

Abstract
Despite the prevalence of anemia in cancer, recombinant erythropoietin (Epo) has declined in use because of recent Phase III trials showing more rapid cancer progression and reduced survival in subjects randomized to Epo. Since Epo receptor (EpoR), Jak2, and Hsp70 are well-characterized mediators of Epo signaling in erythroid cells, we hypothesized that Epo might be especially harmful in patients whose tumors express high levels of these effectors. Because of the insensitivity of immunohistochemistry for detecting low level EpoR protein, we developed assays to measure levels of EpoR, Jak2 and Hsp70 mRNA in formalin-fixed paraffin-embedded (FFPE) tumors. We tested 23 archival breast tumors as well as 136 archival head and neck cancers from ENHANCE, a Phase III trial of 351 patients randomized to Epo versus placebo concomitant with radio-therapy following complete resection, partial resection, or no resection of tumor. EpoR, Jak2, and Hsp70 mRNA levels varied >30-fold, >12-fold, and >13-fold across the breast cancers, and >30-fold, >40-fold, and >30-fold across the head and neck cancers, respectively. Locoregional progression-free survival (LPFS) did not differ among patients whose head and neck cancers expressed above- versus below-median levels of EpoR, Jak2 or Hsp70, except in the subgroup of patients with unresected tumors (n = 28), where above-median EpoR, above-median Jak2, and below-median Hsp70 mRNA levels were all associated with significantly poorer LPFS. Our results provide a framework for exploring the relationship between Epo, cancer progression, and survival using archival tumors from other Phase III clinical trials.
Keywords: Erythropoietin, Erythropoietin receptor, Erythropoiesis stimulating agents, Growth factors, Tumor progression
Introduction
Anemia is common in cancer patients and likely represents an independent poor prognostic factor for survival [1]. Safety concerns associated with transfused blood elevated erythropoietin (Epo) to a mainstay treatment in oncology. However, recent Phase III clinical trials testing new uses for Epo, including targeting higher hemoglobin levels and treating anemia not caused by chemotherapy, showed that Epo reduced cancer survival times. Venous thromboembolism is a well documented risk of Epo [2], however the adverse outcomes in these trials were attributed mainly to accelerated tumor progression [3–7].
Whether Epo can indeed stimulate cancer progression is the subject of an intense controversy [8], and preclinical models have generated conflicting results (reviewed by Arcasoy [9]). Central to the controversy is whether tumor progression reflects an “off-target” interaction between Epo and Epo-responsive tumor cells and/or tumor blood vessels. At issue is whether tumors (or tumor blood vessels) can expropriate signaling pathways known to confer Epo responsiveness in erythroid cells. Epo receptor (EpoR) mRNA and protein are detectable in tumor cells, albeit at levels much lower than in erythroid cells [10, 11]. Notably, a recent study showed that a neuroblastoma cell line expressing fewer than 50 Epo binding sites per cell can still be protected from apoptosis in response to Epo [12]. Thus, the pertinent unanswered question is whether even low-level expression of EpoR or other effectors of Epo-signaling can promote cancer progression in patients treated with Epo.
A direct approach to examining this issue would be to characterize archival tumor specimens from patients who had enrolled in Phase III clinical trials of Epo versus placebo, testing whether randomization to Epo was especially harmful in those patients whose tumors expressed higher levels of EpoR and/or downstream effectors of Epo signaling. A previous study employing this approach characterized 154 archival tumors from ENHANCE, a Phase III trial of 351 patients randomized to Epo versus placebo concomitant with radiotherapy following complete resection, partial resection, or no resection of head and neck cancer [13]. Tumors were evaluated using a commercially available polyclonal antibody raised against a human EpoR peptide (C20), that also cross-reacts with non-EpoR proteins, including heat shock protein 70 (Hsp70) family members [14]. A significant association between Epo assignment and reduced LPFS was observed among patients with C20-positive tumors (p = .003, n = 104) that was not observed in patients with C20-negative tumors. However, the aforementioned cross-reactivity between C20 and non-EpoR proteins obscured the interpretation of this finding.
Because of the inadequacy of reagents for detecting low-level EpoR protein in archival tumors, we measured mRNA. Most clinical tumor specimens are formalin-fixed and paraffin-embedded (FFPE), causing RNA degradation. We therefore developed methods to measure mRNA levels of EpoR and 16 other genes from FFPE tumors. To test whether the adverse effects of Epo might be mediated by increased expression of other genes implicated in Epo-responsiveness, we included Csf2rb, Jak2, and Hsp70. Csf2rb encodes the common beta receptor (βcR), a shared signaling subunit for several cytokine receptors, that has been suggested to enhance Epo signaling in nonerythroid cells [15]. Jak2 is a tyrosine kinase that is an essential mediator of Epo signaling in erythroid cells [16], facilitates cell surface EpoR expression [17], and is also implicated in Epo-mediated neuroprotection [18]. Hsp70 family members are encoded by eight Hspa genes, perform essential roles in protein folding, transport, and degradation [19], and promote cancer cell survival [20]. Hspa1a and Hspa1b encode proteins with one amino acid difference, collectively referred to as the major stress inducible Hsp70. In differentiating erythroid cells, Hsp70 accumulates in the nucleus in response to Epo, where it shields the transcription factor Gata-1 from caspase-3-mediated degradation [21]. Additional markers were included to test whether the adverse effects of Epo might depend on vascular endothelial cell representation (Cdh5, Pecam1, Vegfa), tumor squamous epithelial cell representation (Krt5) [22], or cancer stem cells (Cd44) [23], since a recent study suggested that Epo may increase the self-renewal capacity of CD44+ breast cancer-initiating cells [24]. We also measured transcripts for Epo itself, and seven control genes for normalization (see below). Our results provide a framework for investigating Epo-induced tumor progression.
