29 kDa band as the major product along with a 31 kDa band of uneven intensity depending on the AQP2 variant tested. This 31 kDa band, signature to a glycosylation intermediate, is indicative of an impaired maturation of the protein somehow hindered in its normal maturation process (Deen et al. 2000). As in oocytes only one out of four AQP2 units is estimated to be glycosylated, the remaining 75% is believed to be readily expressed without any post-translational step (glycosylation). Also in oocytes, the wild-type AQP2 is processed rapidly (Hendriks et al. 2004) and consequently, only a faint 31 kDa band can sometimes be evidenced (AQP2-wt lane in this blot). In its place, a mature glycosylated broad band of 39–45 kDa is occasionally found (not shown here). When comparing the 31 kDa and the 29 kDa bands for all three mutants to evaluate post-translational processing, we find that K228E and V24A both have moderate intermediate species (31 kDa) while R187C usually displays a more pronounced 31 kDa band. This underlines possible difficulties in protein maturation for this latter mutant which agrees with the fact that it is the only form with full ER retention in this study, as shown by its absence at the plasma membrane (Fig. 3C). In accordance with this maturation process, we find that only the 29 kDa band successfully reaches the plasma membrane. This is in agreement with the accepted notion that the 31 kDa intermediate band is retained in intracellular stores and most probably destined for degradation. When comparing functionality (Pf values) against plasma membrane expression levels (AQP2 densitometry in Western blots of purified plasma membrane fractions) it is estimated that all AQP2 variants, with the exception of R187C which cannot be evaluated here, display appropriate unitary water transport capacities, similar to AQP2-wt. Also, through densitometry ratio evaluations of plasma membranes over total membranes we have determined that, in oocytes, both K228E and V24A are appropriately targeted to the plasma membrane similar to AQP2-wt (ratio of 1.2 in comparison to wild-type for both mutations) thus indicating that the targeting process for both mutants is not defective, at least in this system. We thus conclude that both K228E and V24A are functional AQP2 variants, similar to the wild-type protein. In comparison to AQP2-wt, when considering functionality (equivalent), plasma membrane targeting (equivalent) and protein abundance (20–40%), we find that K228E and V24A are the two most efficiently expressed AQP2 mutations evidenced so far in oocytes. Previous data, including ours (Guyon et al. 2009a), globally reports overall efficiencies of about 1/10 or less (Marr et al. 2002a; Robben et al. 2006). With such functional characteristics in oocytes, V24A and even more K228E could basically be considered as functional variants of AQP2 with reduced expression levels, which may not satisfactory explain the NDI phenotype observed in compound mutation individuals.
By expressing APQ2 variants in mIMCD-3 cells, a more relevant cell system of medullary origin, we found that the plasma membrane-targeting properties of both K228E and V24A does not correlate with those already determined in oocytes. Despite forskolin treatment, V24A completely fails to reach the membrane while only about 20% of the K228E-expressing cells present positive identification for plasma membrane labelling (Fig. 4, lowest panels) as opposed to the oocyte model in which plasma membrane targeting is readily performed (Fig. 3). AQP2-wt on the other hand is readily targeted at the plasma membrane by the same treatment (over 90% positive plasma membrane labelling, n > 50) as opposed to R187C which remains in intracellular stores after treatment. Our data show that R187C does not reach the plasma membrane in oocytes (Figs 3C and D, and 5) or mIMCD-3 cells (Fig. 4), which is in agreement with other reports using the same systems (Tamarappoo & Verkman, 1998; Yamauchi et al. 1999; Marr et al. 2001, 2002b). Even though oocytes and mammalian cells usually generate similar plasma membrane targeting results for AQP2 mutants (Guyon et al. 2009a), the diverging targeting results found with K228E and V24A underline existing differences between both expression systems and stress the need to confirm key features such as targeting using a more appropriate cell model such as mIMCD-3 cells. Thus, even though both K228E and V24A are functional proteins, the basic defect most probably resides in their profound inability to adequately reach the plasma membrane, worsened by a low protein synthesis level (20–40% in comparison to AQP2-wt, in oocytes) and a possible lack of maturation. In heterozygote individuals bearing one wild-type allele, the functional protein is believed to compensate for the non-functional K228E and V24A mutations which is not possible in compound mutations comprising R187C, as found in the affected individuals in both family cases. Although some reports have presented mutations (A147T, L22V) where defects essentially originate from inadequate synthesis of a functional AQP2 variant (Tamarappoo & Verkman, 1998; Marr et al. 2001), mistargeting is the usual means by which mutated forms of AQP2 operate (Mulders et al. 1998; Marr et al. 2002b; Robben et al. 2006). Although the plasma membrane targeting of AQP2 implicates phosphorylation, we do not know at this time if K228E and V24A present altered phosphorylation or if mistargeting results from some other feature down the normal targeting pathway.
