

Abstract
Smith-Lemli-Opitz syndrome (SLOS) is caused by deficiency in the terminal step of cholesterol biosynthesis, which is catalyzed by 7-dehydrocholesterol reductase (DHCR7). The disorder exhibits several phenotypic traits including dysmorphia and mental retardation with a broad range of severity. Pathogenesis of SLOS is complex due to multiple roles of cholesterol and may be further complicated by unknown effects of aberrant metabolites that arise when 7-dehydrocholesterol (7-DHC), the substrate for DHCR7, accumulates. A viable mouse model for SLOS has recently been developed, and here we characterize cholesterol metabolism in this model with emphasis on changes during the first few weeks of postnatal development. Cholesterol and 7-DHC were measured in “SLOS” mice and compared with measurements in normal mice. SLOS mice had measurable levels of 7-DHC at all ages tested (up to one year), while 7-DHC was below the threshold for detection in normal mice. In perinatal to weaning age SLOS mice, cholesterol and 7-DHC levels changed dramatically. Changes in brain and liver were independent; in brain cholesterol increased several fold while 7-DHC remained relatively constant, but in liver cholesterol first increased then decreased again while 7-DHC first decreased then increased. In older SLOS animals the ratio of 7-DHC/cholesterol, which is an index of biochemical severity, tended to approach, but not reach, normal. While these mice provide the best available genetic animal model for the study of SLOS pathogenesis and treatment, they probably will be most useful at early ages when the metabolic effects of the mutations are most dramatic. To correlate any experimental treatment with improved sterol metabolism will require age-matched controls. Finally, determining the mechanism by which these “SLOS” mice tend to normalize may provide insight into the future development of therapy.
Keywords: Smith-Lemli-Opitz Syndrome, SLOS, cholesterol deficiency, dehydrocholesterol, mutant mouse model, sterol metabolism, Dhcr7, inborn error of metabolism
INTRODUCTION
Smith-Lemli-Opitz syndrome (SLOS) is an inherited disorder resulting in dysmorphias and mental retardation. It is caused by deficient 3β-hydroxysterol-Δ7-reductase (7-dehydrocholesterol reductase, DHCR7, EC 1.3.1.21), which catalyzes the last step in cholesterol synthesis. Thus, SLOS patients have elevated levels of dehydrocholesterol (DHC) and reduced ability to make cholesterol. The molecular and genetic basis of SLOS has been well described [1–3]; however, due to the multiple and essential roles of cholesterol, the pathogenesis of SLOS is complex and not yet well understood. Among recessive inborn errors of metabolism, SLOS has a relatively high incidence varying from 1 in 10,000 to 1 in 60,000 in different regions of Europe and North America [4–10]. The carrier frequency for mutant alleles is estimated as high as 1 in 30 for Caucasian populations and lower for African and Asian populations [3, 9, 11–14]. It is likely that the condition is underdiagnosed because patients having mild disorder without distinctive phenotype may be missed and early fetal demise may be common in the most severely affected [15]. Biochemical diagnosis is based on plasma serum levels of DHC [16], and prenatal diagnosis is possible by measuring DHC in amniotic fluid [17] or chorionic villus cells [15]. More recently, it has been shown that noninvasive prenatal diagnosis of SLOS is possible by measurement of novel steroid metabolites in maternal urine [10, 18].
Current treatment for SLOS is dietary cholesterol supplementation. Anecdotal reports show positive albeit limited effects of exogenous cholesterol on somatic growth and behavior, but developmental outcome does not appear to be altered [19–22]. An effect of dietary cholesterol on behavior is somewhat surprising as the brain is believed to be impervious to external cholesterol [23]. Simvastatin, which inhibits 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase and can cross the blood-brain barrier, is a promising treatment for lowering DHC levels in SLOS patients [1, 24].
The recent development of animal models has made the understanding of SLOS and the possible development of new treatments more feasible. Two mouse models with null mutations in the Dhcr7 gene have been created by homologous recombination [25, 26]. For both of these knockout mutations, homozygous mutant progeny die within one day of birth, which limits experimental usefulness. A more recent “knock-in” mutation resulted in a single amino acid substitution of methionine for threonine in DHCR7 at the amino acid position equivalent to threonine 93 in the human protein. The resulting protein has greatly reduced enzymatic activity [27]. This T93M mutation mimics a known, relatively frequent human mutation, and provides mice with a mild SLOS phenotype [28]. Compound heterozygotes containing one null allele and one T93M allele are viable. Since these are the most severely affected viable SLOS animals available, they have been used in the present study. Although these mice mimic human SLOS at the molecular level, the dysmorphic phenotype is less severe. This may be due to efficient utilization of maternal cholesterol by the mouse fetus [29] in contrast to the human.