METHODS
Cell Lines
All cancer cell lines have been previously described. To prepare Ba/F3-hEpoR cells, Ba/F3 cells [25] were electroporated with pcDNA3.1-hEpoR encoding a human EpoR cDNA (a gift from Joseph Prchal, University of Utah), selected in 1 mg/ml Geneticin (Invitrogen, Carlsbad, CA, http://www.invitrogen.com), and maintained in 1U/ml epoetin alfa (Procrit, Ortho Biotech, Bridgewater, NJ, http://www.orthobiotech.com). COS-hEpoR cells were prepared by transfecting COS cells with pcDNA3.1-hEpoR using Lipofectamine 2000 (Invitrogen) and were collected 48 hours after transfection. AT-2 cells were provided by Janet Rowley (University of Chicago) and ASE2 cells were provided by Chugai Pharmaceuticals (Japan).
Immunohistochemistry
FFPE cell pellets were sectioned (6 micron) and slides were deparaffinized and rehydrated through a graded ethanol series. Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide for 8 minutes, and endogenous biotin sites were blocked using the Avidin/Biotin Blocking Kit (Dako, Glostrup, Denmark, http://www.dako.com). Sections were then incubated with a polyclonal goat anti-EpoR antibody (ab10653, Abcam, Cambridge, MA, http://www.abcam.com) for 60 minutes. Primary antibodies were detected using a biotinylated antigoat secondary antibody (Jackson ImmunoResearch, West Grove, PA, http://www.jacksonimmuno.com) for 30 minutes followed by visualization using the Vector Elite ABC system (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com). Staining was visualized with 3,3′-diamino-benzidine for 7 minutes, and the sections were counterstained with hematoxylin for 2 minutes. Concentration matched isotype controls (Jackson ImmunoResearch) were run for each cell sample.
Flow Cytometric Detection of Cell Surface EpoR
Adherent cell lines were lifted for 15 minutes using 0.02% ethyl-enediaminetetraacetic acid in phosphate buffered saline (PBS), washed with PBS, and filtered through a 70 μm strainer. Cells were blocked for 15 minutes at room temperature in fluorescence-activated cell sorting (FACS) buffer (PBS, 0.1% bovine serum albumin (BSA), 0.02% sodium azide) containing 250 μg/ ml human immunoglobulin G (IgG) (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com). A murine monoclonal antihuman EpoR-phycoerythrin (PE) antibody (FAB307P, R&D Systems, Minneapolis, MN, http://www.rndsystems.com) was then added to 5 μg/ml and cells were incubated for 30 minutes on ice. Cells were also stained with two different murine IgG2b-PE iso-type control antibodies (IC0041P, R&D Systems or 555058, BD Biosciences, Franklin Lakes, NJ, http://www.bdbiosciences.com). After staining, cells were washed, resuspended in FACS buffer, and analyzed by flow cytometry (FACS-Canto, BD Biosciences). Dead cells were excluded from analyses of adherent cells by inclusion of 3.75 μg/ml 7-aminoactinomycin D. For determination of EpoR staining relative to each isotype control, the mean fluorescence value obtained for three replicate isotype control staining reactions was subtracted from the mean fluorescence value obtained for three replicate anti-EpoR staining reactions.
Signal Transducer and Activator of Transcription 5 Phosphorylation
REH (acute lymphoblastic leukemia) and U266 (myeloma) cells were washed twice, starved for 5 hours in Roswell Park Memorial Institute media (RPMI), 0.5% BSA, and stimulated for 15 minutes with either 10 U/ml epoetin alfa (Procrit, Ortho Biotech), or vehicle (Procrit buffer: 2.5 mg/ml human albumin, 1.3 mg/ml sodium citrate, 8.2 mg/ml sodium chloride, 0.11 mg/ml citric acid, 1% benzyl alcohol). For Epo antagonist control reactions, a 225 amino acid recombinant soluble human EpoR extracellular domain (R&D Systems) was added to 2.25 μg/ml. Cells were then fixed in 2% paraformaldehyde (10 minutes, 37°C), washed, permeabilized with 90% methanol in PBS (30 minutes, 4°C), washed, resuspended in FACS buffer, and stained for 20 minutes at room temperature with an Alexa Fluor 647 anti-phospho- signal transducer and activator of transcription 5 (STAT5) phosphotyrosine 464 (PY464) antibody (1:5 dilution, 612599, BD Biosciences). Cells were then washed, resuspended in FACS buffer, and analyzed by flow cytometry (FACS-Canto, BD Biosciences).
Tumor Samples
Permission was obtained from the University of Washington Institutional Review Board to study primary tumors. Breast tumors were from an established repository (Department of Defense grant DAMD 17-02-1-0691) that stores tissues donated by women undergoing surgery for breast cancer (invasive cancer or in situ disease) as FFPE tissue and as snap-frozen tissue. FFPE head and neck tumors were obtained from the local Pathology Department and from ENHANCE, a previously reported [3] multicenter Phase III trial of epoetin beta in 351 patients receiving radiotherapy for head and neck cancer. All ENHANCE samples were among 154 tumors previously examined using the polyclonal C20 anti-EpoR antibody (Santa Cruz Biotechnology, Santa Cruz, CA, http://www.scbt.com) [13]. One hundred thirty-six of the 154 tumors were sent as four micron FFPE sections on deidentified, coded glass slides to the University of Washington for mRNA analysis by an investigator blinded to clinical outcomes.