Coexpression studies
AQP2 is expressed at the plasma membrane as a homotetramer and the relations between subunits may become critical, as displayed with the dominant negative effect. Hence, coexpression analyses combining wild-type and mutant elements may highlight some of the molecular aspects of the protein's functionality and even relate to actual clinical situations. For that reason, coexpression analyses were performed by combining the different APQ2 species to investigate conditions depicted for both the families described here. In a first study, combinations of cRNA were performed based upon respective translation efficiencies (1 unit for AQP2-wt and 5 units for all mutants) in order to evaluate the possible interactions between AQP2 variants, as was done previously in a similar case study (Guyon et al. 2009a). Although a 2:5 ratio could have been considered with respect to results in Fig. 2, choosing a 1:5 ratio seemed more adequate since the 31 kDa high-mannose forms of AQP2 are usually believed to be trapped non-functional forms of the protein and should thus not be considered when designing functional assays. Also, we consider this approach only as indicative since we can merely speculate on the actual expression properties (both quantitatively and qualitatively) of these AQP2 forms, as found in the actual individuals, in pure or combined expressions. The Western blots performed on purified plasma membrane fractions (Fig. 5) indicate that the wild-type protein is adequately synthesized and routed to the plasma membrane in all conditions of expression tested. Through visualization of AQP2-wt using an HA tag (lower blots in Figs 5A and B, and 8B), it is shown that both K228E and V24A not only enable but even seem to promote the plasma membrane insertion of the wild-type protein. These data confirm that the chosen ratio of 1:5 for wild-type over mutant cRNA did not impede the normal expression level of AQP2-wt through synthesis competition, as cautioned previously (Kamsteeg & Deen, 2000). In coexpression analyses where 1 ng AQP2-wt was co-injected with increasing amounts of all three mutants (Fig. 6A and B), we find a gradual increase in water permeability for both K228E and V24A (mean Pf increase = (14.9 ± 2.8) and (14.4 ± 1.0) × 10−4 cm s−1, respectively) which is compatible with the sum of individual activities, indicating again that there is no effective competition for synthesis between wild-type and mutant forms of the protein. The mechanism by which both V24A and K228E promote the plasma membrane insertion of the wild-type form of AQP2 is not known but may implicate heteromeric associations between mutant and wild-type forms.
On the other hand, under the same conditions, R187C seems to impede or restrain AQP2-wt targeting, as seen in both functionality and Western blot studies (Figs 5A and B, and 8B). This behaviour was not expected since this mutant, which does not impede AQP2-wt expression levels under our working conditions (Fig. 8A), is not believed to directly interact with AQP2-wt due to its monomeric nature (Kamsteeg et al. 1999). In the subsequent studies, increasing R187C cRNA loads against 1 ng AQP2-wt was shown to gradually diminish its functionality down to background level (Fig. 6C, mean Pf decrease = (7.0 ± 5.1) × 10−4 cm s−1), a behaviour also found with both K228E and V24A (Fig. 7A and B). Again, this loss of function was not believed to be secondary to direct competition in protein synthesis from overloaded machinery since K228E and V24A fail to do so in similar conditions (Fig. 6A and B). These results indicate that R187C, which is not targeted to the plasma membrane, can impede the functionality of any of the other AQP2 species tested here. Again, the inability of R187C to multimerize basically prevents the dominant negative effect usually proposed to explain the retention of the wild-type protein and, since the coexpression conditions do not seem to significantly impede wild-type protein synthesis (Fig. 8A), other mechanisms leading to interference in the normal processing of AQP2-wt are to be explored, possibly implicating protein maturation and targeting features.
In this study, the two new AQP2 mutations, K228E and V24A, display high levels of functional expression for mutant forms (between 20 and 40% of wild-type) with adequate plasma membrane targeting in oocytes but not in mIMCD-3 cells where they are largely (K228E) or completely (V24A) restrained within internal stores. The lack of response following proper signalling (vasopressin) is thus believed to be responsible for the lack of functionality found with these mutants as well as reduced synthesis levels. With regards to heterozygote family members, we believe that an effective dominant negative effect could have been possible since both mutations can modulate the activity of the wild-type form of AQP2 (Figs 5 and 8), but prevented by low synthesis levels and/or insufficient multimeric associations with AQP2-wt. In that regard, both mutations may be valuable candidates for recovery assays using strategies such as chemical chaperones to promote or restore adequate functionality (Tamarappoo & Verkman, 1998). R187C shows mistargeting in both expression models tested and may also interfere with the normal processing of both mutant and wild-type proteins. Further investigations regarding the synthesis and cellular management of R187C could provide key information in relation to the normal expression of AQP2-wt. Nevertheless heterozygotes of both families are not affected, we can only conclude that the deleterious effects of these mutations, including R187C, do not supersede the basic functionality of AQP2-wt.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR), grant no. MOP-10580, Chair in Genetics of Renal Disease (D.G.B.) and by the FRSQ infrastructure program no. 5252.
