

Abstract
Eosinophil peroxidase (EPO) is an abundant heme protein in eosinophils that catalyzes the formation of cytotoxic oxidants implicated in asthma, allergic inflammatory disorders, and cancer. It is known that some proteins with peroxidase activity (horseradish peroxidase and prostaglandin hydroperoxidase) can catalyze oxidation of bisulfite (hydrated sulfur dioxide), leading to the formation of sulfur trioxide anion radical (·SO3−). This free radical further reacts with oxygen to form peroxymonosulfate anion radical (−O3SOO·) and the very reactive sulfate anion radical (SO4˙̄), which is nearly as strong an oxidant as the hydroxyl radical. However, the ability of EPO to generate reactive sulfur radicals has not yet been reported. Here we demonstrate that eosinophil peroxidase/H2O2 is able to oxidize bisulfite, ultimately forming the sulfate anion radical (SO4˙̄), and that these reactive intermediates can oxidize target proteins to protein radicals, thereby initiating protein oxidation. We used immuno-spin trapping and confocal microscopy to study protein oxidation by EPO/H2O2 in the presence of bisulfite in a pure enzymatic system and in human promyelocytic leukemia HL-60 clone 15 cells, maturated to eosinophils. Polyclonal antiserum raised against the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) detected the presence of DMPO covalently attached to the proteins resulting from the DMPO trapping of protein free radicals. We found that sulfite oxidation mediated by EPO/H2O2 induced the formation of radical-derived DMPO spin-trapped human serum albumin and, to a lesser extent, of DMPO-EPO. These studies suggest that EPO-dependent oxidative damage may play a role in tissue injury in bisulfite-exacerbated eosinophilic inflammatory disorders.
Keywords: Enzymes/Oxidation-Reduction, Methods/Confocal Microscopy, Methods/Electron Paramagnetic Resonance EPR, Oxygen/Radicals, Protein/Metalloproteins, Radicals, Spin Trapping
Introduction
Sulfur dioxide, formed during the combustion of fossil fuels, is a major air pollutant near large cities (1). It can be hydrated to bisulfite in the lung and upon contact with fluids lining the air passages as follows,
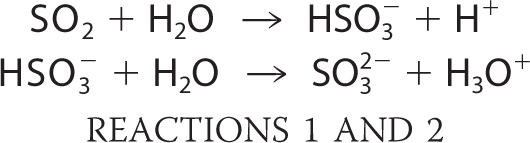 |
where the second pKa = 7.2 (2).
Its two ionized forms in aqueous solution at physiological pH, bisulfite and sulfite (3), are widely used as preservatives and antioxidants to prevent food and beverage spoilage, as bleaching agents for certain products, and as medicine ingredients (4). Bisulfite is used when not referring to a particular sulfite species. Sulfite is generated endogenously during the normal metabolic processing of sulfur-containing amino acids (5). Sulfite is toxic to the lung and can cause allergic reactions in humans, especially in sulfite-hypersensitive individuals, most commonly causing bronchoconstriction in asthmatics (6). It is detoxified to sulfate by sulfite oxidase (7), and deficiency of this enzyme in humans is fatal. Sulfite oxidase is present at high levels in the liver and in lower concentrations in most of the other tissues of the body (e.g. in the lung). The enzymatic oxidation of sulfite proceeds via a two-electron oxidation, but recent studies suggest that the cytotoxicity of bisulfite is mediated by free radicals (8). In fact, free radicals have been demonstrated to be produced by enzymatic initiation of the oxidation of bisulfite by prostaglandin H synthase (9) and horseradish peroxidase (10, 11), with formation of the ·SO3− anion radical as follows:
where k = 4.3 × 107 m−1 s−1 (12), and
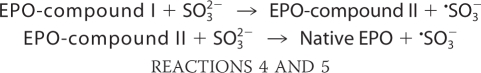 |
The sulfite anion radical reacts very rapidly with oxygen and gives rise to the formation of peroxymonosulfate (−O3SOO·) and sulfate (SO4˙̄) anion radicals through chain propagation steps as follows:
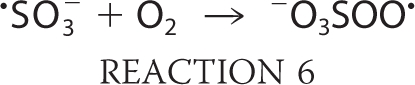 |
where k = 1.5 × 109 m−1 s−1 (13);
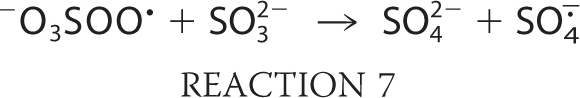 |
where k = 1.3 × 107 m−1 s−1 (13);
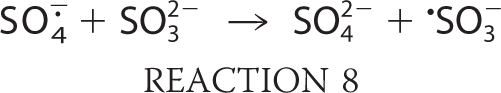 |
where k > 2 × 109 m−1 s−1 (13).
The oxygen transfer reaction of −O3SOO· to form SO4˙̄ is a general reaction of peroxyl radicals and is not limited to oxygen donation to sulfite as in Reaction 7 (2, 14, 15). Furthermore, SO4˙̄ can react with another molecule of bisulfite via Reaction 8, but, being a very strong oxidant (13), it will oxidize almost any biomolecules. Despite the proclivity of peroxidases to serve as initiators in the generation of reactive sulfur-derived species, we can find no reports of bisulfite oxidation mediated by any mammalian peroxidase. Eosinophil peroxidase (EPO)3 is an abundant protein secreted from activated eosinophils (16–18), white blood cells that play a central role in host defense mechanisms, allergic reactions, and asthma (19–24). EPO is a monomer of 70 kDa, composed of heavy and light chains with molecular masses of 55 and 15 kDa, respectively. EPO and the other members of mammalian peroxidase superfamily II have the unique ability to use halides and pseudohalides at plasma levels (SCN−, 20–120 μm; Br−, 20–100 μm; I−, <1 μm; Cl−, 100 mm) to generate hypohalous acids via the reaction H2O2 + X− + H+ → HOX + H2O, where X− represents SCN−, Br−, I−, or Cl−, and HOX is the corresponding hypohalous acid (12, 23–25). Studies thus far on EPO have focused primarily on its preference to oxidize these physiological substrates through a two-electron oxidation pathway. Recently, it has been demonstrated that nitric oxide can serve as a substrate for EPO and compete with plasma levels of bromide, steering the enzyme reaction from a two-electron oxidation to a one-electron oxidation pathway by generation of reactive nitrogen species (25). In a model of nitrite oxidation (24), EPO has been reported to cause nitrotyrosine formation in vivo (26).