ENHANCE Trial Design
Patient selection, treatment, follow-up, evaluation, and baseline characteristics were described previously [3, 13]. Briefly, the main inclusion criteria were squamous cell carcinomas of the head and neck, scheduled definitive or postoperative radiotherapy, and a decreased blood hemoglobin (<13g/dl, men; <12g/dl, women) at randomization. Patients were randomly assigned to 300 IU/kg epoetin beta or placebo three times per week starting 10 to 14 days before radiotherapy, continuing throughout. Prior to randomization, patients were stratified by resection status: 1) complete resection; 2) incomplete resection; or 3) unresected disease. Iron (III) saccharate (200 mg) was administered intravenously once weekly to patients with <25% transferrin saturation. Epoetin beta was stopped if hemoglobin increased more than 2 g/ dl within 1 week or when targets were reached (≥15 g/dl, men; ≥14 g/dl, women) and resumed when hemoglobin fell below target. Locoregional cancer control and survival was assessed at 3-month intervals by an independent oncologist blinded to treatment assignment. The primary endpoint was locoregional progression-free survival (LPFS). Locoregional progression was noted if the tumor recurred or increased by 25%. Baseline serum Epo levels were determined prior to treatment.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
RNA was extracted from cancer cell lines using the RNeasy Mini Kit (Qiagen, Valencia, CA, http://www.qiagen.com) and from FFPE tumor sections using the Absolutely RNA FFPE kit (Strata-gene, La Jolla, CA, http://www.stratagene.com). On-column Dna-seI digestion was performed to remove genomic DNA. First strand cDNA was synthesized with random hexamer primers and Superscript III reverse transcriptase (RT) (Invitrogen), the latter omitted for no-RT control reactions. Next, cDNA targets were amplified using Taqman probes and a 7900HT thermal cycler (Applied Biosystems, ABI, Foster City, CA, http://www.applied-biosystems.com). With the exception of certain intronless members of the Hsp70 family and the candidate reference gene 18s, all probes recognized exon junctions to prevent genomic DNA amplification (supporting information Table 1). Cycle threshold (Ct) values were determined with the Sequence Detection Software (ABI). A coefficient of variance <4% for triplicate Ct determinations was considered acceptable. Where indicated in the text, preamplification of cDNA was performed with the Taqman pre-amplification multiplex system (ABI). Preamplification uniformity (lack of bias) for each Taqman probe was tested by calculating ΔCt values for data obtained with both unamplified and preamplified cDNA, where ΔCt = mean Ct for target gene – mean Ct for reference gene. This was performed using several ENHANCE samples which contained sufficient RNA, erythroid ASE2 cells [26], and a universal human total RNA standard (Stratagene). Uniformity comparisons (ΔCt preamplified –ΔCt unamplified) were considered acceptable to a tolerance of variation of 1.5 cycles per manufacturer’s instructions (ABI). Relative quantification was determined using the comparative Ct method, 2−ΔCT where ΔCt = mean Ct for target gene – mean Ct for reference gene. Reference gene stability was evaluated using the Genorm algorithm [27].
Statistical Analysis
The number of patients included in the analysis of LPFS for each marker depended upon the number of samples yielding sufficient RNA for quantitative reverse transcriptase polymerase chain reaction (RT-PCR). This varied for each marker as a function of the expression level of that gene. Our analyses included all available data for each marker. For LPFS analyses, patients were stratified into above-median or below/equal-median mRNA expression levels. This stratification was done separately for the total population and within each resection stratum for every gene. LPFS was evaluated with the Kaplan-Meier survival estimation. The log-rank test was implemented to test the null hypothesis that the distribution of survival times between patients treated with Epo versus placebo was equal. The STATA statistical software package was used for all analyses (version 10.0, Stata Corporation, College Station, TX, http://www.stata.com). Statistical tests were two-sided and considered significant at p ≤ 0.05. For patients in the placebo group stratified by endogenous serum Epo, ≤11 U/l was defined as low, whereas >11 U/l was defined as high, based on a previous study [28]. Spearman’s correlation coefficients were calculated for C20 staining status (measured as a dichotomous variable) versus EpoR or Hsp70 mRNA levels (measured as a continuous variable). In cell line studies, Spearman’s correlation coefficients were calculated for EpoR mRNA versus surface protein levels.
Results
EpoR mRNA and Surface Protein Levels in Cancer Cell Lines
To characterize the relation between EpoR mRNA and cell surface protein, we tested 32 human cell lines including three high EpoR-expressing positive control cell lines: UT7EPO, ASE2, and OCIM1. For normalization of mRNA levels, we used the three most stable reference genes (Hmbs, Hprt1, Rplp0) among a panel of seven candidates evaluated across all cell lines as determined by the Genorm algorithm (supporting information Table 2). EpoR mRNA levels among the non-control lines ranged from 0.5 to 7.5% (mean 2.0%) of the level in UT7EPO cells (Fig. 1A). For flow cytometry, we compared the fluorescent intensity of cells stained with a monoclonal antibody directed against EpoR with the same cells stained using two different isotype control antibodies. Surface EpoR levels among the noncontrol lines ranged from 1.2 to 25.2% (mean 8.3%) of the level in UT7EPO cells (Fig. 1B). Among all cell lines tested there was a significant correlation between mRNA and surface protein (r = .33, p = .03, n = 32). When positive-control UT7EPO, ASE2, and OCIM1 cells were excluded, the significance of this correlation was maintained among nonadherent cells (r = .58, p = .03, n = 11, Fig. 1C) but no correlation was observed when our analysis was restricted to the adherent cell lines (r = −.19, p = .23, n = 18, Fig. 1D). Of note, our analysis of adherent cell lines (n = 18) required additional processing steps to generate single cell suspensions (see Methods) that were associated with significant cell death and debris (not shown). Moreover, 16 of 18 adherent cell lines produced discordant staining patterns for the two different isotype control antibodies (that is, average deviation in fluorescence values obtained for anti-EpoR antibody staining relative to fluorescence values obtained for two isotype control antibodies exceeded 5% of the mean), whereas only 5 of 11 nonadherent cell lines showed this discrepancy (p < .01) (Fig. 1B). The apparent lack of correlation between EpoR mRNA and surface protein among the adherent cell lines likely results from these technical limitations that preclude accurately estimating levels of EpoR on the cell surface, but may also be influenced by post-transcriptional regulation of EpoR in these lines. Analysis of two of the EpoR-expressing nonerythroid lines (U266 and REH) demonstrated Epo-dependent STAT5 phosphorylation (supporting information Fig. 1), consistent with previous
Figure 1.