Glossary
Abbreviations
- AQP2
aquaporin-2
- ER
endoplasmic reticulum
- HA
haemagglutinin tag
- NDI
nephrogenic diabetes insipidus
- PDI
protein disulphide isomerase
Author contributions
Identification, clinical data and initial evaluations (genotyping, Western) for family members 1 and 2 were performed by A.M.-A. and Devuyst laboratories, respectively. D.G.B.'s laboratory was responsible for confirmation of AQP2 mutations (AVPR2 and AQP2 genotyping), expressions in oocytes and mIMCD-3 cells. All authors contributed equally to the elaboration of the manuscript and approved the final version.
References
- Bissonnette P, Noel J, Coady MJ, Lapointe JY. Functional expression of tagged human Na+-glucose cotransporter in Xenopus laevis oocytes. J Physiol. 1999;520:359–371. doi: 10.1111/j.1469-7793.1999.00359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mattia F, Savelkoul PJ, Kamsteeg EJ, Konings IB, Van Der Sluijs P, Mallmann R, Oksche A, Deen PM. Lack of arginine vasopressin-induced phosphorylation of aquaporin-2 mutant AQP2-R254L explains dominant nephrogenic diabetes insipidus. J Am Soc Nephrol. 2005;16:2872–2880. doi: 10.1681/ASN.2005010104. [DOI] [PubMed] [Google Scholar]
- Deen PM, Croes H, van Aubel RA, Ginsel LA, van Os CH. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest. 1995;95:2291–2296. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen PM, van Balkom BW, Kamsteeg EJ. Routing of the aquaporin-2 water channel in health and disease. Eur J Cell Biol. 2000;79:523–530. doi: 10.1078/0171-9335-00075. [DOI] [PubMed] [Google Scholar]
- Duquette PP, Bissonnette P, Lapointe JY. Local osmotic gradients drive the water flux associated with Na+/glucose cotransport. Proc Natl Acad Sci U S A. 2001;98:3796–3801. doi: 10.1073/pnas.071245198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon C, Lussier Y, Bissonnette P, Leduc-Nadeau A, Lonergan M, Arthus MF, Perez RB, Tiulpakov A, Lapointe JY, Bichet DG. Characterization of D150E and G196D aquaporin-2 mutations responsible for nephrogenic diabetes insipidus: importance of a mild phenotype. Am J Physiol Renal Physiol. 2009a doi: 10.1152/ajprenal.90589.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon C, Lussier Y, Bissonnette P, Leduc-Nadeau A, Lonergan M, Arthus MF, Perez RB, Tiulpakov A, Lapointe JY, Bichet DG. Characterization of D150E and G196D aquaporin-2 mutations responsible for nephrogenic diabetes insipidus: importance of a mild phenotype. Am J Physiol Renal Physiol. 2009b;297:F489–F498. doi: 10.1152/ajprenal.90589.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks G, Koudijs M, van Balkom BW, Oorschot V, Klumperman J, Deen PM, Van Der Sluijs P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J Biol Chem. 2004;279:2975–2983. doi: 10.1074/jbc.M310767200. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Bichet DG, Konings IB, Nivet H, Lonergan M, Arthus MF, van Os CH, Deen PM. Reversed polarized delivery of an aquaporin-2 mutant causes dominant nephrogenic diabetes insipidus. J Cell Biol. 2003;163:1099–1109. doi: 10.1083/jcb.200309017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsteeg EJ, Deen PM. Importance of aquaporin-2 expression levels in genotype-phenotype studies in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2000;279:F778–F784. doi: 10.1152/ajprenal.2000.279.4.F778. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Deen PM. Detection of aquaporin-2 in the plasma membranes of oocytes: a novel isolation method with improved yield and purity. Biochem Biophys Res Commun. 2001;282:683–690. doi: 10.1006/bbrc.2001.4629. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Wormhoudt TA, Rijss JP, van Os CH, Deen PM. An impaired routing of wild-type aquaporin-2 after tetramerization with an aquaporin-2 mutant explains dominant nephrogenic diabetes insipidus. EMBO J. 1999;18:2394–2400. doi: 10.1093/emboj/18.9.