We now demonstrate that EPO uses bisulfite as a one-electron donor substrate to generate reactive intermediates that oxidize the most abundant protein present in plasma, albumin (HSA), to protein radicals via Reactions 3–8. In our study, we used immuno-spin trapping with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (27) and confocal miscroscopy to investigate protein radical formation in the reaction of bisulfite with EPO-H2O2 in cells.
EXPERIMENTAL PROCEDURES
Chemicals
Human EPO was purified from white blood cells (Lee Biosolutions Inc., St. Louis, MO). The purity of the enzyme was assured by the purity ratio (RZ) > 1.0 (A413/A280) and by SDS-PAGE analysis with Coomassie Blue staining. The concentration of the enzyme was calculated using an extinction coefficient of 112 mm−1 cm−1 at 413 nm (28). Human serum albumin (99.99% purity), diethylenetriaminepentaacetic acid, sodium sulfite, sodium thiocyanate, sodium bromide, sodium chloride, sodium iodide, ascorbic acid, glutathione, cysteine, methionine, melatonin, homovanillic acid (HVA), and hydrogen peroxide (obtained as a 30% solution) were from Sigma. The hydrogen peroxide concentration was determined from its absorbance at 240 nm (ϵ = 39.4 m−1 cm−1). DMPO (high purity, ≥99%) from Alexis Biochemicals (San Diego, CA) was sublimed twice under vacuum at room temperature and stored under an argon atmosphere at −80 °C before use. Chelex-100 resin was purchased from Bio-Rad.
Electron Spin Resonance (ESR) Spectroscopy
ESR spectra were obtained at room temperature using a Bruker EMX spectrometer with 100-kHz modulation frequency equipped with an ER 4122 SHQ cavity. Samples were placed in a 10-mm flat cell (200-μl final volume), and recording of the spectra was initiated within 1 min of the start of the reaction. The ESR spectrometer settings were as follows: modulation amplitude, 0.4 G; scan range, 80 G; microwave power, 20 milliwatts; receiver gain, 5 × 104; time constant, 327.68 ms; sweep time, 335.544 s; frequency, 9.80 GHz. The simulation was performed using WinSim version 1.0 software (29).
Kinetic Experiments
Kinetic experiments were carried out with a Cary 100 spectrophotometer (Varian Inc., Palo Alto, CA) using a 500-μl quartz cuvette. Reactions were performed in 100 mm phosphate buffer (Chelex-treated with 25 μm diethylenetriaminepentaacetic acid) at pH 7.4. For reduction of compound II, 1.5 μm EPO was premixed with 1.3 μm HVA and 15 μm hydrogen peroxide. Forty seconds after the mixing, compound II was allowed to react with bisulfite. Pseudo-first order conditions were achieved by keeping the bisulfite concentration at a ≥5-fold excess over the enzyme.
Oxygen Uptake
For oxygen uptake measurements, sodium sulfite and hydrogen peroxide were mixed in a 2-ml chamber equipped with a Clark electrode and a stirrer. The reaction mixture (1.8 ml) was initiated by EPO, and the oxygen uptake curves were obtained at room temperature with an oxygen monitor (YSI Inc., Yellow Springs, OH). The oxygen polarographic electrode was calibrated by the depletion of oxygen during the oxidation of hypoxanthine in the presence of xanthine oxidase and catalase (30).
Chemical Reactions
Typically, reactions of 600 μm HSA, 2 mm Na2SO3, 100 μm H2O2, and 1 μm EPO were carried out in the presence or absence of 1 mm DMPO in 100 mm phosphate buffer (Chelex-treated with 25 μm diethylenetriaminepentaacetic acid) at pH 7.4 in a total volume of 30 μl. After 1 h of incubation at 37 °C, samples were then diluted with deionized H2O for electrophoresis and immuno-spin trapping analyses.
Enzyme-linked Immunosorbent Assay (ELISA)
Rabbit anti-DMPO polyclonal serum, developed in our laboratory, was used for the development of the immunoassays (31–33). The DMPO-HSA nitrone adducts were determined using a standard ELISA in 96-well plates (Greiner Labortechnik, Frickenhausen, Germany) as described previously (34). Optimal concentrations were determined by varying the concentrations of HSA, Na2SO3, H2O2, and DMPO one at a time.
Coomassie Blue Stain and Western Blot
Reaction mixtures were electrophoresed under reducing conditions through duplicate 4–12% BisTris NuPage acrylamide gels (Invitrogen). After electrophoresis, one gel was stained using Coomassie Blue, and the proteins in the other were blotted to a nitrocellulose membrane. Western blotting was performed as described with minor changes (35). In experiments to analyze HSA and EPO immunostain, rabbit polyclonal anti-HSA (Abcam, Cambridge, MA) and anti-EPO (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used at a dilution of 1:10,000, and the secondary antibody used was an alkaline phosphatase-conjugated anti-rabbit IgG (Pierce).