EpoR mRNA and surface protein levels in cancer cell lines. (A): EpoR mRNA levels were determined by quantitative reverse transcriptase polymerase chain reaction. The mRNA level of each cancer cell line is plotted relative to the level in control UT7EPO cells. Error bars represent the standard deviation of results obtained upon normalization to Hmbs, Rplp0, and Hprt1. (B): Differences between the mean fluorescent intensities of cells stained with a phycoerythrin (PE)-conjugated monoclonal anti-EpoR antibody versus each of two different PE-conjugated murine IgG2b-PE isotype controls are depicted. Results are plotted relative to UT7EPO cells. Error bars depict standard deviations of the differences. (C): The rank order of mRNA and protein expression is plotted for all nonadherent cell lines, excluding the positive control lines UT7EPO, ASE2, and OCIM1. (D): The rank order of mRNA and protein expression is plotted for all adherent cell lines. Spearman’s rank order correlation coefficients are indexed above the graphs in (C) and (D).
Development of a Quantitative RT-PCR Assay for EpoR mRNA in Archival Tumor Samples
Most tumors from clinical trials are preserved as FFPE tissue. We found that immunohistochemistry with a specific antibody was not sufficiently sensitive to detect low-level EpoR protein in FFPE tumor cell lines (supporting information Fig. 2). We therefore tested whether EpoR mRNA could be accurately measured in FFPE tumors despite the RNA degradation that accompanies FFPE-processing [30]. Three independent assessments of EpoR mRNA levels from serial sections of 11 FFPE breast tumors demonstrated that our measurements were highly reproducible, and that EpoR mRNA levels varied as much as 34-fold (supporting information Fig. 3). To assess the validity of mRNA measurements from FFPE primary tumors, we compared expression levels for EpoR using 23 breast tumors which were divided and processed both as FFPE and snap-frozen tissue. Because the FFPE and snap-frozen samples represent different pieces of the same tumor, and snap-freezing preserves a higher degree of RNA integrity, this comparison allowed us to simultaneously assess whether RNA degradation influences the accuracy of our measurements as well as the uniformity with which EpoR is expressed across tumors. We also measured mRNA levels of Jak2 and Hsp70, which participate in Epo signaling in erythroid cells [16, 21], Csf2rb, which has been suggested to enhance Epo signaling in nonerythroid cells [15], endothelial-associated genes (Cdh5, Pecam1, Vegfa), the squamous epithelial marker Krt5 (especially relevant for head and neck cancer) [22], the putative cancer stem cell marker Cd44 [23], and Epo itself. Significant correlations between FFPE and snap-frozen mRNA measurements were observed for EpoR, Csf2rb, Jak2, Hsp70, Cd44, Krt5 and Esr1 (estrogen receptor-1, used as a positive control) (Fig. 2). These findings suggest that single tumor sections can be used to gauge the overall expression levels of these markers. Expression levels varied over a wide range for each these genes (Fig. 3A). In contrast, Vegfa, Cdh5, and Pecam1 were not significantly correlated, consistent with regional heterogeneity in tumor vascularity [31] whereas Epo was detected in too few FFPE tumors to permit calculation of a correlation coefficient.
Figure 2.

Analysis of concordance in mRNA measurements between snap frozen and formalin-fixed paraffin-embedded (FFPE) breast tumors. Twenty-three breast tumors are ranked for their level of mRNA expression of the indicated genes (normalized to Hmbs expression). Results using RNA extracted from snap frozen (y-axis) versus FFPE (x-axis) pieces of the same breast tumor are shown. Spearman’s rank order correlation coefficients are indexed above each graph.
Figure 3.

Range of mRNA levels in primary tumors. Relative quantification values are shown as fold differences, with the value from the lowest-expressing tumor assigned a value of 1. Numbers at the bottom of each graph indicate the number of tumors for which data were obtained. (A): Results for breast tumors. (B): Results for head and neck tumors from ENHANCE.
Assessing EpoR mRNA Levels in Head and Neck Cancers from ENHANCE
We assayed EpoR mRNA levels in 136 archival FFPE head and neck tumors from ENHANCE, a subset of the 154 evaluated previously by immunohistochemistry using the C20 antibody [13]. Since most samples consisted of only a single microscope slide with minimal tissue, we employed a non-biased target-specific cDNA preamplification method for all genes (supporting information Table 3). We included 7 candidate reference genes for normalization (Hprt1, Ppia, Ipo8, Hmbs, Gapdh, Tfrc, and Rplp0). These reference genes were included based on their stability among 16 candidates tested by the Genorm algorithm [27] in a panel of 8 breast cancers and 8 head and neck cancers (supporting information Table 2). Results for Hprt1 were excluded because of high or no Ct values in many samples. For tumor samples with sufficient RNA (representing 123 different tumors), there were strong positive correlations in Ct values among all reference genes (r ≥ 0.88 for all pairwise comparisons, p < .001).