Clearly, effective cholesterol biosynthesis is especially critical at certain times during development and continues to be important throughout life. Cholesterol levels and the balance between endogenous and exogenous sources of cholesterol can be expected to change substantially with different stages of development, but patterns of change are remarkably similar among mammals [30]. Furthermore, because of the blood-brain barrier, cholesterol metabolism in the brain is independent of systemic (liver) cholesterol metabolism. In the mouse model for SLOS we can trace these changes with great sensitivity and accuracy by measuring sterol levels in different tissues at different developmental stages. This is important for understanding SLOS pathogenesis and for developing potential therapies. Here we have compared 7-DHC and cholesterol levels in SLOS mice and normal mice at different developmental ages from late fetus to mature adult. Our results confirm and extend previously reported results [28] and show that there were dramatic changes in 7-DHC and total cholesterol concentrations with developmental age. Also, the pattern of change in liver was quite distinct from the pattern of change in brain.
EXPERIMENTAL
Reagents and chemicals
Cholesterol was from Sigma-Aldrich (St. Louis, MO, USA), 7-dehydrocholesterol was from Steraloids (Newport, RI, USA), and cholesterol-26,26,26,27,27,27-d6 was from CDN Isotopes (Pointe-Claire, Canada). Ultrapure water with a resistivity of 18.2 MΩcm was obtained using a MilliQ purification system (Millipore, Molsheim, France). N,O-bis(trimethylsilyl)-trifluoroacetamida (BSTFA ) was from Pierce (Rockford, IL, USA). All other chemicals were of reagent grade and purchased from Merck (Darmstadt, Germany).
Preparation of standard solutions
Ethanolic stock solutions at 1 mg/mL of each sterol were prepared by dissolving 10 mg of each substance in 10 mL of ethanol in a volumetric flask. Working solutions of different concentrations were prepared by appropriate ethanolic dilution of stock solutions. All solutions were stored in the dark at −20°C until used.
Mice
Two lines of mutant mice developed at NIH (by author F.D. Porter and colleagues) were established at the CHORI Animal Facility. The first was a knockout mutation incorporating a deletion of coding exons 3, 4, and 5 (Δ3–5) into the Dhcr7 gene [26]. Heterozygous progeny had a completely normal phenotype, which is consistent with the recessive nature of DHCR7 mutations in humans. Because homozygous Δ3–5/Δ3–5 mutants died shortly after birth, the Δ3–5 mutation was maintained by crossing heterozygous (+/Δ3–5, normal phenotype) with normal C57BL/6 (+/+) mice. Resulting progeny were screened for the +/Δ3–5 genotype by PCR [26]. Due to a minimum of 6 generations of backcrossing, mice carrying the Δ3–5 mutation had a predominantly C57BL/6 genetic background. The second mutant line contained a point mutation, T93M, in Dhcr7 [28]. Homozygous (T93M/T93M) mice were viable and could be bred and maintained as homozygotes. These interbred mice had a mixed genetic background derived from strains 129 and C57BL/6. Compound heterozygous mice (Δ3–5/T93M) were generated by crossing +/Δ3–5 females with T93M/T93M males. Thus, the fetal environment of compound heterozygotes and their littermates was always a phenotypically normal mother having a predominantly C57BL/6 background. Progeny were genotyped by PCR as before [26, 28]. Timed pregnancies were based on evidence of a copulatory plug to establish fetal day one, and pregnant mice were checked daily for new litters. Mice were weaned 4 weeks after birth and were maintained on normal, cholesterol-free chow (Teklad irradiated rodent diet 2918; Harlan, Madison, WI). All animal work conformed to NIH guidelines and was approved by the Institutional Animal Care and Use Committee.
Tissue collection
Mice were killed at indicated ages, and dissected livers, brains and tails were immediately frozen in liquid nitrogen. For fetal, newborn and young (up to and including 33 days old) animals, entire litters were killed at one time. Tail tissue was used for DNA extraction and genotyping by PCR [26], while livers and brains were used for sterol analysis (see below). All brains and livers from timed litters were analyzed regardless of genotype so that phenotypically normal (+/T93M) and SLOS (Δ3–5/T93M) littermates could be compared. For older SLOS animals normal littermates were not available and C57BL/6 mice were used as normal controls.
