

Abstract
The mechanism of chain selection and trimerization of fibril-associated collagens with interrupted triple helices (FACITs) differs from that of fibrillar collagens that have special C-propeptides. We recently showed that the second carboxyl-terminal non-collagenous domain (NC2) of homotrimeric collagen XIX forms a stable trimer and substantially stabilizes a collagen triple helix attached to either end. We then hypothesized a general trimerizing role for the NC2 domain in other FACITs. Here we analyzed the NC2 domain of human heterotrimeric collagen IX, the only member of FACITs with all three chains encoded by distinct genes. Upon oxidative folding of equimolar amounts of the α1, α2, and α3 chains of NC2, a stable heterotrimer with a disulfide bridge between α1 and α3 chains is formed. Our experiments show that this heterotrimerization domain can stabilize a short triple helix attached at the carboxyl-terminal end and allows for the proper oxidation of the cystine knot of type III collagen after the short triple helix.
Keywords: Collagen, Extracellular Matrix Proteins, Protein Domains, Protein Folding, Protein Self-assembly, Collagen IX, FACITs, NC2 Domain, Heterotrimerization Domain
Introduction
The fibril-associated collagens with interrupted triple helices (FACITs)2 include types IX, XII, XIV, XVI, XIX, XX, XXI, and XXII. Collagen IX is a heterotrimer composed of three distinct α chains, and all others are homotrimers whose α chains are characterized by short collagenous domains (COL) interrupted by several non-collagenous domains (NC) (1, 2). Unlike the fibril-forming collagens, the FACITs have significantly shorter NC1 domains (the carboxyl-terminal NC domains) (75 residues for human collagen XII, fewer than 30 residues for human collagen IX, and even fewer than 20 residues for human collagen XIX), whereas those of fibrillar collagens are of a different type and comprise about 260 residues. The FACITs share a remarkable sequence homology at their COL1/NC1 junctions by having two strictly conserved cysteine residues separated by four residues in the NC1 domain. These cysteines form interchain disulfide bonds, a so-called cystine knot, but only after the triple helix is formed (3–6). In other words, the NC1 domain cannot trimerize itself and requires exogenous alignment of three chains. It has been suggested that the NC2 domain in all FACITs is able to form an α-helical coiled coil, thus bearing an ability to trimerize those collagens (7). Experimental evidence for that was recently reported by us for the NC2 domain of collagen XIX (6).
Collagen IX contains three collagenous domains (COL1–COL3) and four noncollagenous domains (NC1–NC4) (Fig. 1). The molecule is covalently associated with the surface of interstitial collagen fibrils in cartilage (8), where it plays a role in maintaining the long term structural integrity of this tissue (9). Loss of collagen IX is associated with such diseases as osteoarthritis, rheumatoid arthritis, intervertebrate disk degeneration, ocular defects, loss of hearing, and others (9–13).
FIGURE 1.

Domains of collagen IX. A, schematic presentation of collagen IX with three collagenous (COL1–COL3) and four non-collagenous (NC1–NC4) domains, numbered from the carboxyl terminus. B, the amino acid sequences of NC3, NC2, and NC1 of human collagen IX of all three chains (Swiss-Prot numbers P20849, Q14055, and Q14050). Cysteines in NC3 or NC1 form two cystine knots, each covalently cross-linking three chains (22).
Collagen IX is the most intriguing FACIT collagen in terms of its chain selection and heterotrimerization properties. Several attempts were made to decipher its code. Reassociation of the chains of a pepsin-resistant low molecular weight fragment of bovine collagen IX was tested in vitro (14). The low molecular weight fragment includes the sequence of COL1 and the beginning of NC1 with intact disulfides. Upon reduction and reassociation, followed by the formation of disulfide-bonded multimers, only a negligible amount of α1α2α3 was observed (14). Another in vitro study was focused on either NC1 sequences or NC1 sequences extended with short fragments of COL1 (15). Although experiments with just NC1 sequences did not produce any significant amount of multimers, the extended sequences were partially successful and yielded ∼10% of disulfide-bonded heterotrimeric α1α2α3 (15). On the other hand, a recent study of full-length protein and several deletion mutants expressed in insect cells showed that COL1 and NC1 are not required for trimerization of collagen IX, although the COL1-NC1 region might be important for chain specificity (16). Additionally, the authors reported that the COL2-NC2 region of collagen IX is not sufficient for trimerization (16).
To explore the trimerization potential of the collagen IX, we have studied folding and stability of the NC2 domain alone as well as in junction with a collagenous sequence containing the type III collagen cystine knot. The cystine knot naturally found in type III collagen is located at the end of the collagenous domain and forms interchain disulfide bonds. Exploiting the cystine knot allowed detection of the trimer formation under non-reducing SDS-PAGE and subsequent unambiguous mass spectroscopic analysis. The NC2 domain appeared to be an effective heterotrimerization domain that promotes chain selection and folding of the triple helix. Moreover, it must play an important postfolding role in stabilizing the triple helix. Matrix metalloproteinase-3 (MMP-3) cleavage of the collagen IX NC2 domain (17) should then initiate degradation of the molecule. Protection of the collagen IX NC2 domain against MMP-3 cleavage can thus stabilize the integrity of cartilage and prevent onsets of cartilage diseases.
EXPERIMENTAL PROCEDURES
Cloning of Trx_α1NC2, Trx_α2NC2, and Trx_α3NC2
To facilitate expression of short sequences comprising the NC2 domain of human collagen IX, they were cloned as parts of fusion molecules with a His-tagged thioredoxin with a thrombin cleavage site (HisTag-Trx-thr) to cleave off products later. Initially, DNA encoding HisTag-Trx-thr was recloned from the vector pHisTrx2 (18) into pET23d(+) (Novagen) using NcoI and BamHI restriction sites. The resulting plasmid, pET23-HisTrx, had multiple cloning sites just after the HisTag-Trx-thr gene. All constructs in this study were cloned and expressed using the plasmid pET23-HisTrx.
The plasmid (clone ID 5248739, NCBI accession number BC041479), containing an incomplete sequence of the human collagen IX α2 chain was purchased from Open Biosystems and used as a template for PCR. Two other templates, encoding α1 or α3 NC2 domains, were synthetic oligonucleotides: 5′-GGTAGAGCACCGACAGATCAGCACATTAAGCAGGTTTGCATGAGAGTCATACAAGAACATTTTGCTGAGATGGCTGCCAGTCTTAAGCGTCCAGACTCAGGTGCCACT-3′ for α1 and 5′-GGGAAGGAGGCCAGCGAGCAGCGCATCCGTGAGCTGTGTGGGGGGATGATCAGCGAACAAATTGCACAGTTAGCCGCGCACCTACGCAAGCCTTTGGCACCCGGGTCCATT-3′ for α3. The latter contained two modified codons (underlined sequences) for arginine, and they replaced codons that are rare in Escherichia coli.