We tested normalization of EpoR values with each of the reference genes and assessed the extent to which relative quantification values might be influenced by RNA abundance/integrity. Specifically, a phenomenon was reported by Cronin et al. in which greater age of FFPE blocks (and the lower RNA abundance/integrity) was associated with higher relative quantification values even after normalization [32]. This effect was attributed to differential degradation of target versus endogenous control gene transcripts and was reduced by minimizing the size and range of target and control gene assay amplicon sizes. In our data set, higher reference gene Ct values (less RNA abundance/integrity) were indeed associated with higher relative EpoR quantification upon normalization to Ppia levels as evidenced by the strong positive correlation between Ppia Ct values and normalized EpoR relative quantification values (supporting information Fig. 4A). Similar results were obtained for normalization with Gapdh, Rplp0, Ipo8 or Tfrc. Whereas a similar pattern of normalized EpoR expression within subgroups of tumor samples with similar amounts of RNA abundance/integrity was observed, the overall systematic effect of RNA abundance/integrity on normalization would have precluded comparisons among all patients. Consistent with Cronin et al., this systematic effect was alleviated upon normalization to Hmbs, which had the shortest amplicon size among all reference gene assays tested (64 bp) (supporting information Fig. 4B). Five samples with low RNA abundance/integrity produced relative quantification values greater than mean +1 standard deviation and were omitted. For 30 tumors, EpoR mRNA levels could not be determined relative to other samples because of undetermined endogenous control gene (Hmbs) and/or EpoR Ct values. Among the remaining 101 tumors, we observed a >30-fold range of EpoR mRNA, and expression levels also varied widely for the other genes examined (Fig. 3B).
mRNA Levels and Locoregional Progression-Free Survival
We evaluated LPFS within each resection stratum for patients with tumors expressing above- versus below-median levels of each marker (determined separately for each stratum). Significant associations between transcript level, Epo treatment and adverse outcome were observed only in the no resection stratum (n = 28) (Table 1). Significantly poorer LPFS was observed for Epo-treated subjects with above-median but not below-median levels of EpoR (Fig. 4A) or Jak2 (Fig. 4B) (EpoR: above-median p = .02, n = 14, below-median p = .8, n = 14; Jak2: above-median p = .04, n = 17, below-median p = .34, n = 18). In addition, we found a significant association between Epo treatment, poor outcome, and below-median but not above-median levels of Hsp70 family members in aggregate (Fig. 4C) (Hsp70 below-median p = .01, n = 20, above-median p = .38, n = 19) and individually (supporting information Table 4). The significance of these associations was not further increased by dichotomizing mRNA at higher thresholds (that is, highest 10% vs. the rest) (not shown). Combinations of above-median EpoR, above-median Jak2, and below-median Hsp70 did not increase the significance of the association between treatment assignment (Epo versus placebo) and LPFS compared to each marker individually (not shown).
Table 1.
Analysis of exogenous erythropoietin administration and locoregional progression-free survival by mRNA marker statusa
| mRNA Marker/Resection Stratum | Below Medianb Marker Value | Above Medianb Marker Value | ||||
|---|---|---|---|---|---|---|
| Number of Patients | Log Rank p Valuec | Number of Patients | Log Rank p Valuec | |||
| Epo | Placebo | Epo | Placebo | |||
| EpoR | ||||||
| All patients | 24 | 27 | 0.22 | 23 | 27 | 0.31 |
| Complete | 11 | 13 | 0.90 | 14 | 10 | 0.47 |
| Incomplete | 5 | 8 | 0.17 | 6 | 6 | 0.82 |
| No resection | 7 | 7 | 0.80 | 4d | 10d | 0.02d |
| Jak2 | ||||||
| All patients | 29 | 29 | 0.65 | 26 | 31 | 0.15 |
| Complete | 14 | 14 | 0.12 | 14 | 13 | 0.15 |
| Incomplete | 7 | 6 | 0.33 | 4 | 8 | 0.71 |
| No resection | 8 | 10 | 0.34 | 8d | 9d | 0.04d |
| Hsp70e | ||||||
| All Patients | 31 | 31 | 0.98 | 29 | 32 | 0.40 |
| Complete | 15 | 15 | 0.26 | 15 | 14 | 0.74 |
| Incomplete | 6 | 7 | 0.79 | 5 | 7 | 0.16 |
| No resection | 9d | 11d | 0.01d | 10 | 9 | 0.38 |
| Csf2rb | ||||||
| All patients | 24 | 29 | 0.55 | 26 | 27 | 0.83 |
| Complete | 9 | 17 | 0.57 | 17 | 8 | 0.34 |
| Incomplete | 5 | 8 | 0.78 | 6 | 6 | 0.18 |
| No resection | 9 | 6 | 0.35 | 4 | 11 | 0.26 |
| Cd44 | ||||||
| All Patients | 30 | 32 | 0.73 | 30 | 31 | 0.50 |
| Complete | 16 | 14 | 0.70 | 14 | 15 | 0.96 |
| Incomplete | 5 | 8 | 0.52 | 6 | 6 | 0.89 |
| No resection | 9 | 11 | 0.12 | 10 | 9 | 0.10 |
| Krt5 | ||||||
| All Patients | 30 | 27 | 0.84 | 27 | 30 | 0.28 |
| Complete | 17 | 10 | 0.86 | 10 | 16 | 0.61 |
| Incomplete | 4 | 9 | 0.89 | 7 | 5 | 0.65 |
| No resection | 10 | 8 | 0.26 | 9 | 9 | 0.08 |
Stratification was above versus below/equal to the median.
The median was calculated separately for all patients and within each resection stratum.
The p value is two sided and is based on the log rank test to compare differences in Kaplan Meier distributions in response to Epo versus placebo.
Groups with significant adverse effects of Epo.