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara M, Iwai K, Ooeda T, Igarashi T, Ogawa E, Katsushima Y, Shinbo I, Uchida S, Terada Y, Arthus MF, Lonergan M, Fujiwara TM, Bichet DG, Marumo F, Sasaki S. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am J Hum Genet. 2001;69:738–748. doi: 10.1086/323643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc-Nadeau A, Lahjouji K, Bissonnette P, Lapointe JY, Bichet DG. Elaboration of a novel technique for the purification of plasma membranes from Xenopus laevis oocytes. Am J Physiol Cell Physiol. 2006;292:C1132–C1136. doi: 10.1152/ajpcell.00136.2006. [DOI] [PubMed] [Google Scholar]
- Levin MH, Haggie PM, Vetrivel L, Verkman AS. Diffusion in the endoplasmic reticulum of an aquaporin-2 mutant causing human nephrogenic diabetes insipidus. J Biol Chem. 2001;276:21331–21336. doi: 10.1074/jbc.M101901200. [DOI] [PubMed] [Google Scholar]
- Lin SH, Bichet DG, Sasaki S, Kuwahara M, Arthus MF, Lonergan M, Lin YF. Two novel aquaporin-2 mutations responsible for congenital nephrogenic diabetes insipidus in Chinese families. J Clin Endocrinol Metab. 2002;87:2694–2700. doi: 10.1210/jcem.87.6.8617. [DOI] [PubMed] [Google Scholar]
- Marr N, Bichet DG, Hoefs S, Savelkoul PJ, Konings IB, De Mattia F, Graat MP, Arthus MF, Lonergan M, Fujiwara TM, Knoers NV, Landau D, Balfe WJ, Oksche A, Rosenthal W, Muller D, van Os CH, Deen PM. Cell-biologic and functional analyses of five new aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J Am Soc Nephrol. 2002a;13:2267–2277. doi: 10.1097/01.asn.0000027355.41663.14. [DOI] [PubMed] [Google Scholar]
- Marr N, Bichet DG, Lonergan M, Arthus MF, Jeck N, Seyberth HW, Rosenthal W, van Os CH, Oksche A, Deen PM. Heteroligomerization of an aquaporin-2 mutant with wild-type aquaporin-2 and their misrouting to late endosomes/lysosomes explains dominant nephrogenic diabetes insipidus. Hum Mol Genet. 2002b;11:779–789. doi: 10.1093/hmg/11.7.779. [DOI] [PubMed] [Google Scholar]
- Marr N, Kamsteeg EJ, van Raak M, van Os CH, Deen PM. Functionality of aquaporin-2 missense mutants in recessive nephrogenic diabetes insipidus. Pflugers Arch. 2001;442:73–77. doi: 10.1007/s004240000498. [DOI] [PubMed] [Google Scholar]
- Mulders SM, Bichet DG, Rijss JP, Kamsteeg EJ, Arthus MF, Lonergan M, Fujiwara M, Morgan K, Leijendekker R, Van Der Sluijs P, van Os CH, Deen PM. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest. 1998;102:57–66. doi: 10.1172/JCI2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–F270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- Shinbo I, Fushimi K, Kasahara M, Yamauchi K, Sasaki S, Marumo F. Functional analysis of aquaporin-2 mutants associated with nephrogenic diabetes insipidus by yeast expression. Am J Physiol Renal Physiol. 1999;277:F734–F741. doi: 10.1152/ajprenal.1999.277.5.F734. [DOI] [PubMed] [Google Scholar]
- Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–2267. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umenishi F, Narikiyo T, Schrier RW. Effect on stability, degradation, expression, and targeting of aquaporin-2 water channel by hyperosmolality in renal epithelial cells. Biochem Biophys Res Commun. 2005;338:1593–1599. doi: 10.1016/j.bbrc.2005.10.127. [DOI] [PubMed] [Google Scholar]
- Wu B, Steinbronn C, Alsterfjord M, Zeuthen T, Beitz E. Concerted action of two cation filters in the aquaporin water channel. EMBO J. 2009;28:2188–2194. doi: 10.1038/emboj.2009.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K, Fushimi K, Yamashita Y, Shinbo I, Sasaki S, Marumo F. Effects of missense mutations on rat aquaporin-2 in LLC-PK1 porcine kidney cells. Kidney Int. 1999;56:164–171. doi: 10.1046/j.1523-1755.1999.00523.x. [DOI] [PubMed] [Google Scholar]