Sequences encoding human collagen IX NC2 domain were PCR-amplified using the following set of oligonucleotide pairs (forward and reversed, respectively): 5′-TGCGGATCCGGTAGAGCACCGACAGATCAGCACAT-3′ and 5′-GTCAGTCGACTTAAGTGGCACCTGAGTCTGGACGCTT-3′ for α1, 5′-TGCGGATCCGGCCGGGATGCCACTGACCAGCAC- 3′ and 5′-GTCAGTCGACTTACACCGCACCCAGGGCTTCCCGCTT-3′ for α2, 5′-TGCGGATCCGGGAAGGAGGCCAGCGAGCAGCGC-3′ and 5′-GTCAGTCGACTTAAATGGACCCGGGTGCCAAAGGCTT-3′ for α3. Underlined sequences are BamHI and SalI restriction sites for forward and reversed primers, respectively.
PCR products were cloned into the pET23-HisTrx vector using the restriction sites BamHI and SalI. The DNA inserts were verified by Sanger dideoxy-DNA sequencing.
Cloning of Trx_α1NC2-(GPP)5CC, Trx_α2NC2-(GPP)5CC, and Trx_α3NC2-(GPP)5CC
Constructs with collagenous sequences were prepared as follows. A DNA fragment encoding the collagenous sequence and the collagen III cystine knot was PCR-amplified using the oligonucleotides 5′-GTCAGGATCCGGTGCTAGCGGTCCGCCAGGACCACCGGGT-3′ (forward; BamHI site is underlined, and the NheI site is in boldface type) and 5′-GTCAGTCGACTTAAACACCACCACAGCACGGGCCTGGTGGACCAGGAGG-3′(reversed; SalI site is underlined) and a synthetic oligonucleotide as a template: 5′-GGGCCCCCTGGTCCGCCAGGACCACCGGGTCCACCTGGTCCTCCTGGTCCACCAGGCCCG-3′. The PCR product was cloned into the pET23-HisTrx vector using restriction sites BamHI and SalI. The DNA insert was verified by Sanger dideoxy-DNA sequencing. The resulting plasmid, pET23-HisTrx-GPP5CC, was used to clone three chains of NC2 using the restriction sites BamHI and NheI. For that, fragments of the NC2 chains were PCR-amplified using the same forward primers for α2 and α3, the new forward primer for α1 (TGCGGATCCGGCTATCCGGGTAGAGCACCGACAGATCAGCACAT (the BamHI site is underlined, and the extra sequence encoding tripeptide unit GYP is in boldface type), and the following reversed primers (the NheI site is underlined): 5′-GTCAGCTAGCACCAGTGGCACCTGAGTCTGGACGCTT-3′ for α1, 5′-GTCAGCTAGCACCCACCGCACCCAGGGCTTCCCGCTT-3′ for α2, and 5′-GTCAGCTAGCACCAATGGACCCGGGTGCCAAAGGCTT-3′for α3.
Expression of Proteins and Initial Purification
The recombinant proteins were expressed separately in the E. coli BL21(DE3) host strain (Novagen). Colonies from freshly transformed competent cells were resuspended in 2xTY medium (16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl per liter), grown to A600 ∼0.6–0.8, and induced by adding isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm. The constructs without collagenous sequence were expressed at 25 °C for 16–20 h. Cells containing the constructs with collagenous sequence were initially grown at 25 °C, transferred to 4 °C, and expressed for 7–10 days.
Each construct was initially purified separately. Cells were harvested by centrifugation, resuspended in 20 mm Tris/HCl buffer, pH 8, and disrupted by ultrasonication. After the sonication, the buffer was adjusted to 100 mm Tris/HCl buffer, pH 8, containing 200 mm NaCl, 10 mm imidazole, by adding appropriate amounts of stock solutions. Debris was removed by centrifugation at 15,000 × g for 30 min, and the lysate was incubated with the Ni2+-NTA resin (Qiagen) at room temperature for 30 min. The Ni2+-NTA resin with bound protein was loaded into a column, allowed to drain, and thoroughly washed with the wash buffer (50 mm sodium phosphate buffer, pH 8, containing 500 mm NaCl, 20 mm imidazole). The protein was eluted with the elution buffer (50 mm sodium phosphate buffer, pH 8, containing 500 mm NaCl and 500 mm imidazole).
Oxidative Folding
Initially, the purified constructs either with or without the collagenous part were folded under the same oxidative conditions. The three chains of approximately equal concentrations were mixed and diluted with water, and the buffer was adjusted to 100 mm Tris/HCl buffer, pH 8.6, containing 15 mm sodium phosphate, 150 mm NaCl, 150 mm imidazole, 10 mm reduced glutathione, 1 mm oxidized glutathione at 25 °C. The final concentration of each chain was ∼10 μm. The solution was sequentially incubated at 37 °C for 24 h, 30 °C for 24 h, and 25 °C for 24 h, and the pH value was periodically checked and adjusted to be not lower than 8.3. Finally, the solution was extensively dialyzed against 50 mm Tris/HCl buffer, pH 8, containing 150 mm NaCl, at room temperature to remove imidazole and reducing agents.
Thrombin Cleavage and Removal of Thioredoxin
Thrombin cleavage was performed at 4 °C for 48 h with recombinant thrombin protease (BaculoGoldTM; BD Biosciences) in 50 mm Tris/HCl buffer, pH 8.0, supplemented with 150 mm NaCl. The final concentration of thrombin was 1 unit/ml or 17 μg/ml (based on the information of the manufacturer). The resulting fragments of interest had two additional amino acid residues GS before the native amino acid sequence (Table 1). Thrombin-cleaved material was run over the Ni2+-NTA resin to separate NC2-containing fragments from His-tagged thioredoxin or uncleaved material. The NC2-containing fragments were eluted with 20 mm imidazole, 50 mm sodium phosphate, 500 mm NaCl, pH 7.2.
TABLE 1.
Sequences, calculated molar masses, and pI values of the individual peptides
The sequences are shown for individual peptides after the cleavage of the thioredoxin part. Molar masses (M), are calculated for reduced cysteines.