Hsp70 mRNA represents the cumulative expression of all 8 family members. Results for individual family members are presented in Supporting information Table 4.
Figure 4.
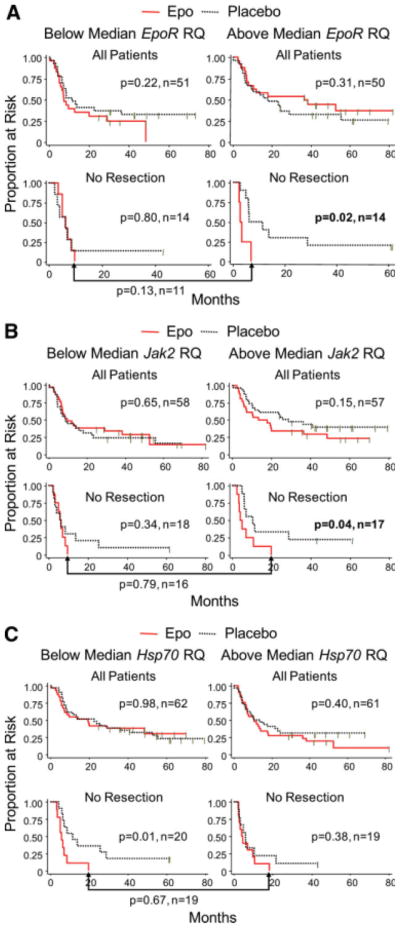
Effects of exogenous Epo on LPFS with stratification by mRNA status. Outcomes in response to Epo versus placebo are shown in Kaplan-Meier plots. The log-rank p value is two sided. Comparisons of outcomes of patients randomized to Epo are indexed below the brackets. (A): Results for EpoR. (B): Results for Jak2. (C): Results for Hsp70. Hsp70 mRNA measurements reflect the cumulative expression of all eight family members.
We also compared Epo-treated patients with above-median EpoR expression to Epo-treated patients with below-median expression in the no resection stratum (see bracket below the graphs in Fig. 4A). A trend toward worse LPFS in Epo-treated patients with above-median levels of tumor EpoR mRNA was not significantly different from patients whose tumors expressed below-median EpoR mRNA (p = .13, n = 11). Analogous comparisons for Jak2 and Hsp70 mRNA levels also showed no significant differences among Epo-treated patients.
Relationship to Endogenous Erythropoietin Levels
If exogenous Epo can stimulate tumor progression, endogenous Epo might also stimulate tumor progression. The ENHANCE study documented single-time-point pretreatment serum Epo levels [3], and we obtained these results for 147 of the 154 patients reported previously [13] from the trial sponsor. Confining analyses to subjects enrolled in the placebo group, we did not find an association between LPFS and baseline hemoglobin level or LPFS and baseline serum Epo level (not shown). Additionally, baseline hemoglobin levels and serum Epo levels did not correlate (not shown). Finally, we tested whether elevated endogenous Epo levels were associated with LPFS in subjects with above-median versus below-median levels of EpoR, Jak2 or Hsp70 mRNA. Because of the small number of patients available, we combined the incomplete and no resection strata into a new category called “residual tumor.” However, there was no association between endogenous Epo level and LPFS based on tumor EpoR, Jak2 or Hsp70 mRNA levels (supporting information Table 5).
Correlations with C20 Staining
The tumors we evaluated were among the 154 previously characterized using the C20 antibody [13], which was raised against a human EpoR sequence but cross-reacts with other proteins, including Hsp70 family members [14]. Notably, we found no significant correlation between C20 status and EpoR mRNA (r = −0.11, p = .26, n = 100), or Hsp70 family member mRNA in aggregate (r = 0.06, p = .54, n = 122) or individually (supporting information Fig. 5).
Discussion
As one of the most prominent drugs in oncology, the unexpected association between Epo and increased cancer death rates has created concern and uncertainty. One of the central questions is whether Epo induced “off-target” signaling in tumors or tumor blood vessels can hasten cancer progression. Preclinical models may bring insight to this issue, however definitive answers can only come from studies in humans. The substantial challenge presented by the very low level of EpoR present in nonerythroid cells is counter-balanced in part by an extensive body of literature surrounding Epo signaling in erythroid cells. Using this knowledge, we developed methods to characterize human tumors for their potential competency to respond to Epo. Since existing reagents for detecting EpoR protein in tumor sections are insufficiently sensitive and specific [14], we measured mRNA. Laying the foundation for this effort, we show that EpoR mRNA levels can be estimated despite the extensive RNA degradation that accompanies FFPE processing and, barring results of adherent cell lines that are difficult to accurately assess by flow cytometry, we show that EpoR mRNA levels appear to reasonably estimate levels of EpoR cell surface protein.
We found a >30-fold range of EpoR mRNA across a series of breast cancers and head and neck cancers. This finding does not necessarily contradict a previous study documenting the lack of significant differences in EpoR mRNA levels between tumors and normal tissues [33]. A preferential susceptibility to Epo-induced signaling in malignant versus normal tissue is not a prerequisite for a direct effect of Epo on tumors, for example, a heritable basis for variation in EpoR expression levels has been proposed in swine [34]. Measurements of total tumor EpoR mRNA levels cannot distinguish the cellular origin of the EpoR transcript, and the extent to which the various cell types within tumors might respond to Epo and contribute to tumor progression remains undetermined. Future efforts directed at laser capture microdissection of various cell types from tumor samples will help resolve this issue. Wide variations in Jak2, Hsp70, and Csf2rb mRNA levels were also found across our series of breast cancers and head and neck cancers.