Preparation of calibration and quality control samples
Calibration curves were prepared daily for each analytical batch by spiking appropriate amounts of ethanolic solutions of each compound. The final calibration curve for cholesterol contained five concentration levels at 1, 5, 10, 17 and 25 μg. For 7-DHC, the calibration samples were prepared at 0.05, 0.1, 0.5, 1 and 4 μg. The quality control sample used to check for precision was an aliquot of the brain homogenate corresponding to a 14 days-old Δ 3–5/T93M heterozygous mouse.
Sample preparation
Brains and livers were homogenized in a mixture of methanol/chloroform (1:1 v/v). To a small portion of this homogenate was added 1μg of internal standard (10 μl of a 100 μg/mL ethanolic solution of cholesterol-d6). Samples were then saponified by adding 500 μl of ethanol together with 300 μl of a 33% KOH solution, and heating at 55°C for 45 minutes. After reaching room temperature 1 mL of water was added, and samples were extracted with 5-mL of hexane by vortexing for 1 min. Organic layers were transferred to new tubes, evaporated and derivatized with 50 μl of BSTFA (55°C for 45 minutes) to form the corresponding TMS derivatives. Extracts were finally diluted with 500 μl of cyclohexane, transferred to autosampler vials and analyzed by GC/MS. To avoid conversion of 7-DHC to previtamin D3, all tubes were covered with foil, and the whole procedure was conducted under minimum lighting conditions.
GC/MS apparatus and conditions
Gas chromatography/mass spectrometry (GC/MS) was carried out on a PolarisQ ion trap mass spectrometer (Thermofinnigan, San Jose, CA, US) interfaced with a TraceGC (Carlo Erba, Milan, Italy) gas chromatograph. Samples were injected with an AI3000 autosampler (Carlo Erba). The sterols were separated on a DB-1 cross-linked methyl-silicone column, 15 m × 0.25 mm i.d., film thickness 0.25 μm (J&W Scientific, Folsom, CA, USA). Helium was used as the carrier gas at a constant flow of 1.8 ml/min. A 2 μl aliquot of the final derivatized extract was injected into the system operated in splitless mode (valve opened at 2 min). The GC temperature was ramped as follows: initial 50°C, held for 3 min, increased to 230°C at 30°C min−1, thereafter increased to 285°C at 2°C min−1. The injector, transfer line, and ion source were kept at 260°C, 280°C and 225°C respectively. The mass spectrometer was operated in the electron impact mode. The mass range scanned was from 100 to 800 amu.
Method validation
To establish that the procedure was dependable, especially due to the instability of 7-DHC, intra- and inter-assay validation was carried out. The protocol validated three criteria, recovery, linearity and precision. For each a calibration curve was prepared at five concentration levels, each in duplicate. Quality control (QC) samples were added in each assay.
Recovery was evaluated by analyzing the calibration samples and pure standards. Extraction of cholesterol with hexane is current practice and recoveries are well established. Recoveries greater than 80% were obtained both for cholesterol and 7-DHC.
Linearity was evaluated over the calibration range in duplicate samples spiked at five different levels. Peak-area ratios (between each compound and the internal standard) were used for calculations. Regression analysis was performed using SPSS for Windows. To correct for the heteroscedasticity [31] of the data, the model was weighted by the inverse of the variance (1/x2). A linear correlation was found in all cases with mean determination coefficients (r2) better than 0.98.
Intra-assay precision (repeatability) as well as inter-assay reproducibility were evaluated at the concentrations of 7-DHC and cholesterol present in the QC sample. Five replicates were used for intra-assay comparisons, and three for inter-assay comparisons. Performance results were expressed as the relative standard deviation (%) of the detector response (i.e., the ratio between peak areas of the analyte and internal standard for their selected base ions). Values for intra-assay repeatability and inter-assay reproducibility were 8.3% and 10.5% respectively for the quantification of cholesterol. For the quantification of 7-DHC the intra- and inter-assay precisions were 7.4% and 14.5% respectively.
RESULTS
To determine the time course of cholesterol biosynthesis and accumulation during development, we compared the concentrations of cholesterol and 7-DHC in brains and livers from SLOS mice (T93M/Δ3–5) and their non-affected littermates (T93M/+). The youngest mice were 18-day old fetuses (one day before expected birth) and the oldest were 10 or 12 months old. Emphasis was on development through the age of weaning. The numbers of animals tested at different time points varied depending on litter size and the number of SLOS vs. normal pups within the litter. When possible, 2 litters at a given time point were analyzed. Males and females did not differ noticeably for either cholesterol or 7-DHC, so data for both sexes were pooled for each time point. 8-DHC, the isomer of 7-DHC, was always substantially less than 7-DHC and often was not significantly above baseline. Therefore, rather than include marginal measurements of 8-DHC, only 7-DHC and cholesterol measurements are reported here.