Final Purification
Two additional purification steps were applied for the NC2-containing products, namely, cation- and anion-exchange columns. First, the starting material was extensively dialyzed against 50 mm HEPES buffer, pH 7, loaded onto the SP-Sepharose column (GE Healthcare) and eluted with a linear gradient of NaCl (0–0.6 m). The major peak was observed at 0.25–0.3 m NaCl, and its fractions were pooled for the next purification step. The fractions were combined and extensively dialyzed against 20 mm Tris/HCl, pH 8, loaded onto the Q-Sepharose column (GE Healthcare), and eluted with a linear gradient of NaCl (0–200 mm). The major peak was eluted at 40–50 mm NaCl, and its fractions were pooled. To eliminate proteolytic contamination, an extra purification step was applied to the α123NC2 complex. Fractions after the anion-exchange column were combined and loaded onto the phenyl-Sepharose column (GE Healthcare) in 50 mm sodium phosphate buffer (pH 7.2), supplemented with 1 m ammonium sulfate. The complex was eluted with 0.5 m ammonium sulfate in 50 mm sodium phosphate buffer (pH 7.2), the rest of the material was eluted with much lower concentrations of ammonium sulfate, and the majority of proteolytic contamination was eluted only with 8 m urea. Amino acid compositions and protein concentrations were determined in triplicate after hydrolysis in 6 m HCl (22 h at 110 °C) using a Hitachi L-8800A amino acid analyzer.
HPLC and MS Analysis
HPLC analysis was performed on a HP 1090 Liquid Chromatograph with a detection wavelength of 215 nm. Chromatographic separation was achieved by gradient elution on a 5-μm pore size 2.1 × 150-mm Zorbax 300SB-C18 column. Liquid chromatography-MS analysis was performed on a Waters Q-TOF Micro Mass Spectrometer with an electrospray ionization source coupled to a Waters nanoAcquity HPLC system. Samples were loaded onto a 5-μm pore size 180 μm × 20-mm Symmetry C18 trapping column. Chromatographic separation was achieved by gradient elution off of the trapping column onto a 1.7-μm pore size 100 μm × 100-mm BEH130 C18 analytical column at a flow rate of 0.8 μl/min. Raw MS data were processed using Waters MassLynx software and deconvoluted using the maximum entropy algorithm MaxEnt 1.
Analytical Ultracentrifugation
Sedimentation equilibrium measurements were performed with a Beckman model XLA analytical ultracentrifuge. Absorbance was measured at 240 nm. Runs were carried out at 20 °C in an An60-Ti rotor using 12-mm cells and Epon two channel centerpieces. Speeds used were 22,000 and 25,000 r.p.m. for α123NC2-(GPP)5CC and α123NC2, respectively. Data analysis was done using Ultrascan II (version 9.3). Partial specific volumes were calculated using individual sequences of peptides and averaged; the values were 0.725 and 0.732 cm3/g for α123NC2-(GPP)5CC and α123NC2, respectively.
Circular Dichroism Analysis
CD spectra were recorded on an AVIV model 202 spectropolarimeter (AVIV Instruments, Inc.) with thermostatted quartz cells of 1–5-mm path length. The spectra were normalized for concentration and path length to obtain the mean molar residue ellipticity after subtraction of the buffer contribution. Thermal scanning curves were recorded at 222 nm for the α123NC2 complex to monitor the α-helical secondary structure transition or at 230 nm for α123NC2-(GPP)5CC to monitor the collagen triple helix transition. Peptide concentrations were determined by amino acid analysis.
Evaluation of the Thermodynamic Data
CD transition curves of the α123NC2 complex were interpreted based on a two-state mechanism where two unfolded chains, U13 (α1-α3) and U2 (α2), associate into a native complex, N.
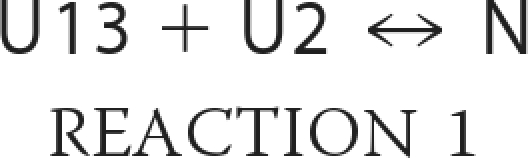 |
The equilibrium constant KN is calculated as follows,
where [N] is concentration of the native complex, and [U13] and [U2] are concentrations of unfolded α1-α3 and α2, respectively.
The two mass conservations are defined by c013 = [U13] + [N] and c02 = [U2] + [N]. For the complex with [U13] = [U2], the two total concentrations are equal, c0 = c013 = c02. Equation 1 can be rewritten as follows,
where F is the fraction of folded complex,
From Equation 2, we derive the following,
where w = 1 + 1/(2KNc0).
The measured CD signal is connected with F by the relation,
where parameters aN and bN and parameters aU and bU account for the linear temperature dependencies of dichroism signals of the native and unfolded state, respectively.
The equilibrium constant is related with the standard Gibbs free energy ΔG0, the standard enthalpy ΔH0, and the standard entropy ΔS0 of the transition by the following,
Assuming that ΔH0 and ΔS0 are constant within the temperature interval of the transition, the global fit of Equation 5 using relations from Equations 4 and 6 allowed us to determine the standard enthalpy, ΔH0, and the standard enthropy, ΔS0. The parameters aN, bN, aU, bU, ΔH0, and ΔS0 were fitted simultaneously.
From Equations 2 and 6, the midpoint of the transition (Tm), where F = 0.5, follows.
RESULTS
Design of Constructs
Constructs containing the NC2 regions of three human collagen IX chains (α1, α2, or α3) either extended or not with a collagen triple helical sequence ending with the cystine knot of collagen III (Table 1) were cloned as part of a fusion molecule. The fusion molecule comprised a His-tagged thioredoxin followed by a thrombin cleavage sequence and a fragment of interest (18). The cystine knot of collagen III (19) was used as a folding marker for the triple helix formation. It was shown earlier that two cysteines in each chain form interchain disulfide bonds only after the triple helix is folded (20). Covalently linking three collagenous chains allows an easy detection of a trimeric band on SDS-PAGE under non-reducing conditions. As a collagenous sequence, we used a short stretch of only five GPP units (Table 1).
Bacterial Expression of Fusion Proteins
Temperature optimization was required to obtain similar expression levels of different constructs. The most problematic were constructs containing the α2 chain sequences. Finally, constructs without collagenous sequence were expressed at 25 °C, whereas constructs with collagenous sequence required prolonged expression at 4 °C. Although all constructs were expressed separately, they produced only soluble proteins. The yields were sufficient and were estimated to be 20–50 mg of a fusion protein starting from 1 liter of bacterial medium.