Cell Culture and Treatment
HL-60 (clone 15) cells (ATCC, Manassas, VA) were grown in RPM1–1640 medium supplemented with 10% fetal bovine serum and optimal antibiotics. The cultures were treated with 0.5 mm butyric acid for 5 days for their differentiation into mature eosinophils. The cells were harvested and checked for viability by trypan blue dye exclusion. Harvested 2 × 106 cells/ml of viable cells were seeded in a 24-well culture plate and incubated with DMPO, bisulfite, and 5 mm glucose plus 50 milliunits/ml glucose oxidase (G/GOx) for 1 h. This concentration initially produced 5 μm H2O2/min and required mixing because the reaction is oxygen-dependent. Following incubation, the cells were washed once with PBS and lysed using radioimmune precipitation buffer containing proteinase inhibitors. The cell lysates were used immediately or stored at −80 °C until further use.
Confocal Microscopy
For confocal microscopy studies, treated HL-60 (clone 15) cells were allowed to adhere on polylysine-coated plates (MatTek, Ashland, MA) for 30 min on ice. The cells were fixed with 0.4% paraformaldehyde and permeabilized with 0.01% Surfact-Amps X-100 (Thermo Scientific, Waltham, MA). After blocking with 0.1% bovine serum albumin in phosphate-buffered saline, EPO and DMPO-nitrone adducts were stained using primary anti-EPO (mouse host) (Abcam, Cambridge, MA) and anti-DMPO (rabbit host) antisera. After a 1-h incubation, the cells were washed, and secondary Alexafluor anti-mouse and Alexafluor anti-rabbit conjugated with fluorescein (Invitrogen) were added.
RESULTS
ESR, Optical Spectroscopy, and Oxygen Uptake
Incubations of bisulfite (500 μm) with eosinophil peroxidase (3 μm) plus H2O2 (3 μm) in the presence of DMPO (100 mm) yielded an ESR spectrum of a sulfur trioxide anion radical (·SO3−) radical adduct (Fig. 1A). The hyperfine coupling constants of the assigned DMPO/·SO3− radical adduct (aN = 14.7 G and aβH = 16.0 G) are consistent with previous reports (9, 15). When EPO, bisulfite, or H2O2 was omitted, no radical adduct was formed (Fig. 1, spectra C, D, and E, respectively).
FIGURE 1.

Formation of radical adducts in reaction between sulfite (Na2SO3) and EPO/hydrogen peroxide (H2O2) in the presence of DMPO. Spectrum A, reaction mixture containing Na2SO3 (0.5 mm), DMPO (100 mm), and H2O2 (3 μm) in 100 mm phosphate buffer, pH 7.4. After initiation with EPO (3 μm), the mixture was immediately placed into the flat cell. Spectrum B, simulation of DMPO/·SO3− radical adduct (aβH = 16.0 G and aN = 14.7 G). Spectrum C, same as spectrum A without EPO. Spectrum D, same as spectrum A except H2O2 was not added. Spectrum E, same as spectrum A without Na2SO3.
The proposed mechanism of enzymatic oxidation of bisulfite to the ·SO3− radical by the eosinophil peroxidase/H2O2 system proceeds in two sequential, one-electron reduction reactions of compounds I and II by sulfite, similar to the oxidation of bisulfite by horseradish peroxidase and prostaglandin H synthase (9, 11, 15, 36). To determine the reduction of EPO compound II by bisulfite (compound I of eosinophil peroxidase has been reported to be very unstable (12)), EPO was premixed with a substoichiometric concentration of HVA and a 10-fold excess of hydrogen peroxide. Under these conditions, a spectrum of compound II was obtained with peaks at 433, 537, and 565 nm (Fig. 2A), and the corresponding time trace (at 433 nm) clearly indicated that compound II was stable for at least 1 min (Fig. 2B, inset). It has been shown that HVA readily reduces compound I but is an extremely poor substrate for compound II (37). Consequently, we premixed EPO (1.5 μm), HVA (1.3 μm), and hydrogen peroxide (15 μm) to generate compound II, and after 40 s, we then followed its reaction with various concentrations of bisulfite. The corresponding spectral changes indicated a transition of compound II to ferric EPO (with peaks at 413, 500, and 639 nm) (Fig. 2A). For each sulfite concentration, the loss of absorbance at 433 nm displayed monoexponential character. Typical time traces for the reaction of sulfite with compound II are shown in the inset of Fig. 2B. The pseudo-first-order rate constants (kobs) were obtained from these traces and plotted against the concentration of bisulfite (Fig. 2B). The slope of this secondary plot yielded the second-order rate constant for the reduction of compound II at pH 7.4. It was calculated to be (2.1 ± 0.6) × 102 m−1 s−1. The calculated rate is very similar to that reported for horseradish peroxidase. The second-order rate constants for the reactions between HRP compounds I and II with bisulfite at neutral pH are (7.6 ± 0.8) × 10 m−1 s−1 and (1.8 ± 0.06) × 102 m−1 s−1, respectively (36). Although these reactions are relatively slow, only 1.4 × 10−13 m ·SO3− is necessary to propagate this enzymatically initiated, free radical chain reaction (38).
FIGURE 2.

Reduction of EPO compound II by sulfite. A, spectral changes upon the addition of 15 μm Na2SO3 to compound II. EPO compound II was formed by mixing 1.5 μm ferric EPO with 1.3 μm homovanillic acid (HVA) and 15 μm H2O2 and waiting for 40 s. The first spectrum was taken 10 s after mixing, and subsequent spectra were taken at 2, 10, and 15 min. The arrows show the direction of absorbance changes with time. B, pseudo-first-order rate constants for reduction of compound II by sulfite. The second-order rate constant is calculated from the slope. The inset shows the time traces and fits of the reduction of compound II at pH 7.4 by Na2SO3. The concentrations of sulfite for each time trace were 0 μm (a), 7.5 μm (b), 15 μm (c), 30 μm (d), and 75 μm (e).