Cholesterol and 7-DHC in brain
In normal mice cholesterol concentrations in whole brain homogenates increased more than 6 fold from fetus to mature adult with much of the increase occurring within the first 4 weeks (Fig. 1,a). 7-DHC concentrations in normal mice were not detectable under the conditions used. Cholesterol levels in SLOS mice were lower than in their normal littermates, but followed a similar increasing curve and reached normal levels by 8 months to a year. 7-DHC concentrations in SLOS mice followed an entirely different time course than cholesterol; they were relatively high (equal to cholesterol) at early time points, but did not increase. Rather, they remained remarkably stable through the first 33 days of age, and then decreased to low, but measurable, levels in older mice. Summing 7-DHC and cholesterol in SLOS mice indicated that the increase in the total sterol level with age was not significantly different between normal and SLOS (P=0.84) (Fig. 1,b). These changes approximated a linear increase over the first few weeks and appeared to plateau in the mature adults. Non-parametric regression suggested linearity up to 33 days of age (P=0.02 for normal and P=0.008 for SLOS). Correlation coefficients for sterol concentration versus age (up to 33 d) were 0.88 and 0.92 for normal and SLOS respectively.
Figure 1.

Cholesterol and 7-DHC concentrations in whole brain homogenates. Data are expressed as μg sterol/mg tissue (wet weight). (a) Cholesterol in normal mice (○) and SLOS mice (●), and 7-DHC in SLOS mice (■) are compared. Bars indicate standard deviations among animals of the same genotype and age. (b) The sum of cholesterol plus 7-DHC concentrations for normal (○) and SLOS (△) mice.
Using the ratio of 7-DHC/cholesterol as an indicator of SLOS severity (normal mice had a ratio of essentially zero), it was clear that the youngest animals had the most severe biochemical imbalance (Fig. 2,a). As animals aged, the 7-DHC/cholesterol ratio dropped, and by one year of age the ratio was only 0.01. Thus, the ratio tended to normalize over time.
Figure 2.
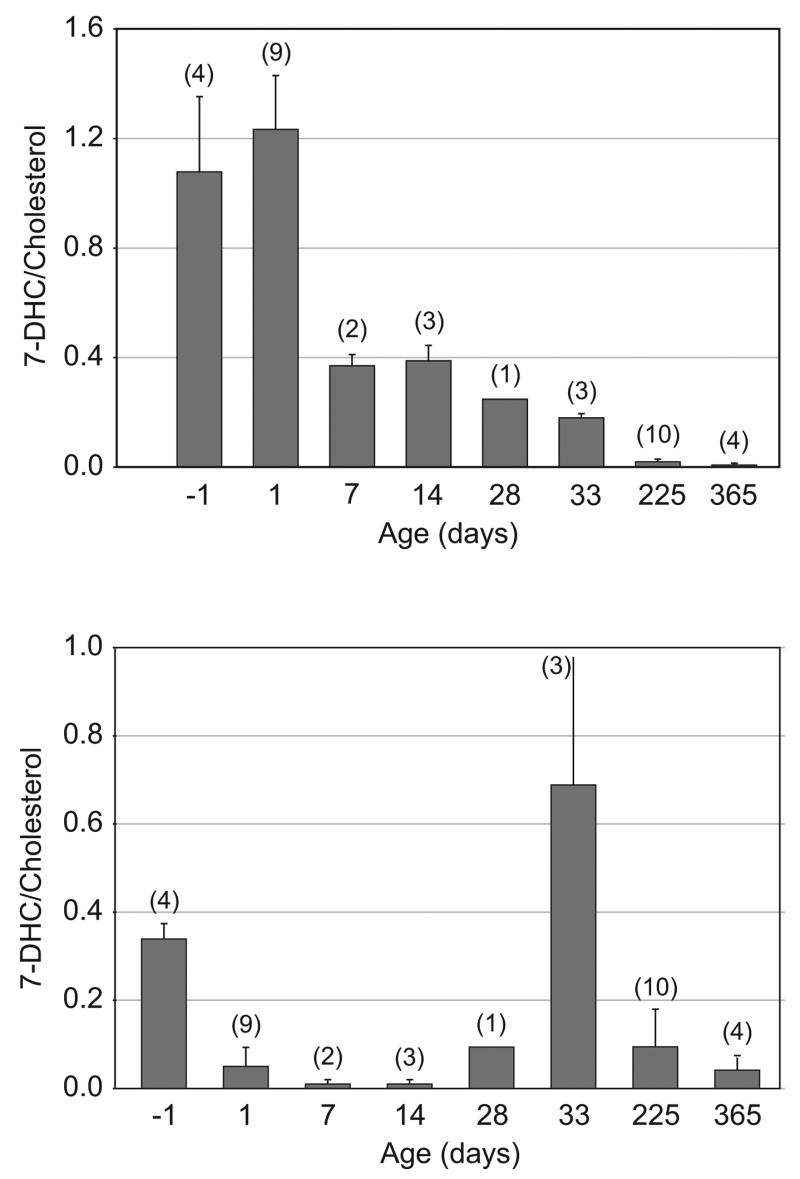
7-DHC/cholesterol ratios in SLOS mice. (a) Brain. (b) Liver. The number of animals for each determination is indicated (n), and bars indicate standard deviations. The higher the ratio, the greater the biochemical imbalance. For normal mice the ratio did not deviate measurably from zero.