We used these tools to hunt for an association between Epo exposure, a tumor’s inferred competency to respond to Epo based on mRNA levels of Epo-associated signaling molecules and patient outcomes. The ideal testing grounds for this effort are the archival tumors of patients who were randomized in clinical trials of Epo versus placebo and whose outcomes are known. For this first study, we examined available tumors from ENHANCE [3]. Above-median levels of mRNA for EpoR, and its tethered signaling intermediate Jak2, emerged as candidate predictors of reduced LPFS in unresected patients treated with Epo compared to placebo. In contrast, we found no significant association between Csf2rb, Krt5, or Cd44 mRNA levels and outcome. Since Hsp70 mediates Epo signaling in erythroid cells [21] and is detected by the C20 antibody [14], we predicted correlations between Hsp70 mRNA levels, Epo and LPFS analogous to those observed with EpoR and Jak2. To the contrary, we found a strong association between below-median levels of all Hsp70 family members, Epo treatment and poor outcome. Importantly, we did not find a correlation between EpoR mRNA levels and prior staining of the same tumors using the C20 antibody [13]. These findings are consistent with the interpretation that the C20 staining does not correlate with EpoR expression [14]. Confirmation of C20 staining as a predictor of tumor susceptibility to Epo may lead to the identification of other proteins involved in Epo responsiveness.
Whether these tentative associations are reflective of underlying tumor biology is unknown, and the interpretation of our findings must be tempered by several limitations. First, contrasting the disparate outcomes of subjects randomized to Epo versus placebo, mRNA levels were not associated with significant differences in LPFS when restricting our analysis to patients in the Epo-treated group (see the brackets beneath the Kaplan Meier plots in Fig. 4). Thus, differences in LPFS associated with above- versus below-median mRNA levels cannot be accounted for entirely by differences in outcomes in response to Epo. Similarly, we found no association between these markers and adverse outcome in the presence of elevated levels of endogenous Epo in patients enrolled in the placebo arm of ENHANCE. Second, the statistically significant associations that we observed were confined to patients with unresected tumors, and did not extend to patients with incomplete or complete resection of their tumors. This discrepancy might be explained by the prediction that, absent resection, a larger amount of tumor would be available for Epo stimulation. In support of this interpretation, the original ENHANCE trial also did not find an association between Epo treatment and worse outcomes in patients with completely resected tumors [3]. Third, the small number of patients with unresected tumors also precludes further stratification to adjust for potential baseline imbalances or confounding clinical characteristics. Fourth, in view of the exploratory nature of our hypothesis, we did not adjust for multiple comparisons. This increases the likelihood that the observed significant p values represent false positives [35]. Most importantly, our findings are constrained by lack of access to additional tumors from other Phase III clinical trials of Epo. Because these were large multicenter trials lacking centralized tumor repositories, obstacles to obtaining these tumors likely can only be surmounted by the trial sponsors. Tapping this little explored resource using the methods described here may bring new insight to Epo and cancer progression.
Supplementary Material
Acknowledgments
We thank Roche for providing baseline serum Epo levels for patients enrolled in ENHANCE, Janet Rowley for AT-2 cells, and Chugai Pharmaceutical for ASE2 cells. We gratefully acknowledge Erica Jonlin for assistance with human subjects issues, Elizabeth Wayner for antibody development, Julie Randolph Habecker for immunohistochemistry, Margaret Pepe and Ruth Etzioni for statistical advice, Kerstin Edlefsen and Keith Loeb for assistance with pathology specimens, Kathy O’Briant, Leah Sabacan and J. David Beatty for breast cancer samples and helpful discussions, and Whitney Hitchcock, Philamer Calses, and Marianna Rudakova for assistance with flow cytometry analyses. This work was supported by the National Institutes of Health [1R01CA135357 to C.A.B.]; and the Department of Defense [CDMRP, DAMD17-02-0691 to N.U.].
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
C. Anthony Blau owned stock in and served as an officer or member of the board for CellNexus, LLC.
Author contributions: C.P.M.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; K.A.L. and N.U.: data analysis and interpretation, final approval of manuscript; K.V.-S. and J.F.K.: collection and assembly of data; D.M.: provision of study materials; M.H.: conception and design, provision of study materials, data analysis and interpretation, manuscript writing, final approval of manuscript; C.A.B.: conception and design, financial support, assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
References
- 1.Caro JJ, Salas M, Ward A, et al. Anemia as an independent prognostic factor for survival in patients with cancer: A systemic, quantitative review. Cancer. 2001;91:2214–2221. [PubMed] [Google Scholar]
- 2.Bennett CL, Silver SM, Djulbegovic B, et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. JAMA. 2008;299:914–924. doi: 10.1001/jama.299.8.914. [DOI] [PubMed] [Google Scholar]
- 3.Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: Randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255–1260. doi: 10.1016/S0140-6736(03)14567-9. [DOI] [PubMed] [Google Scholar]
- 4.Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol. 2005;23:5960–5972. doi: 10.1200/JCO.2005.06.150. [DOI] [PubMed] [Google Scholar]
- 5.Wright JR, Ung YC, Julian JA, et al. Randomized, double-blind, placebo-controlled trial of erythropoietin in non-small-cell lung cancer with disease-related anemia. J Clin Oncol. 2007;25:1027–1032. doi: 10.1200/JCO.2006.07.1514. [DOI] [PubMed] [Google Scholar]
- 6.Smith RE, Jr, Aapro MS, Ludwig H, et al. Darbepoetin alfa for the treatment of anemia in patients with active cancer not receiving chemotherapy or radiotherapy: Results of a phase III, multicenter, randomized, double-blind, placebo-controlled study. J Clin Oncol. 2008;26:1040–1050. doi: 10.1200/JCO.2007.14.2885. [DOI] [PubMed] [Google Scholar]
- 7. [Accessed March 13, 2008.];Oncologic Drugs Advisory Committee meeting briefing materials. at http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4345b2-01-FDA.pdf.