The resulting sulfur trioxide anion radical (·SO3−) is known to react further with molecular oxygen and form peroxymonosulfate (−O3SOO·) and sulfate (SO4˙̄) anion radicals in the free radical chain mechanism reported previously (2, 14, 15). We therefore investigated the consumption of oxygen by this system. When the primary ·SO3− radical reacted with oxygen in the absence of DMPO (Fig. 3A), the consumption of oxygen increased strongly with the bisulfite concentration, with 200 μm bisulfite notably stimulating oxygen uptake (Fig. 3A, trace b) and causing total oxygen consumption within 2 min at 2 mm bisulfite (Fig. 3A, trace e). When we examined the DMPO concentration dependence of oxygen consumption experiments using 2 mm bisulfite, 100 μm H2O2, and 50 nm EPO as the initiator, the prior addition of 100 mm DMPO (the same amount used for the ESR data) almost completely prevented oxygen uptake (Fig. 3B, trace a) (i.e. there were no radical chain reactions forming −O3SOO· and SO4˙̄ radicals. At lower DMPO concentrations of 0.3 mm and 1 mm DMPO, oxygen uptake was inhibited after 10 min by ∼40 and ∼60%, respectively. The ESR detection of the DMPO/·SO3− radical adduct by spin trapping in aerobic conditions and oxygen uptake experiments indicates that there is a strong competition between the spin trap and the oxygen for the primary ·SO3− radical. In order to allow the radical chain reaction to proceed, the concentration of DMPO has to be decreased so that a significant fraction of the primary ·SO3− radical is not trapped by DMPO, allowing its reaction with oxygen to produce secondary −O3SOO· and tertiary SO4˙̄ radicals via Reactions 6–8. This result demonstrates that to trap protein radicals formed by −O3SOO· and/or SO4˙̄ will require DMPO concentrations of 10 mm or less (Scheme 1).
FIGURE 3.

Oxygen uptake curves in the EPO-H2O2-bisulfite system. A, oxygen uptake as a function of sulfite concentration. Na2SO3 was placed in a chamber with 100 μm hydrogen peroxide in 100 mm phosphate buffer, pH 7.4, and the reaction was initiated with 50 nm EPO at the times indicated. The concentrations of sodium sulfite for each curve were 0 mm (a), 0.2 mm (b), 0.5 mm (c), 1 mm (d), 2 mm (e), and 20 mm (f). B, oxygen uptake as a function of DMPO concentration. DMPO was placed in a chamber with sodium sulfite (2 mm) and hydrogen peroxide (100 μm) in 100 mm phosphate buffer, pH 7.4, and the reaction was initiated with 50 nm EPO at the times indicated. The concentrations of DMPO for each curve were 100 mm (a), 10 mm (b), 1 mm (c), and 0.3 mm (d).
SCHEME 1.

Proposed mechanism of protein oxidative damage induced by the eosinophil peroxidase-H2O2-bisulfite system.
Formation of HSA-DMPO Nitrone Adducts Induced by the Eosinophil Peroxidase-H2O2-Bisulfite System as Determined by Immuno-spin Trapping
To characterize the ability of the SO4˙̄ radical (or possibly other sulfite-derived radicals such as −O3SOO·) to oxidize target proteins, the most abundant plasma protein (human serum albumin) was incubated with the complete eosinophil peroxidase-H2O2-bisulfite system in the presence of DMPO, and the reaction products were analyzed by Western blotting using an anti-DMPO polyclonal antibody. Initially, samples containing 600 μm HSA (plasma concentration) were mixed with 2 mm bisulfite and 100 μm H2O2 in the absence or presence of different concentrations of DMPO, and the reactions were initiated by the addition of 1 μm eosinophil peroxidase. The reaction products were then subjected to gel electrophoresis and Western blot. Coomassie Blue staining verified equal amounts of HSA in all treatments and showed the presence of a single band at 60 kDa due to albumin together with a small amount of HSA dimer at ∼120 kDa (Fig. 4A). No additional bands were observed, indicating that at 1 μm, eosinophil peroxidase was undetectable.
FIGURE 4.

Concentration-dependent effects of DMPO on the formation of HSA radical-derived nitrone adducts. A, Coomassie Blue staining. B, anti-DMPO immunostain as shown with Western blot. Reactions, including HSA (600 μm), H2O2 (100 μm), Na2SO3 (2 mm), and DMPO as indicated, were initiated with 1 μm EPO, and the mixtures were incubated for 1 h at 37 °C in 100 mm phosphate buffer (pH 7.4). Each lane contained 3.8 μg of HSA.
Immunochemical detection of HSA-DMPO nitrone adducts was also performed (Fig. 4B). Samples lacking eosinophil peroxidase, H2O2, or Na2SO3 contained negligible anti-DMPO cross-reacting material. Incubation of HSA with 0.1 or 0.3 mm DMPO produced nitrone adducts at background levels, whereas increasing the DMPO concentration up to 1 mm resulted in a significant increase in HSA-DMPO-derived nitrone adducts. Further increases in the spin trap concentration up to 3 mm produced modest increases in adduct formation, whereas 10 mm caused a decrease. The addition of 100 mm DMPO totally inhibited DMPO-nitrone adduct production, preventing the radical chemistry in Reactions 6–8 by trapping the primary ·SO3− radical, thus diminishing the formation of the damaging radical intermediates.
Production of HSA-derived nitrone adducts also depended on Na2SO3 concentration (supplemental Fig. 1A). When 1 and 10 μm bisulfite and 600 μm albumin were oxidized in the presence of 1 mm DMPO and EPO (1 μm)/H2O2 (100 μm), no DMPO-nitrone adducts were detected. Western blotting performed on reactions containing from 100 μm to 2 mm bisulfite showed increased production of DMPO-HSA radical-derived nitrone adducts and faint bands of DMPO-HSA dimer at the higher bisulfite concentrations.