Cholesterol and 7-DHC in liver
Cholesterol biosynthesis and accumulation in liver was quite different from brain. Firstly, as expected, concentrations of cholesterol and 7-DHC were generally lower in liver. Secondly, developmental changes in liver sterols followed a different time course (Fig. 3,a). Cholesterol levels in both normal and SLOS mice increased rapidly for the first several days after birth, but then dropped again around 4 weeks of age. Final cholesterol concentrations (8 months to a year of age) were similar to those in 1-day old mice. In contrast, 7-DHC concentrations in SLOS mice dropped after birth and increased temporarily around 4 to 5 weeks of age. Eventually, they also reached levels approximating the 7-DHC concentrations in 1-day old mice. As in brain, the sum of cholesterol and 7-DHC in SLOS mice was nearly indistinguishable from cholesterol alone in normal mice (Fig. 3,b). However, in liver the kinetics of accumulation were more complex than in brain.
Figure 3.

Cholesterol and 7-DHC concentrations in liver. Livers from normal and SLOS mice at the indicated ages were analysed for cholesterol and 7-DHC. Data are expressed as μg sterol/mg tissue (wet weight). (a) Cholesterol in normal mice (○) and SLOS mice (●), and 7-DHC in SLOS mice (■) are compared. Bars indicate standard deviations among animals of the same genotype and age. (b) The sum of cholesterol plus 7-DHC concentrations for normal (○) and SLOS (△) mice.
With cholesterol and 7-DHC concentrations in SLOS mice following roughly mirror image time courses, the 7-DHC/cholesterol ratios changed dramatically during development (Fig. 2,b). Severity of the biochemical imbalance was greatest in the fetal mice and in the 33-day old mice. Ratios temporarily approached normal at 7 and 14 days of age. In mature SLOS mice, the ratio was again low, but not as low as in 7–14 day old pups.
DISCUSSION
It is reassuring that independent sterol measurements reported here are consistent with previous measurements reported for SLOS mutant mice [28]. Here, results provide a more detailed picture of changes in sterol levels during postnatal development. Under the assay conditions used, all SLOS mice, regardless of age, had measurable concentrations of 7-DHC in brain and liver. Conversely, none of the phenotypically normal mice (either T93M/+ or +/+) had measurable levels of 7-DHC. Thus, biochemically, all of the mice with a T93M/Δ3–5 genotype had an abnormal phenotype. However, the extent of this biochemical imbalance varied greatly with age and whether the tissue was brain or liver. In general, mutant mice tended to normalize their cholesterol synthesis with age. There are at least three possible explanations for this. First, the mice might employ an alternative synthetic source of cholesterol, other than the established biosynthetic pathway from HMG CoA with intermediate 7-DHC. Cholesterol concentrations in cells are regulated by complicated feedback mechanisms that work at both transcriptional and posttranslational levels with HMG CoA reductase playing a key role [32]. Increased cholesterol from an alternate pathway might reduce HMG CoA reductase and decrease the amount of 7-DHC synthesized. At this point, however, we have no direct evidence for an alternate pathway that bypasses the need for DHCR7. Second, the low level of DHCR7 activity in mutants might increase over time through some compensatory mechanism such as increased DHCR7 gene expression or decreased decay rate of the enzyme. Correa-Cerro et al. [28] suggest that residual activity of mutant DHCR7 could be upregulated over time in mice, although this does not appear to be the case in humans. Third, exogenous cholesterol from the diet might contribute to the total cholesterol pool, thus contributing to feedback regulation at HMG CoA reductase.