Initial Purification of Fusion Proteins Trx_α1NC2-(GPP)5CC, Trx_α2NC2-(GPP)5CC, and Trx_α3NC2-(GPP)5CC and Their Reoxidation
Soluble fractions of cell lysates were separately purified over the Ni2+-NTA resin and analyzed on a gel (Fig. 2A, lanes 1–3). In addition to bands corresponding to monomeric species around 20 kDa, dimeric species, trimeric species, and ladders of higher multimers were observed, indicating formation of multiple intermolecular disulfide bonds, presumably due to misfolding. When equimolar amounts (∼10 μm) of all three constructs were combined and reoxidized using reduced and oxidized glutathiones as reshuffling agents, one predominant trimeric band was observed on a gel under non-reducing conditions (Fig. 2A, lane 4). Further purification steps included cleavage and removal of the His-tagged thioredoxin partner and final purification through the cation- and anion-exchange columns prior to the MS analysis.
FIGURE 2.

Initial purification, reoxidation, cleavage, and separation of α123NC2-(GPP)5CC. A, non-reduced samples analyzed on 4–12% NuPAGE MOPS (Invitrogen). Lanes 1–3, separate elutions of α1NC2-(GPP)5CC, α2NC2-(GPP)5CC, and α3NC2-(GPP)5CC from the Ni2+-NTA resin (Qiagen); lane 4, the reoxidized mix of α1NC2-(GPP)5CC, α2NC2-(GPP)5CC, and α3NC2-(GPP)5CC; lanes 5 and 6, thrombin cleavage products of the reoxidized mix at 30 min and 16 h. 0, non-cleaved trimer; −1, trimer without a single copy of thioredoxin; −2, trimer without two copies; star, final trimeric product without thioredoxin moieties; T, cleaved thioredoxin. B, thrombin cleavage products analyzed on 15% Tris/glycine SDS-PAGE under non-reducing conditions. Lane 1, non-cleaved material; lanes 2 and 3, cleaved products at 16 and 36 h. C, the purification of the thrombin cleaved products over the Ni2+-NTA resin (Qiagen) and the analysis on 15% Tris/glycine SDS-PAGE under non-reducing conditions. Lane 1, loading material; lanes 2 and 3, flow-through and wash with loading buffer; lane 4, 20 mm imidazole elution, which presumably contains α123NC2-(GPP)5CC; lanes 5–7, 40, 60, and 500 mm imidazole elutions.
Thrombin Cleavage of Oxidized Trx_α123NC2-(GPP)5CC and Thioredoxin Removal
Thrombin digestion of the oxidized material (labeled with 0) showed gradual cleavage of one (−1), two (−2), and finally all three thioredoxin parts from the trimeric band (Fig. 2, A (lanes 5 and 6) and B (lanes 2 and 3)). The resulting band corresponding to a trimer without thioredoxin moieties (with an apparent mass of ∼20 kDa) is marked with a star. Separation of the cleaved His-tagged thioredoxin part from the NC2-containing trimer was performed using the Ni2+-NTA resin (Fig. 2C). The moderate binding of the NC2-containing trimer to the Ni2+-NTA resin was presumably due to several histidine residues in the NC2 sequences (Table 1).
Purification and MS Analysis of α123NC2-(GPP)5CC
It was concluded that different calculated pI values of the NC2 sequences (Table 1) might be effectively used to separate possible different combinations of α chains. A single major peak was observed in consequent runs over the cation- and anion-exchange columns (Figs. 3 and 4). Finally, the MS analysis of the purified trimer showed a molar mass of 18,650.5Da, which is only consistent with the oxidized heterotrimeric complex, α123NC2-(GPP)5CC (Fig. 5 and Table 2).
FIGURE 3.

Purification of α123NC2-(GPP)5CC using SP-Sepharose. A, chromatogram. Fractions labeled with a, b, c, and d were analyzed on a gel. B, analysis of the fractions on 4–12% NuPAGE MOPS (Invitrogen) under non-reducing conditions. Lane 1, loading; lane 2, flow-through; lane 3, fraction a; lanes 4–11, fractions b; lanes 12–14, fractions c; lane 15, fraction d.
FIGURE 4.

Purification of α123NC2-(GPP)5CC using Q-Sepharose. A, chromatogram. Fractions labeled with a, b, and c were analyzed on a gel. B, analysis of the fractions on 4–12% NuPAGE MOPS (Invitrogen) under non-reducing conditions. Lane 1, loading; lane 2, flow-through; lanes 3–8, fractions a; lane 9, fraction b; lanes 10, fraction c.
FIGURE 5.

Deconvoluted mass spectra of α123NC2-(GPP)5CC. The 18,651 peak corresponds to α123NC2-(GPP)5CC.
TABLE 2.
Mass spectrometry and sedimentation equilibrium data
Molar masses were calculated for oxidized cysteines. Sedimentation equilibrium runs were performed at 20 °C in 50 mm sodium phosphate, 150 mm NaCl, pH 8. Concentrations of complexes were 0.25 and 0.12 mg/ml for α123NC2-(GPP)5CC and α123NC2, respectively.
| Complex | Calculated molar mass | Mass spectrometry | Sedimentation equilibrium |
|---|---|---|---|
| Da | Da | kDa | |
| α123NC2-(GPP)5CC | 18,651.1 | 18,650.5 | 18.1 ± 3.6 |
| α123NC2 | 8226.3 + 3977.5 = 12,203.8 | 8226.0; 3977.0 | 10.8 ± 2.0 |
Production and Analysis of α123NC2
The same strategy was applied for constructs without a collagenous sequence and the collagen III cystine knot. Because these constructs lacked the ability to form covalently linked trimers, only monomeric and dimeric bands were observed on a denaturing gel under non-reducing conditions (data not shown). After the thrombin cleavage, the NC2-containing complex also showed binding to the Ni2+-NTA resin and was eluted using the same imidazole concentration (Fig. 6A). Again, two bands were observed under non-reducing conditions, one at ∼4 kDa and another at ∼9 kDa (Fig. 6A, lane 4). The complex was further purified using the cation- and anion-exchange columns, analogously to α123NC2-(GPP)5CC. An additional purification step using the phenyl-Sepharose column was necessary to remove impurities and/or proteolytic fragments (Fig. 6B). Most of the contamination was only eluted with 8 m urea (Fig. 6B, lane 10). The complex was run over the analytical C18 HPLC, and two major peaks were observed following the absorbance of peptide bonds at 215 nm (Fig. 7A). The ratio of areas for those peaks was 2:1. The MS analysis of the peaks identified molar masses of 8226.0 and 3977.0 Da, respectively (Fig. 7, B and C), which corresponds to disulfide-bonded α1NC2-α3NC2 and dissociated α2NC2 (Table 2). These data supports the formation of the heterotrimeric α123NC2 complex with disulfide-linked α1 and α3 chains and the right stoichiometry of chains.