The effect of increasing concentrations of H2O2 was also determined (supplemental Fig. 1B). Omission of HSA, DMPO, or H2O2 resulted in no immunostaining above the background level. In the presence of 1 mm DMPO, 2 mm Na2SO3, and eosinophil peroxidase (1 μm), 10 μm H2O2 produced DMPO-HSA radical-derived nitrone adducts detectable by Western blot. Although the addition of smaller amounts of H2O2 had no observable effect, 50 and 100 μm H2O2 significantly increased production of DMPO-nitrone adducts, which appeared as HSA monomers and, in the case of 100 μm H2O2, HSA dimers.
Eosinophil Peroxidase-catalyzed Bisulfite Oxidation Results in HSA Fragmentation
As shown in Fig. 5A, the omission of HSA resulted in no staining on the gel, whereas the sample not treated with eosinophil peroxidase confirmed the presence of the single HSA band at 60 kDa together with a small amount of a 120-kDa dimer. Incubation of HSA with 5 and 10 μm EPO resulted in visualization of the heavy chain of EPO (∼55 kDa). The light chain (∼15 kDa) of the peroxidase was much less pronounced but still visible on the stained gel. Anti-EPO immunodetection of the same samples indicated the presence of eosinophil peroxidase (Fig. 5B), exhibiting two bands corresponding to the heavy and light chains.
FIGURE 5.

Concentration-dependent effects of eosinophil peroxidase on the formation of HSA radical-derived nitrone adducts. A, Coomassie Blue staining. B, anti-EPO Western blotting. C, anti-DMPO Western blotting. D, anti-HSA Western blotting. Reactions, including HSA (600 μm), Na2SO3 (2 mm), DMPO (1 mm), and H2O2 (100 μm), were initiated with EPO as indicated, and the mixtures were incubated for 1 h at 37 °C in 100 mm phosphate buffer (pH 7.4). Each lane contained 3.8 μg of HSA.
When the samples were analyzed with anti-DMPO, no immunostain was observed in the controls (without HSA or EPO) except for a faint band in the sample without EPO (Fig. 5C). When the EPO concentration was increased from 1 to 5 μm, anti-DMPO staining also increased. With 10 μm EPO, a sharp increase in staining and drastic oxidative damage to HSA were observed. DMPO-HSA radical-derived nitrone adducts were observed not only as monomers but also as dimers and multiple fragments of HSA. Interestingly, with 10 μm EPO, immunostained samples showed a small but detectable amount of nitrone adducts on EPO, clearly visible on the light EPO chain (the heavy chain DMPO-EPO nitrone adducts overlapped with the DMPO-HSA complexes). To test the structural damage on HSA due to the bisulfite oxidation catalyzed by EPO, we examined the same samples using anti-HSA polyclonal antibody. Untreated HSA appeared essentially as the monomer with some dimer and even trimer and smaller amounts of aggregates and fragments (Fig. 5D). When HSA was incubated with EPO (1–10 μm), we observed EPO-dependent fragmentation of HSA and decreased HSA dimer, trimer, etc. The pattern of HSA fragmentation suggests the possibility of specific sites for oxidative cleavage to the protein by bisulfite oxidation.
Effect of Halides and Pseudohalides in Their Physiological Concentrations on HSA-DMPO Radical Formation
Because thiocyanate and halides are considered to be physiological substrates for EPO, we used ELISA and Western blot analysis to determine and compare their effect on the production of bisulfite-induced DMPO-HSA nitrone adducts (Fig. 6). The addition of 100 μm thiocyanate (plasma concentration) totally inhibited production of DMPO-nitrone adducts, confirming that thiocyanate is the preferred substrate for EPO and a strong competitor of bisulfite. Surprisingly, in the presence of bromide (100 μm) alone, the nitrone adduct production was increased by ∼70%, but in the presence of 2 mm bisulfite, the yield of DMPO-nitrone adducts was similar to the control sample with bisulfite only, although this absence of effect is only apparent because bromide independently caused HSA-derived radical formation. ELISA analysis was also performed with sodium chloride (100 mm) and sodium iodide (0.5 μm), but they yielded no nitrone adduct generation by themselves. Sulfite-dependent HSA radical production was inhibited in the presence of these halides by 26 and 6%, respectively. Western blot results paralleled those from the ELISA analysis (Fig. 6, A and B). However, the addition of l-methionine (20 mm) partially inhibited the production of DMPO-HSA-derived nitrone adducts in the presence of bromide alone, which is consistent with the common usage of Met as a scavenger of HOBr (39, 40) (Fig. 6C). Therefore, we also examined the effect of l-methionine on adduct formation in the absence of bisulfite. Although the addition of 5 mm Met had little effect, adduct production was completely inhibited at concentrations greater than 20 mm Met (Fig. 6D). In contrast, only partial inhibition by l-methionine was obtained in the presence of bisulfite, implying that bisulfite oxidation also caused radical damage to HSA even in the presence of bromide.
FIGURE 6.

Effect of halides and pseudohalides on the formation of DMPO-HSA-derived radical nitrone adducts. A, Western blotting. B, ELISA. Reaction mixtures containing HSA (600 μm), Na2SO3 (2 mm), DMPO (1 mm), and H2O2 (100 μm) with and without the indicated concentrations of NaSCN, NaBr, NaCl, and NaI were initiated by EPO (1 μm). ELISA data presented are the means ± S.D. from three independent determinations using fresh preparations of all reaction components. C, Western blotting effect of halides and pseudohalides on the formation of DMPO-HSA-derived radical nitrone adducts in the presence of 20 mm methionine. D, concentration-dependent effect of methionine on the formation of DMPO-HSA-derived radical nitrone adducts in the absence of bisulfite. For Western blotting, reactions, including albumin (600 μm), NaBr (100 μm), DMPO (1 mm), H2O2 (100 μm), and methionine as indicated, were initiated with 1 μm EPO, and the mixtures were incubated for 1 h at 37 °C in 100 mm phosphate buffer (pH 7.4). Each lane contained 3.8 μg of HSA. E, effect of halides and pseudohalides on the formation of DMPO/·SO3− radical adduct. Spectrum a, reaction mixture containing Na2SO3 (100 μm), DMPO (100 mm), and H2O2 (100 μm) in 100 mm phosphate buffer, pH 7.4. After initiation with EPO (1 μm), the mixture was immediately placed into the flat cell. Na2SO3 (100 μm) was used for the remaining experiments. The spectrum was attenuated in the presence of 100 μm NaSCN (spectrum b), 100 μm NaBr (spectrum c), 100 mm NaCl (spectrum d), and 0.5 μm NaI (spectrum e). Control without EPO did not form DMPO/·SO3− radical adduct (spectrum f).