Dietary cholesterol seems a likely explanation for the contrasting cholesterol and 7-DHC concentrations in liver as compared to brain. Even animals tested at day 1 were observed to have milk spots, indicating that they had already been suckling. Since milk contains cholesterol, this may have contributed to the early rise in liver cholesterol accompanied by the decline in 7-DHC. Among mammals, circulating cholesterol, which can be taken up by liver but not by brain, tends to be high in animals that are suckling [30]. By three weeks of age the mice could eat cholesterol-free solid food, although they could also continue to suckle until they were weaned. For the experiments conducted, mice were weaned at 28 days. This abruptly ended the source of dietary cholesterol and forced the mice to rely on endogenously synthesized cholesterol via either residual DHCR-7, or possibly an undefined alternate pathway. Thus, the apparent peak in 7-DHC at 33 days might reflect a reactivation of the cholesterol biosynthetic pathway from HMG CoA in the absence of milk cholesterol. This possibility needs to be tested by further experimentation. In general cholesterol synthesis and accumulation in the brain is considered to be separated from systemic cholesterol by the blood-brain barrier [30]. It has been shown elsewhere that adding cholesterol to the diet of adult SLOS mice decreased DHC in peripheral tissues, but not in brain [28].
In brains of both normal and SLOS mice, total sterol concentrations approximated a linear increase for the first 4 weeks. In normal mouse brain it has been shown that the rate of cholesterol accumulation is equivalent to the rate of cholesterol synthesis [30]. Thus, the results suggest that the rate of early cholesterol synthesis is constant and relatively high. These results also imply a very low turnover rate, which is consistent with an estimated half-life of at least 5 years for cholesterol in the human brain [33]. Since most of the cholesterol is incorporated into stable structures (e.g., myelin and cell membranes), it may not be uniformly available for feedback regulation of HMG CoA reductase. Note that in spite of high and increasing total cholesterol concentrations, 7-DHC in SLOS brain did not decrease significantly during the first 4 weeks. In mature SLOS mice, however, 7-DHC was reduced and the 7-DHC/cholesterol ratio approached normal. This is consistent with a much higher need for de novo cholesterol synthesis during the period of brain growth and development.
It was also noted that levels of total sterol (cholesterol plus its precursor 7-DHC) in both liver and brain were indistinguishable between SLOS and normal animals. This implies that the Dhcr7 mutations did not affect cholesterol metabolism at the level of HMG CoA reductase. However, using newborn null mutants, Fitzky et al. [25] reported that total sterol biosynthesis was reduced by approximately 50% (41% in brain, 68% in liver) and that this appeared to be due to a 7-DHC-mediated decrease in the half-life of HMG CoA reductase. The apparent difference in the effect on sterol synthesis may be due to the more severe consequences of homozygous null mutants as compared to the T93M/Δ3–5 mutants used here.
Although the T93M/Δ3–5 mouse model phenotype mimics human SLOS in general, there are significant differences. First, dysmorphia seems less severe in the mouse. This may be due to more efficient fetal utilization of maternal cholesterol by rodents [29]. And second, as demonstrated here and elsewhere [28], SLOS mice tend to normalize their 7-DHC/cholesterol ratios as they age. A similar biochemical correction has not been observed in human patients. Defining the mechanism of this correction in the SLOS mice should provide insight into potential therapies for SLOS. At the biochemical level, younger rather than older SLOS mice seem to be a closer approximation of the human disease and a superior experimental model. Because of the demonstrated changes in 7-DHC/cholesterol with age, experiments testing potential therapeutic interventions will need to be accompanied by age-matched, preferably littermate, controls. The study described here is a precursor to our major research goal, increasing cholesterol synthesis in the SLOS mice using a viral vector to deliver a functional DHCR7 gene to liver or brain cells. This could be particularly useful in manipulating the high 7-DHC/cholesterol ratios observed in the brains of pre-weaning SLOS mice, and in evaluating the postnatal, neurological and behavioral consequences of 7-DHC/cholesterol imbalance. Thus, we anticipate that T93M/Δ3–5 mutant mice will provide a valuable tool for studying the complex pathogenesis of SLOS and for developing potential therapies.