FIGURE 6.

Purification of α123NC2. A, separation of thrombin cleavage products using the Ni2+-NTA resin (Qiagen) analyzed on 4–12% NuPAGE MES (Invitrogen) under non-reducing conditions. Lane 1, loading, thioredoxin is labeled with T, NC2 products (dimer and monomer) are labeled with arrowheads, and bands corresponding to thioredoxin and disulfide-linked dimer of NC2 are partially overlapped. Lanes 2 and 3, flow-through and washing with loading buffer; lane 4, 20 mm imidazole elution, which contains α123NC2 (seen as two bands); lanes 5 and 6, 40 and 500 mm imidazole elutions (the latter contains thioredoxin). B, final purification of α123NC2 using the phenyl-Sepharose column (GE Healthcare) analyzed on 4–12% NuPAGE MES under non-reducing conditions. Lane 1, loading with 1 m ammonium sulfate; lane 2, flow-through; lanes 3 and 4, elutions with 0.5 and 0.3 m ammonium sulfate; lanes 5–8, elutions with 0.2, 0.1, 0.05, and 0 m ammonium sulfate, respectively; lanes 9 and 10, elutions with 1 and 8 m urea. Two bands of the α123NC2 complex observed under non-reducing conditions presumably correspond to a single chain of α2NC2 and a disulfide-cross-linked product of chains α1NC2 and α3NC2.
FIGURE 7.

Analytical HPLC and mass spectroscopy of α123NC2. HPLC analysis of α123NC2 produced two major peaks (A). Liquid chromatography-MS was performed on the sample; the mass spectrum obtained for peak I (B) corresponds to α1NC2-α3NC2, and that of peak II (C) corresponds to α2NC2. The inset in B shows the absence of masses corresponding to α1NC2-α1NC2 and α3NC2-α3NC2.
Sedimentation Equilibrium Analysis
The oligomeric state of the purified heterotrimeric complexes, α123NC2-(GPP)5CC or α123NC2, were analyzed by analytical ultracentrifugation. Sedimentation equilibrium runs at 20 °C in phosphate-buffered saline, pH 8, for 24 h revealed trimeric organization for both complexes (Table 2). Although the determined trimeric masses were within the error limits of the experiment, the values were slightly less then expected in both cases. This discrepancy could probably be due to an underestimation of the partial specific volumes used for the analysis. The calculations of partial specific volumes were based on the amino acid composition, whereas disulfide bonds are known to notably increase the value of the partial specific volume. In the case of the α123NC2 complex, where α1NC2-α3NC2 and α2NC2 are in a kinetically controlled equilibrium, such a prolonged run can result in a bigger deviation from the ideal mass value (toward lower mass). Indeed, there is the kinetic equilibrium between the complex and a small population of dissociated α1NC2-α3NC2 and α2NC2. The kinetically supplied dissociated products behave differently from the complex during sedimentation-diffusion that changes an ideal 1:1 ratio between α1NC2-α3NC2 and α2NC2 due to their different physical properties (α1NC2-α3NC2 is more then twice as heavy as α2NC2 (Table 2)).
Secondary Structure Content and Thermal Transitions
The far UV CD spectra of α123NC2-(GPP)5CC or α123NC2 in buffers with two different pH values are shown in Fig. 8. Notably, they are similar to the spectra reported previously for (GPP)10-containing NC2 or just NC2 of homotrimeric collagen XIX (6). The α123NC2 complex has predominantly an α-helical structure (Fig. 8B), whereas α123NC2-(GPP)5CC demonstrates superimposition of α-helical and collagen triple-helical structures (Fig. 8A). Equimolar subtraction of the α123NC2 spectrum from the α123NC2-(GPP)5CC spectrum and subsequent adjustment of the mean molar ellipticity demonstrate the presence of the collagen triple helical structure (Fig. 8C). It is worth mentioning that the calculated triple helical structure does include a contribution from two stretches of amino acids (i.e. GAS and GPCCGGV) (10 residues in total) and thus cannot be directly compared with known CD spectra of ideal (GPP)n peptides. Moreover, the interface between the triple helix and the cystine knot was shown to deviate from a perfect triple helix (19).
FIGURE 8.

Circular dichroism spectroscopy of the NC2-containing complexes. A, CD spectra of α123NC2-(GPP)5CC recorded in 50 mm sodium phosphate buffer, pH 8 (black circles), and in 50 mm sodium acetate buffer, pH 4.5 (patterned circles), using 7 μm complex concentrations and a 1-mm path length quartz cuvette equilibrated at 20 °C. B, CD spectra of α123NC2 recorded in 50 mm sodium phosphate buffer, pH 8 (black circles), and in 50 mm sodium acetate buffer, pH 4.5 (patterned circles), using 18.7 μm complex concentrations and a 1-mm path length quartz cuvette equilibrated at 20 °C. C, calculated spectra of the collagenous part of α123NC2-(GPP)5CC in two buffers, respectively.
The thermal stability of the complexes was studied at pH 4.5 to prevent disulfide bond reshuffling upon denaturation. Thermal denaturations were also observed at pH 8 with similar transitions upon heating, but refolding curves upon cooling deviated significantly. Transitions at pH 4.5 showed full reversibility and were further analyzed (Fig. 9).
FIGURE 9.

Thermal transitions of the NC2-containing complexes. A, thermal transition curves of α123NC2-(GPP)5CC were recorded in 50 mm sodium acetate buffer, pH 4.5, supplemented with 0 m (green circles), 1 m (red circles for heating and blue circles for cooling), and 2 m (cyan circles) guanidine hydrochloride using 7 μm complex concentrations and a 1-mm path length quartz cuvette. The change in collagen triple helical and α-helical contents was monitored at 230 nm with a scan rate of 1 °C/min. Heating and cooling transition curves are shown for the sample with 1 m guanidine hydrochloride to demonstrate the reversibility of the transition. The first transition shown by a decrease of the CD signal is associated with the unfolding of the collagen triple helix, whereas the second transition is associated with the unfolding of the NC2 domain. B, thermal transition curves of α123NC2 were recorded in 50 mm sodium acetate buffer, pH 4.5, using two complex concentrations, 1.87 (yellow circles) and 18.7 μm (brown circles), and 5- or 1-mm path length quartz cuvettes, respectively. The change in α-helical content was monitored at 222 nm with a scan rate of 0.25 °C/min. The curves were globally fitted (white lines) as described under “Experimental Procedures.”