To support the results from the immunological detection and to show that the enzyme-initiated free radical oxidative cycle in which bisulfite is oxidized to ·SO3− (Reactions 6–8) takes place in the presence of halides and pseudohalides, we recorded ESR spectra using the EPO-H2O2-bisulfite system (Fig. 6E). Reactions containing 100 mm DMPO to trap the primary sulfur trioxide anion radical (·SO3−), 100 μm bisulfite, 100 μm H2O2, and plasma concentrations of halides or thiocyanate were initiated with 1 μm EPO. As shown in Fig. 6E, the ESR signal of ·SO3− was nearly abolished by the addition of thiocyanate, the best substrate for EPO compound I (41), whereas incomplete inhibition was detected in the presence of bromide or iodide. The presence of chloride in these reactions had no effect on the ESR intensity. The absence of EPO resulted in no radical formation.
Effect of Antioxidants and Inhibitors on DMPO-HSA Radical Formation
To characterize the effect of antioxidants/nucleophilic scavengers and inhibitors on the generation of DMPO-HSA nitrone adducts, samples were incubated with and without bisulfite, and the reactions were initiated by EPO (1 μm) plus H2O2 (100 μm) in the presence of 1 mm DMPO (supplemental Fig. 2) and selected inhibitors. Control experiments without bisulfite produced only background amounts of nitrone adducts. ELISA results showed that the addition of 1 mm ascorbic acid in the presence of 2 mm bisulfite inhibited HSA radical production by 77%. Likewise, the addition of reduced thiols, such as glutathione, was also very effective at inhibiting adduct production with 5 mm GSH, decreasing the levels of the adducts by 88%. Another potent radical scavenger, cysteine, was tested at its plasma concentration level (100 μm) and inhibited HSA radical production by 34%. The effect of melatonin, one of the reported inhibitors of the catalytic activity of EPO (42), was also tested. In the presence of this hormone (200 μm), less than 50% of the nitrone adducts were detected. Western blot analysis also supported the ELISA data (supplemental Fig. 2).
EPO Radical Formation in HL-60 Cells (Clone 15) Induced by Bisulfite
To demonstrate that the immunological detection of DMPO-HSA and DMPO-EPO nitrone adducts is not limited to a pure enzymatic system, we investigated production of protein-DMPO adducts in HL-60 cells (clone 15), known to differentiate primarily to eosinophils (43), by employing immuno-spin trapping coupled with confocal microscopy. Reactions were carried out for Western blot analysis by incubation of 2 × 106 cells/ml with DMPO, bisulfite, and G/GOx to generate H2O2. As shown in Fig. 7A, only the complete system produced immunoreactivity with anti-DMPO, demonstrating band-specific anti-DMPO. Controls without bisulfite, G/GOx, or both failed to form any significant DMPO-protein adduct formation. Anti-EPO Western blot of the same samples showed that EPO was present in each lane in approximately the same amounts and thus served as the appropriate protein-loading control (Fig. 7B). The localization of the major anti-DMPO protein blot on the membrane matched the corresponding most abundant anti-EPO band, suggesting that the anti-DMPO band might be EPO, although other eosinophil proteins were oxidized to free radicals in a sulfite- and hydrogen peroxide-dependent manner (see the additional bands in Fig. 7A).
FIGURE 7.

Immuno-spin trapping and confocal microscopy images of the colocalization of protein-DMPO adducts obtained by treating HL-60 (clone 15) cell cytosol with Na2SO3. Shown are anti-DMPO (A) and anti-EPO (B) Western blotting of HL-60 (clone 15) cells. Cells (2 × 106/ml) were treated with 10 mm DMPO and 100 μm bisulfite, and the reaction was initiated with 5 mm glucose and 50 milliunits/ml glucose oxidase, which was added last. The reaction was incubated on a plate stirrer for 1 h at 37 °C. In both Western blots, 30 μg of protein was loaded into each lane. C, confocal microscopy images of the colocalization of protein-DMPO adducts (red stain) and EPO obtained by treating HL-60 cells with bisulfite (100 μm) and glucose (5 mm) plus glucose oxidase (50 milliunits/ml) in the presence of 10 mm DMPO. D, same as C except without bisulfite and G/GOx. Clockwise, the quadrants in each picture represent anti-EPO (green stain), anti-DMPO nitrone adducts (red stain), and overlaid pictures obtained from anti-DMPO and anti-EPO (yellow shade). Gray, transmission image.
Parallel confocal fluorescence microscopy experiments were also performed. After fixation and permeabilization, cells were stained with primary anti-DMPO and anti-EPO, followed by Alexafluor-conjugated secondary anti-rabbit (red) and anti-mouse (green) antisera to stain the DMPO-nitrone adducts and EPO, respectively. The data showed that the cells exposed to bisulfite treatment followed by G/GOx have an extensive fluorescence signal throughout the cytosol, confirming the intracellular formation of protein radicals (Fig. 7C). In the absence of bisulfite or G/GOx, EPO green staining was easily detectable, but no red staining was observed due to the absence of anti-DMPO antibody binding (Fig. 7D). These data indicate that protein radical formation was a consequence of the bisulfite-induced protein oxidation to free radicals, which were trapped by DMPO.