Acknowledgments
We thank Dorothy Tabron and Ayberk Akat for help with the animals and analyses, Dr. Berna Atik for statistical analysis, and Betsy Lathrop and Tom McElderry for help with the manuscript. All of these people are affiliated with CHORI. This work was funded in part by NIH grants R03 HD045302, R01 HD053036 and S10 RR017854, the intramural research program of the National Institute of Child Health and Human Development, CHORI and the SLO Research Fund.
This work was funded in part by NIH grants R03 HD045302, R01 HD053036 and S10 RR017854, the intramural research program of the National Institute of Child Health and Human Development, CHORI and the SLO Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Correa-Cerro LS, Porter FD. 3•-hydroxysterol •7-reductase and the smith-lemli-opitz syndrome. Mol Genet Metab. 2005;84:112–26. doi: 10.1016/j.ymgme.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 2.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 3.Kelley RI, Hennekam RC. The smith-lemli-opitz syndrome. J Med Genet. 2000;37:321–35. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Opitz JM. Rsh (so-called smith-lemli-opitz) syndrome. Curr Opin Pediatr. 1999;11:353–62. doi: 10.1097/00008480-199908000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Bzduch V, Behulova D, Skodova J. Incidence of smith-lemli-opitz syndrome in slovakia. Am J Med Genet. 2000;90:260. doi: 10.1002/(sici)1096-8628(20000131)90:3<260::aid-ajmg17>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 6.Ryan AK, Bartlett K, Clayton P, Eaton S, Mills L, Donnai D, Winter RM, Burn J. Smith-lemli-opitz syndrome: A variable clinical and biochemical phenotype. J Med Genet. 1998;35:558–65. doi: 10.1136/jmg.35.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowry RB, Yong SL. Borderline normal intelligence in the smith-lemli-opitz (rsh) syndrome. Am J Med Genet. 1980;5:137–43. doi: 10.1002/ajmg.1320050205. [DOI] [PubMed] [Google Scholar]
- 8.Nowaczyk MJ, McCaughey D, Whelan DT, Porter FD. Incidence of smith-lemli-opitz syndrome in ontario, canada. Am J Med Genet. 2001;102:18–20. doi: 10.1002/1096-8628(20010722)102:1<18::aid-ajmg1376>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 9.Nowaczyk MJ, Waye JS, Douketis JD. Dhcr7 mutation carrier rates and prevalence of the rsh/smith-lemli-opitz syndrome: Where are the patients? Am J Med Genet A. 2006;140:2057–62. doi: 10.1002/ajmg.a.31413. [DOI] [PubMed] [Google Scholar]
- 10.Craig WY, Haddow JE, Palomaki GE, Kelley RI, Kratz LE, Shackleton CH, Marcos J, Stephen Tint G, MacRae AR, Nowaczyk MJ, Kloza EM, Irons MB, Roberson M. Identifying smith-lemli-opitz syndrome in conjunction with prenatal screening for down syndrome. Prenat Diagn. 2006;26:842–9. doi: 10.1002/pd.1518. [DOI] [PubMed] [Google Scholar]
- 11.Porter FD. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr Opin Pediatr. 2003;15:607–13. doi: 10.1097/00008480-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Nowaczyk MJ, Nakamura LM, Eng B, Porter FD, Waye JS. Frequency and ethnic distribution of the common dhcr7 mutation in smith-lemli-opitz syndrome. Am J Med Genet. 2001;102:383–6. doi: 10.1002/ajmg.1441. [DOI] [PubMed] [Google Scholar]
- 13.Ciara E, Popowska E, Piekutowska-Abramczuk D, Jurkiewicz D, Borucka-Mankiewicz M, Kowalski P, Goryluk-Kozakiewicz B, Nowaczyk MJ, Krajewska-Walasek M. Slos carrier frequency in poland as determined by screening for trp151x and val326leu dhcr7 mutations. Eur J Med Genet. 2006 doi: 10.1016/j.ejmg.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Battaile KP, Battaile BC, Merkens LS, Maslen CL, Steiner RD. Carrier frequency of the common mutation ivs8-1g>c in dhcr7 and estimate of the expected incidence of smith-lemli-opitz syndrome. Mol Genet Metab. 2001;72:67–71. doi: 10.1006/mgme.2000.3103. [DOI] [PubMed] [Google Scholar]
- 15.Kratz LE, Kelley RI. Prenatal diagnosis of the rsh/smith-lemli-opitz syndrome. Am J Med Genet. 1999;82:376–81. [PubMed] [Google Scholar]