The α123NC2-(GPP)5CC complex was monitored at 230 nm to maximize a change in the collagen triple helix content upon transitions. Nevertheless, a change in the α-helical content remained significant and allowed us to simultaneously monitor both possible transitions (Fig. 9A). The second transition was not completed at 90 °C (Fig. 9A, green) and required the addition of guanidine hydrochloride to fully resolve both transitions in the available temperature range. The midpoint transition temperature values, Tm, of both transitions were shifted to lower temperature due to the denaturing effect of guanidine hydrochloride. The first transition indicated by a decrease in the signal upon heating is associated with the melting of the collagen triple helix (6) (the Tm values are ∼59, ∼44, or ∼27 °C with 0, 1, or 2 m guanidine hydrochloride, respectively), whereas the second transition is linked to the unfolding of the α-helical NC2 domain (the Tm values are ∼90, ∼78, or ∼65 °C with 0, 1, or 2 m guanidine hydrochloride, respectively). According to the change in the α-helical content (the second transition) in buffer supplemented with either 1 or 2 m guanidine hydrochloride, only about a half of the transition of the NC2 domain was observed in the plain buffer; thus, the Tm value of the NC2 domain in the α123NC2-(GPP)5CC complex is ∼90 °C. The Tm value of the collagen triple helix is ∼59 °C, which demonstrates impressive dual stabilizing effect of the NC2 domain on one side and the cystine knot on the opposite side. Compare this with Tm = 58 °C for the NC2 domain of collagen XIX linked to (GPP)10, NC2(GPP)10 (6), where the stabilizing role of the NC2 domain is possibly the same, but the collagenous part is much longer and lacks the cystine knot.
In contrast to the α123NC2-(GPP)5CC complex, where no dependence on concentration for the Tm values was observed, the melting transitions of the α123NC2 complex showed a remarkable decrease in Tm upon decreasing the concentration (Fig. 9B). This dependence demonstrated that the loss of α-helicity was coupled with the dissociation of α1NC2-α3NC2 and α2NC2. Because the α1NC2 chain is disulfide-linked to the α3NC2 chain, only two products dissociate from the complex upon denaturation, and the folding reaction should be considered as bimolecular. According to this and other assumptions (see “Experimental Procedures”), two transitions using a 10-fold difference in concentrations of the complex were separately globally fitted and yielded similar values of the standard enthalpy, ΔH0, and the standard entropy, ΔS0 (namely for 18.7 μm, Tm = 65.8 °C, ΔH0 = −202.8 kJ/mol of complex, ΔS0 = −502 J/mol of complex K; for 1.87 μm, Tm = 51.2 °C, ΔH0 = −199.6 kJ/mol of complex, ΔS0 = −500 J/mol of complex K. To achieve the Tm value of ∼90 °C observed for the α123NC2-(GPP)5CC complex, the concentration of the α123NC2 complex was estimated to be ∼2 mm based on Equation 7. This again emphasizes the stabilizing role of the cystine knot within the α123NC2-(GPP)5CC complex despite the collagenous sequence separating it from the NC2 domain. Similar stabilizing effects are expected from the natural cystine knots located within the NC3 and NC1 domains.
DISCUSSION
It is shown for the first time that the NC2 domain of the heterotrimeric collagen IX promotes chains selection and trimerization in a highly specific and effective manner. Previous attempts to attribute this role to either COL1 (14) or NC1 (15) showed only small amounts of the heterotrimer formed. The discovery of the heterotrimerization properties for the NC2 domain of collagen IX further extends our finding of the homotrimerization properties for the NC2 domain of collagen XIX (6); the NC2 domain in all FACIT collagens can play a universal role in chain selection, chain registration, initiation of the triple helix formation and its stabilization. Interestingly, single tripeptide unit deletions within the COL1 domain of the α3(IX) chain are known to not co-segregate with any disease phenotype and do not affect the formation of correctly folded heterotrimeric collagen IX, whereas similar deletions in type I collagen are lethal (23). With the primary role of NC2 in the folding initiation of collagen IX, this discrepancy is now eliminated. Nevertheless, the deletion in COL1 should affect the fold of NC1, especially the cystine knot patterning. As in collagen XIX, the NC2 domain of collagen IX is also predominantly α-helical and can represent a special type of α-helical coiled-coil (6) with the unusual heptad repeat patterns found in all FACITs (7).
Availability of an effective collagen-specific heterotrimerization domain with three distinct chains opens the prospect of easy production of short native collagen fragments with chain composition control. There is a possibility that in addition to chain selectivity, the NC2 domain of collagen IX can determine a stagger of the collagen triple helix, but further experiments are required. Acquiring a three-dimensional structure of the NC2 domain would be of great importance to this study; thus, crystallization trials are in progress. Due to the heterotrimeric nature of the complex, NMR techniques are also applicable for structure determination. The study of the detailed mechanism of chain selection under reducing conditions and disulfide bond formation is in progress. Whereas the present study addressed the heterotrimerization properties of the NC2 domain of collagen IX under conditions where equimolar amounts of all three chains were present, other conditions with no or significantly decreased levels of any one or two chains should be studied to cover different pathologies. Preliminary results showed that α1NC2 was able to form a homotrimer with the prominent α-helical content, although thermal denaturation experiments did not reveal a cooperative unfolding transition, indicating a more labile structure. Further detailed and accurate studies have to be performed on individual α1NC2 and other individual or mixed NC2 chains.