In addition to the immunological detection of protein-DMPO in HL-60 (clone 15) cell cytosol, we also examined the effect of halides, thiocyanate, and ascorbate in their physiological concentrations on the yield of DMPO nitrone adducts (Fig. 8). The addition of mixture containing thiocyanate, bromide, chloride, and ascorbic acid significantly inhibited DMPO-protein formation, confirming the competition between bisulfite and EPO substrates. The effect of individual inhibition by the addition of thiocyanate, chloride, and ascorbate separately was also shown by ELISA (Fig. 8B). Similarly to the albumin model system, the addition of bromide in the presence of bisulfite had a net negligible effect on radical formation. EPO was still significantly oxidized to its protein radical in eosinophils incubated with 100 μm thiocyanate (Fig. 8), although thiocyanate was the strongest inhibitor of sulfite anion radical formation (Fig. 6E, spectrum b).
FIGURE 8.

Effect of halides, thiocyanate, and ascorbic acid (as radical scavenger) on the formation of protein-DMPO nitrone adducts in HL-60 (clone 15) cell cytosol. A, Western blotting. B, ELISA. Reaction mixtures containing HL-60 (clone 15) cells (2 × 106/ml), Na2SO3 (100 μm), and DMPO (10 mm) with and without the indicated concentrations of NaSCN, NaBr, NaCl, and ascorbic acid were initiated with 5 mm glucose and 50 milliunits/ml glucose oxidase, which was added last. The reaction was incubated on a plate stirrer for 1 h at 37 °C. Each lane contained 30 μg of protein.
DISCUSSION
Our spin-trapping ESR results showed that bisulfite is oxidized to sulfur trioxide anion radical (·SO3−) by the bisulfite-eosinophil peroxidase-H2O2 system. Once the ·SO3− radical is formed via the enzymatic oxidation of bisulfite, it reacts very rapidly with oxygen and generates −O3SOO· and SO4˙̄ radicals (2). These radicals are powerful oxidants (E−O3SOO·/−O3SOOH = 1.1 V and ESO4˙̄/SO42− = 2.43 V) that can attack target proteins to form protein radicals (e.g. HSA in plasma) (13, 44). We used eosinophil peroxidase-H2O2-bisulfite as a source for generation of oxidants (−O3SOO· and SO4˙̄ anion radicals) and demonstrated their ability to oxidize the most abundant plasma protein to protein radicals (Scheme 1).
In the radical chain chemistry of bisulfite oxidation, the sulfur trioxide anion radical (·SO3−) is the primary radical, and −O3SOO· and SO4˙̄ are the secondary and tertiary radicals, respectively (15). Previous work on the oxidation of bisulfite by the horseradish peroxidase-H2O2 system and ESR spin-trapping experiments showed that a decrease of the DMPO concentration to 3 mm or lower allowed the authors to begin trapping the tertiary SO4˙̄ anion radical (15). Our oxygen consumption experiments are in agreement with the radical mechanism for the enzymatic oxidation of bisulfite. The sensitivity of oxygen uptake to bisulfite concentration demonstrated that only 50 nm eosinophil peroxidase was sufficient to start the radical chain reaction (Fig. 3). The addition of DMPO to the system decreases the oxygen consumption, as is consistent with DMPO competing with oxygen for ·SO3−. Moreover, our DMPO concentration-dependent oxygen uptake indicates that a spin trap range between 0.3 and 10 mm decreased the trapping of ·SO3− to the point that the radical chain reaction with formation of −O3SOO· and SO4˙̄ radicals could proceed.
The second-order rate constant for EPO compound I formation has been measured at (4.3 ± 0.4) × 107 m−1 s−1 (pH 7, 15 °C) (12), which is significantly higher than the rates published for the other members of the mammalian peroxidase superfamily (myeloperoxidase, lactoperoxidase, and thyroid peroxidase) (45, 46). In the case of horseradish peroxidase, the rate constants for the one-electron reductions of compound I and II by bisulfite are relatively slow (36), but recent studies have shown that only a very low concentration of the primary ·SO3− is necessary to initiate the radical chain Reactions 6–8 (38). On the other hand, DMPO successfully competes with oxygen for the ·SO3− radical (the rate constants are presented in Scheme 1), and when the spin trap is added in high concentrations (100 mm), the chain reaction is inhibited (Fig. 3B). However, the polyclonal anti-DMPO antibody identifies the oxidation product of the DMPO radical adduct. This means that, first, compounds I and II must oxidize bisulfite to ·SO3− radical to initiate the radical chain reaction (the standard redox potential of EPO compound I has been determined to be 1.10 ± 0.01 V (47) versus 0.63 V for the ·SO3− (13, 48)). Second, the tertiary radical from the chain reaction and strong oxidant (SO4˙̄) must oxidize amino acid(s) from the target protein (HSA) to protein radicals. Third, the concentration of DMPO must be sufficient for the protein radicals to react with DMPO, and, finally, these DMPO radical adducts must be oxidized to their nitrone form. As a result, the overall high yield of DMPO nitrone adducts was achieved by reducing the DMPO concentration to 1 mm in the presence of plasma level albumin (600 μm).
In the studies reported here, the formation of HSA-derived radicals in the reaction of the protein with the sulfate and, perhaps, peroxymonosulfate anion radicals generated by the eosinophil peroxidase-H2O2-bisulfite system is detected by immuno-spin trapping (Scheme 1). Our Western blot experiments showed that in the presence of DMPO, the eosinophil peroxidase-H2O2-bisulfite system produced sulfite-derived radicals that oxidized not only albumin but also eosinophil peroxidase itself (Fig. 5). These protein target- and initiator-derived radicals are trapped by the nitrone spin trap DMPO and detected as DMPO-HSA and DMPO-EPO nitrone adducts, respectively. Production of HSA nitrone adducts was also strongly dependent on bisulfite and H2O2 concentrations. It is interesting to note that positive results were detected on the anti-DMPO Western blot with 2 mm bisulfite, which is approximately half of the amount used as a preservative in wines (up to 5–6 mm) (4, 49).