- 8.Blau CA. Erythropoietin in cancer: Presumption of innocence? Stem Cells. 2007;25:2094–2097. doi: 10.1634/stemcells.2007-0229. [DOI] [PubMed] [Google Scholar]
- 9.Arcasoy MO. Erythropoiesis-stimulating agent use in cancer: Preclini-cal and clinical perspectives. Clin Cancer Res. 2008;14:4685–4690. doi: 10.1158/1078-0432.CCR-08-0264. [DOI] [PubMed] [Google Scholar]
- 10.Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res. 2001;61:3561–3565. [PubMed] [Google Scholar]
- 11.Sinclair AM, Todd MD, Forsythe K, et al. Expression and function of erythropoietin receptors in tumors: Implications for the use of erythropoiesis-stimulating agents in cancer patients. Cancer. 2007;110:477–488. doi: 10.1002/cncr.22832. [DOI] [PubMed] [Google Scholar]
- 12.Um M, Gross AW, Lodish HF. A “classical” homodimeric erythropoie-tin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal. 2007;19:634–645. doi: 10.1016/j.cellsig.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Henke M, Mattern D, Pepe M, et al. Do erythropoietin receptors on cancer cells explain unexpected clinical findings? J Clin Oncol. 2006;24:4708–4713. doi: 10.1200/JCO.2006.06.2737. [DOI] [PubMed] [Google Scholar]
- 14.Elliott S, Busse L, Bass MB, et al. Anti-Epo receptor antibodies do not predict Epo receptor expression. Blood. 2006;107:1892–1895. doi: 10.1182/blood-2005-10-4066. [DOI] [PubMed] [Google Scholar]
- 15.Brines M, Grasso G, Fiordaliso F, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit hetero-receptor. Proc Natl Acad Sci U S A. 2004;101:14907–14912. doi: 10.1073/pnas.0406491101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihle JN, Gilliland DG. Jak2: normal function and role in hematopoietic disorders. Curr Opin Genet Dev. 2007;17:8–14. doi: 10.1016/j.gde.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8:1327–1338. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 18.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 19.Morano KA. New tricks for an old dog: The evolving world of Hsp70. Ann N Y Acad Sci. 2007;1113:1–14. doi: 10.1196/annals.1391.018. [DOI] [PubMed] [Google Scholar]
- 20.Sherman M, Multhoff G. Heat shock proteins in cancer. Ann N Y Acad Sci. 2007;1113:192–201. doi: 10.1196/annals.1391.030. [DOI] [PubMed] [Google Scholar]
- 21.Ribeil JA, Zermati Y, Vandekerckhove J, et al. HSP70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. 2007;445:102–105. doi: 10.1038/nature05378. [DOI] [PubMed] [Google Scholar]
- 22.Becker MT, Shores CG, Yu KK, et al. Molecular assay to detect meta-static head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2004;130:21–27. doi: 10.1001/archotol.130.1.21. [DOI] [PubMed] [Google Scholar]
- 23.Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a sub-population of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips TM, Kim K, Vlashi E, et al. Effects of recombinant erythro-poietin on breast cancer-initiating cells. Neoplasia. 2007;9:1122–1129. doi: 10.1593/neo.07694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palacios R, Steinmetz M. Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 1985;41:727–734. doi: 10.1016/s0092-8674(85)80053-2. [DOI] [PubMed] [Google Scholar]
- 26.Inoue Y, Tsushima H, Ando K, et al. Chemokine expression in human erythroid leukemia cell line AS-E2: Macrophage inflammatory protein-3alpha/CCL20 is induced by inflammatory cytokines. Exp Hema-tol. 2006;34:19–26. doi: 10.1016/j.exphem.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul I, Lappin TR, Maxwell P, et al. Pre-operative plasma erythropoietin concentration and survival following surgery for non-small cell lung cancer. Lung Cancer. 2006;51:329–334. doi: 10.1016/j.lungcan.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 29.Fu P, Jiang X, Arcasoy MO. Constitutively active erythropoietin receptor expression in breast cancer cells promotes cellular proliferation and migration through a MAP-kinase dependent pathway. Biochem Biophys Res Commun. 2009;379:696–701. doi: 10.1016/j.bbrc.2008.12.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonov J, Goldstein DR, Oberli A, et al. Reliable gene expression measurements from degraded RNA by quantitative real-time PCR depend on short amplicons and a proper normalization. Lab Invest. 2005;85:1040–1050. doi: 10.1038/labinvest.3700303. [DOI] [PubMed] [Google Scholar]
- 31.Grizzi F, Colombo P, Taverna G, et al. Geometry of human vascular system: is it an obstacle for quantifying antiangiogenic therapies? Appl Immunohistochem Mol Morphol. 2007;15:134–139. doi: 10.1097/01.pai.0000213105.18569.fa. [DOI] [PubMed] [Google Scholar]
- 32.Cronin M, Pho M, Dutta D, et al. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. Am J Pathol. 2004;164:35–42. doi: 10.1016/S0002-9440(10)63093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinclair AM, Rogers N, Busse L, et al. Erythropoietin receptor transcription is neither elevated nor predictive of surface expression in human tumour cells. Br J Cancer. 2008;98:1059–1067. doi: 10.1038/sj.bjc.6604220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallet JL, Freking BA, Leymaster KA, et al. Allelic variation in the erythropoietin receptor gene is associated with uterine capacity and litter size in swine. Anim Genet. 2005;36:97–103. doi: 10.1111/j.1365-2052.2005.01233.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang R, Lagakos SW, Ware JH, et al. Statistics in medicine–reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357:2189–2194. doi: 10.1056/NEJMsr077003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.