A recent study of different deletion mutants of collagen IX expressed in a baculovirus system showed that α chains can associate in the absence of COL1 and NC1, although the COL1-NC1 region is important for chain specificity (16). Another conclusion was that the COL2-NC2 region alone is not sufficient for trimerization (16), which directly contradicts our present finding of the role of the NC2 domain. It is worth mentioning that even for the full-length chains, the yield of heterotrimeric collagen in the baculovirus system was no more than 10% of total chain production, with most of material trapped in monomers and dimers as analyzed on a denaturing gel under non-reducing conditions (16, 21). This indicates protein misfolding that leads to aggregation. Whereas the heterotrimerization of “successful candidates” was assessed by monitoring a small fraction of the disulfide-linked heterotrimer (due to the cystine knots in NC3 and/or NC1 (22)), the “unsuccessful candidate,” COL2-NC2 (which lacked any cystine knot), was initially treated with a covalent cross-linker, BS3, to stabilize oligomers, but only monomeric, dimeric, or aggregated molecules with very high molecular mass, and not trimers, were detected (16). We believe that the heterotrimer formation for COL2-NC2 fragments was not detected simply due to overall problems with folding and possible aggregation that hindered the covalent fixation of trimers using BS3. Unfortunately, the data are not shown for the NC2-COL2 fragment, neither a non-reducing gel (that could demonstrate or not the single disulfide bond formation within the NC2 domain between cysteines of α1 or α3) nor a gel with BS3 fixed products.
Collagen IX was shown to be cleaved by MMP-3 (stromelysin-1) (24), and the cleavage site was later identified within the NC2 domain (17). Elevated levels of MMP-3 were observed in synovial fluid of patients with both rheumatoid arthritis and osteoarthritis (25). Taking into account the role of the NC2 domain in trimerization and triple helix stabilization, the MMP-3 cleavage must greatly destabilize the collagen IX molecule and make it prone to further degradation by other proteases (gelatinases). Loss of collagen IX is linked with loss of the structural integrity of the cartilage and development of a severe degenerative joint disease resembling human osteoarthritis (9). Additionally, because collagen IX molecules are localized on the surface of collagen II-containing fibrils (26, 27), their loss will render naked collagen II fibrils more accessible for MMPs, which can induce the cascade of events leading to arthritis (25). In the case of rheumatoid arthritis, a role of collagen II fragments in generating the autoimmune response is well established (28). Moreover, patients with recent onset rheumatoid arthritis have significantly elevated levels of autoantibodies to collagen IX (29), possibly indicating the antigenicity of degraded collagen IX fragments as well. MMP-3 cleavage experiments using the isolated recombinant NC2 domain are in progress. An experimental system for testing small molecules as potential protectors of the NC2 domain (but not general inhibitors of MMP-3) against the MMP-3 cleavage can now be developed.
Cartilage oligomeric matrix protein (COMP) is a pentameric glycoprotein found in the extracellular matrix of cartilage (30), tendon (31), and ligament, where it is thought to play an important role in tissue development and homeostasis through interactions with cells (32) and collagens I and II (33). Mutations in COMP or collagen IX are known to result in phenotypes within the multiple epiphyseal dysplasia disease spectrum and suggested their interaction. Indeed, COMP was shown to interact with collagen IX, and the binding sites were mapped to regions within or close to all four NC domains of collagen IX (34, 35). Statistical analysis of COMP binding sites along collagen IX molecules using electron microscopy showed the highest frequency of occupation of the NC2 domain in the long isoform of collagen IX from cartilage and an even higher frequency for the short isoform (lacking the NC4 domain) from vitreous (34). The isolated heterotrimeric collagen IX NC2 domain is now available for COMP binding experiments. Moreover, taking into account the small size of the NC2 domain, there might be an interesting competing interplay between COMP binding and MMP-3 cleavage. The chain selection and heterotrimerization properties of the NC2 domain can be used as a tool for a recombinant production of the correctly folded NC3 and NC1 domains, the other binding candidates for COMP.
Glycosaminoglycans play important roles in cell adhesion and extracellular matrix assembly. A remarkable binding of heparin to collagen IX was reported (36) and further analyzed (37, 38). The full-length recombinant collagen IX has an apparent Kd of 3.6 nm for the heparin binding, and electron microscopy suggests the presence of four heparin-binding sites located within or near all four NC domains (37). The heparin-binding ability of the NC4 domain was found to be rather moderate with a Kd of 0.6 μm (37), which emphasized the importance of other heparin-binding sites along the molecule. Whereas the NC4 domain is a product of the single α1 chain, all other domains are heterotrimeric, and their production will again require the usage of the NC2 domain.
The binding affinity of the matrilin-3 A-domain for type IX collagen was shown to be a few nm, and the binding site was mapped to the amino-terminal part of COL3 (39). Detailed structural insight into this interaction can now be gained by an adequate design of heterotrimeric collagenous peptides spanning COL3 using the folding role of the NC2 domain.
This work was supported by a grant from the Shriners Hospital for Children.
- FACIT
- fibril-associated collagen with interrupted triple helices
- MMP
- matrix metalloproteinase
- NTA
- nitrilotriacetic acid
- HPLC
- high pressure liquid chromatography
- MS
- mass spectrometry
- COMP
- cartilage oligomeric matrix protein
- MOPS
- 4-morpholinepropanesulfonic acid
- MES
- 4-morpholineethanesulfonic acid
- BS3
- Bis[sulfosuccinimidyl] suberate.