- 16.Kelley RI. Diagnosis of smith-lemli-opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin Chim Acta. 1995;236:45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- 17.Abuelo DN, Tint GS, Kelley R, Batta AK, Shefer S, Salen G. Prenatal detection of the cholesterol biosynthetic defect in the smith-lemli-opitz syndrome by the analysis of amniotic fluid sterols. Am J Med Genet. 1995;56:281–5. doi: 10.1002/ajmg.1320560309. [DOI] [PubMed] [Google Scholar]
- 18.Shackleton CH, Roitman E, Kratz L, Kelley R. Dehydro-oestriol and dehydropregnanetriol are candidate analytes for prenatal diagnosis of smith-lemli-opitz syndrome. Prenat Diagn. 2001;21:207–12. doi: 10.1002/1097-0223(200103)21:3<207::aid-pd27>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 19.Irons M, Elias ER, Abuelo D, Bull MJ, Greene CL, Johnson VP, Keppen L, Schanen C, Tint GS, Salen G. Treatment of smith-lemli-opitz syndrome: Results of a multicenter trial. Am J Med Genet. 1997;68:311–4. [PubMed] [Google Scholar]
- 20.Elias ER, Irons MB, Hurley AD, Tint GS, Salen G. Clinical effects of cholesterol supplementation in six patients with the smith-lemli-opitz syndrome (slos) Am J Med Genet. 1997;68:305–10. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 21.Tierney E, Nwokoro NA, Porter FD, Freund LS, Ghuman JK, Kelley RI. Behavior phenotype in the rsh/smith-lemli-opitz syndrome. Am J Med Genet. 2001;98:191–200. doi: 10.1002/1096-8628(20010115)98:2<191::aid-ajmg1030>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 22.Sikora DM, Ruggiero M, Petit-Kekel K, Merkens LS, Connor WE, Steiner RD. Cholesterol supplementation does not improve developmental progress in smith-lemli-opitz syndrome. J Pediatr. 2004;144:783–91. doi: 10.1016/j.jpeds.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 23.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Jira PE, Wevers RA, de Jong J, Rubio-Gozalbo E, Janssen-Zijlstra FS, van Heyst AF, Sengers RC, Smeitink JA. Simvastatin. A new therapeutic approach for smith-lemli-opitz syndrome. J Lipid Res. 2000;41:1339–46. [PubMed] [Google Scholar]
- 25.Fitzky BU, Moebius FF, Asaoka H, Waage-Baudet H, Xu L, Xu G, Maeda N, Kluckman K, Hiller S, Yu H, Batta AK, Shefer S, Chen T, Salen G, Sulik K, Simoni RD, Ness GC, Glossmann H, Patel SB, Tint GS. 7-dehydrocholesterol-dependent proteolysis of hmg-coa reductase suppresses sterol biosynthesis in a mouse model of smith-lemli-opitz/rsh syndrome. J Clin Invest. 2001;108:905–15. doi: 10.1172/JCI12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wassif CA, Zhu P, Kratz L, Krakowiak PA, Battaile KP, Weight FF, Grinberg A, Steiner RD, Nwokoro NA, Kelley RI, Stewart RR, Porter FD. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of rsh/smith--lemli--opitz syndrome. Hum Mol Genet. 2001;10:555–64. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 27.Wassif CA, Krakowiak PA, Wright BS, Gewandter JS, Sterner AL, Javitt N, Yergey AL, Porter FD. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in smith-lemli-opitz syndrome fibroblasts. Mol Genet Metab. 2005;85:96–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Correa-Cerro LS, Wassif CA, Kratz L, Miller GF, Munasinghe JP, Grinberg A, Fliesler SJ, Porter FD. Development and characterization of a hypomorphic smith-lemli-opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet. 2006;15:839–51. doi: 10.1093/hmg/ddl003. [DOI] [PubMed] [Google Scholar]
- 29.Willnow TE, Hilpert J, Armstrong SA, Rohlmann A, Hammer RE, Burns DK, Herz J. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93:8460–4. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dietschy JM, Turley SD. Thematic review series: Brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–97. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann C, Smeyers-Verbeke J, Massart DL, McDowall RD. Validation of bioanalytical chromatographic methods. J Pharm Biomed Anal. 1998;17:193–218. doi: 10.1016/s0731-7085(97)00198-2. [DOI] [PubMed] [Google Scholar]
- 32.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 33.Björkhem I, Lutjohann D, Diczfalusy U, Stahle L, Åhlborg G, Wahren J. Cholesterol homeostasis in human brain: Turnover of 24s-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39:1594–600. [PubMed] [Google Scholar]