REFERENCES
- 1.Myllyharju J., Kivirikko K. I. (2004) Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 2.Ricard-Blum S., Ruggiero F. (2005) Pathol. Biol. 53, 430–442 [DOI] [PubMed] [Google Scholar]
- 3.Mazzorana M., Gruffat H., Sergeant A., van der Rest M. (1993) J. Biol. Chem. 268, 3029–3032 [PubMed] [Google Scholar]
- 4.Lesage A., Penin F., Geourjon C., Marion D., van der Rest M. (1996) Biochemistry 35, 9647–9660 [DOI] [PubMed] [Google Scholar]
- 5.Mazzorana M., Cogne S., Goldschmidt D., Aubert-Foucher E. (2001) J. Biol. Chem. 276, 27989–27998 [DOI] [PubMed] [Google Scholar]
- 6.Boudko S. P., Engel J., Bächinger H. P. (2008) J. Biol. Chem. 283, 34345–34351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAlinden A., Smith T. A., Sandell L. J., Ficheux D., Parry D. A., Hulmes D. J. (2003) J. Biol. Chem. 278, 42200–42207 [DOI] [PubMed] [Google Scholar]
- 8.Eyre D. R., Wu J. J. (1995) J. Rheumatol. Suppl. 43, 82–85 [PubMed] [Google Scholar]
- 9.Fässler R., Schnegelsberg P. N., Dausman J., Shinya T., Muragaki Y., McCarthy M. T., Olsen B. R., Jaenisch R. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 5070–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diab M. (1993) Orthop. Rev. 22, 165–170 [PubMed] [Google Scholar]
- 11.Asamura K., Abe S., Imamura Y., Aszodi A., Suzuki N., Hashimoto S., Takumi Y., Hayashi T., Fässler R., Nakamura Y., Usami S. (2005) Neuroscience 132, 493–500 [DOI] [PubMed] [Google Scholar]
- 12.Boyd L. M., Richardson W. J., Allen K. D., Flahiff C., Jing L., Li Y., Chen J., Setton L. A. (2008) Arthritis Rheum. 58, 164–171 [DOI] [PubMed] [Google Scholar]
- 13.Carter E. M., Raggio C. L. (2009) Curr. Opin. Pediatr. 21, 46–54 [DOI] [PubMed] [Google Scholar]
- 14.Labourdette L., van der Rest M. (1993) FEBS Lett. 320, 211–214 [DOI] [PubMed] [Google Scholar]
- 15.Mechling D. E., Gambee J. E., Morris N. P., Sakai L. Y., Keene D. R., Mayne R., Bächinger H. P. (1996) J. Biol. Chem. 271, 13781–13785 [DOI] [PubMed] [Google Scholar]
- 16.Jäälinoja J., Ylöstalo J., Beckett W., Hulmes D. J., Ala-Kokko L. (2008) Biochem. J. 409, 545–554 [DOI] [PubMed] [Google Scholar]
- 17.Wu J. J., Lark M. W., Chun L. E., Eyre D. R. (1991) J. Biol. Chem. 266, 5625–5628 [PubMed] [Google Scholar]
- 18.Kammerer R. A., Schulthess T., Landwehr R., Lustig A., Fischer D., Engel J. (1998) J. Biol. Chem. 273, 10602–10608 [DOI] [PubMed] [Google Scholar]
- 19.Boudko S. P., Engel J., Okuyama K., Mizuno K., Bächinger H. P., Schumacher M. A. (2008) J. Biol. Chem. 283, 32580–32589 [DOI] [PubMed] [Google Scholar]
- 20.Boudko S. P., Engel J. (2004) J. Mol. Biol. 335, 1289–1297 [DOI] [PubMed] [Google Scholar]
- 21.Pihlajamaa T., Perälä M., Vuoristo M. M., Nokelainen M., Bodo M., Schulthess T., Vuorio E., Timpl R., Engel J., Ala-Kokko L. (1999) J. Biol. Chem. 274, 22464–22468 [DOI] [PubMed] [Google Scholar]
- 22.Bruckner P., Mendler M., Steinmann B., Huber S., Winterhalter K. H. (1988) J. Biol. Chem. 263, 16911–16917 [PubMed] [Google Scholar]
- 23.Paassilta P., Pihlajamaa T., Annunen S., Brewton R. G., Wood B. M., Johnson C. C., Liu J., Gong Y., Warman M. L., Prockop D. J., Mayne R., Ala-Kokko L. (1999) J. Biol. Chem. 274, 22469–22475 [DOI] [PubMed] [Google Scholar]
- 24.Okada Y., Konomi H., Yada T., Kimata K., Nagase H. (1989) FEBS Lett. 244, 473–476 [DOI] [PubMed] [Google Scholar]
- 25.Burrage P. S., Mix K. S., Brinckerhoff C. E. (2006) Front. Biosci. 11, 529–543 [DOI] [PubMed] [Google Scholar]
- 26.Vaughan L., Mendler M., Huber S., Bruckner P., Winterhalter K. H., Irwin M. I., Mayne R. (1988) J. Cell Biol. 106, 991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Rest M., Mayne R. (1988) J. Biol. Chem. 263, 1615–1618 [PubMed] [Google Scholar]
- 28.Rowley M. J., Nandakumar K. S., Holmdahl R. (2008) Mod. Rheumatol. 18, 429–441 [DOI] [PubMed] [Google Scholar]
- 29.Jäälinoja J., Nissilä M., Kauppi M. J., Hakala M., Laiho K., Karttunen R., Hörkkö S., Ala-Kokko L. (2008) J. Rheumatol. 35, 745–751 [PubMed] [Google Scholar]
- 30.Hedbom E., Antonsson P., Hjerpe A., Aeschlimann D., Paulsson M., Rosa-Pimentel E., Sommarin Y., Wendel M., Oldberg A., Heinegård D. (1992) J. Biol. Chem. 267, 6132–6136 [PubMed] [Google Scholar]
- 31.DiCesare P., Hauser N., Lehman D., Pasumarti S., Paulsson M. (1994) FEBS Lett. 354, 237–240 [DOI] [PubMed] [Google Scholar]
- 32.Mörgelin M., Heinegård D., Engel J., Paulsson M. (1994) Biophys. Chem. 50, 113–128 [DOI] [PubMed] [Google Scholar]
- 33.Rosenberg K., Olsson H., Mörgelin M., Heinegård D. (1998) J. Biol. Chem. 273, 20397–20403 [DOI] [PubMed] [Google Scholar]
- 34.Holden P., Meadows R. S., Chapman K. L., Grant M. E., Kadler K. E., Briggs M. D. (2001) J. Biol. Chem. 276, 6046–6055 [DOI] [PubMed] [Google Scholar]
- 35.Thur J., Rosenberg K., Nitsche D. P., Pihlajamaa T., Ala-Kokko L., Heinegård D., Paulsson M., Maurer P. (2001) J. Biol. Chem. 276, 6083–6092 [DOI] [PubMed] [Google Scholar]
- 36.Munakata H., Takagaki K., Majima M., Endo M. (1999) Glycobiology 9, 1023–1027 [DOI] [PubMed] [Google Scholar]
- 37.Pihlajamaa T., Lankinen H., Ylöstalo J., Valmu L., Jäälinoja J., Zaucke F., Spitznagel L., Gösling S., Puustinen A., Mörgelin M., Peränen J., Maurer P., Ala-Kokko L., Kilpelaïnen I. (2004) J. Biol. Chem. 279, 24265–24273 [DOI] [PubMed] [Google Scholar]
- 38.Leppänen V. M., Tossavainen H., Permi P., Lehtiö L., Rönnholm G., Goldman A., Kilpelaïnen I., Pihlajamaa T. (2007) J. Biol. Chem. 282, 23219–23230 [DOI] [PubMed] [Google Scholar]
- 39.Fresquet M., Jowitt T. A., Ylöstalo J., Coffey P., Meadows R. S., Ala-Kokko L., Thornton D. J., Briggs M. D. (2007) J. Biol. Chem. 282, 34634–34643 [DOI] [PMC free article] [PubMed] [Google Scholar]