In addition, we used HL-60 cells (clone 15), which differentiate into eosinophils following co-incubation with butyric acid, to investigate whether DMPO-protein adducts could be detected in eosinophils. This model of bisulfite-driven, EPO-initiated protein oxidation in cells supports our findings that the eosinophil peroxidase-H2O2-bisulfite system, in which bisulfite is oxidized to form sulfite-derived radicals, oxidizes a set of well distinguished bands of target proteins (probably including EPO) in eosinophils (Fig. 7). Bisulfite is one of the few sulfating agents approved by the Food and Drug Administration as a food preservative and antioxidant to prevent or reduce spoilage (4). It also appears as an ingredient of many medications, such as antibiotics, analgesics, anesthetics, etc. However, sulfites have been associated with adverse allergic and asthmatic type reactions experienced by sulfite-hypersensitive individuals (it is estimated that up to 500,000 sulfite-sensitive individuals live in the United States (6)). The most frequent sulfite reaction symptoms are difficulties in breathing, food intolerance symptoms, asthma, and, occasionally, anaphylactic shock. There is no specific treatment for sulfite toxicity, and in general, to our knowledge, the mechanisms of the potentially toxic reactions of bisulfite are poorly understood. The mean serum sulfite concentration in healthy individuals reported by Ji et al. (50) is ∼5 μm. However, in their study, after oral metabisulfite loading with vegetable juice, there was a rapid rise of sulfite concentration in plasma (112 μm) in 30 min. The toxic potential of bisulfite is most clearly indicated by the loss of sulfite oxidase, the molybdenum-containing enzyme that oxidizes sulfite to sulfate (SO42−) (51). In fact, in humans, the loss of sulfite oxidase is fatal in infancy or early childhood (52). It is noteworthy that in cases of sulfite oxidase deficiency, the concentration of sulfite in plasma is abnormal (10 μm to ∼2 mm) (5, 53). The capacity of sulfite oxidase for sulfite oxidation is normally extremely high (54–56); the reaction proceeds via a one-step, two-electron oxidation to sulfate with no free radical intermediates (7). It has been shown that the presence of sulfite oxidase is substantially reduced as compared with that in normal and sulfite-sensitive asthmatic subjects (57). The enzyme is present at high levels in the liver and in lower concentrations in most of the other tissues of the body (e.g. in the lung). Sulfite reserves in serum or plasma, occurring as protein and low molecular weight S-sulfonates, which form by a nucleophilic reaction of bisulfite with disulfides (58, 59).
Another radical mechanism of oxidation of bisulfite is xanthine-dependent oxidation in the presence of xanthine oxidase, which has been proposed by Fridovich and Handler (60). The authors conclude that xanthine oxidase, in catalyzing the aerobic oxidation of xanthine, generates superoxide anion, which serves to initiate the bisulfite chain reaction. A previous report from this laboratory (11) demonstrated that incubation of bisulfite with horseradish peroxidase and H2O2 is not sensitive to the presence of superoxide dismutase, confirming that the peroxidase-catalyzed pathway does not involve a superoxide-initiated chain reaction. Furthermore, recent studies of the horseradish peroxidase/H2O2 system showed that initial DMPO/·SO3− formation is twice as fast as the initial consumption of O2 and H2O2, which is in agreement with the radical chain mechanism of Reactions 6–8 and the expected stoichiometry (38). Once the chain reaction dominates, the total consumption of O2 and the formation of −O3SOOH greatly exceed the initial concentration of H2O2. This chemistry is an enzymatically initiated, free radical chain reaction. We have strong evidence that eosinophil peroxidase-catalyzed oxidation of bisulfite to ·SO3−, −O3SOO·, and SO4˙̄ radicals is responsible for the albumin oxidation we observed, and this evidence suggests the possibility of free radical metabolism of bisulfite and subsequent protein damage in vivo.
All of the members of the mammalian peroxidase superfamily share the ability to oxidize pseudohalides to pseudohypohalous acids via a two-electron reduction step of compound I to the ferric enzyme. The preferred substrates for EPO are reported to be thiocyanate (SCN−), followed by iodide (I−) and bromide (Br−) (12, 16, 24, 61, 62). The relative rates of EPO compound I reduction are SCN− > I− > Br− ≫ Cl−, and the reduction is about 10 times more effective than that of myeloperoxidase, except for chloride (41). When we added plasma concentrations of thiocyanate, iodide, and chloride to our DMPO-trapping experiments, we detected no oxidation of albumin in the absence of bisulfite (Fig. 6). Although iodide is a good donor for EPO compound I, its low physiological concentration in blood (< 1 μm) makes its oxidation by EPO of little importance. In addition to the effect of EPO physiological substrates on the yield of DMPO-HSA-derived nitrone adducts, the Western blot from HL-60 cell cytosol indicated the presence of a DMPO-EPO-containing band in the systems with halides and thiocyanate (Fig. 8). ELISA results showed that the addition of 100 μm thiocyanate in the presence of bisulfite inhibited the EPO radical production by ∼75%, but the protein was still oxidized to its radical, due, perhaps, to the lower intracellular thiocyanate concentration (Fig. 8B). The plasma concentration of thiocyanate can be as low 20 μm (12, 23) and is probably lower yet within cells. In addition, under conditions where unusually high concentrations of hydrogen peroxide are generated, thiocyanate and bromide may be depleted. In this regard, it is pertinent that the vast majority of humans suffer no ill effects from sulfite exposure, and unrecognized, relatively rare factors must be involved, such as unusually low thiocyanate and bromide concentrations.
In summary, our results show that the EPO-H2O2-bisulfite system provides an enzymatic pathway for production of −O3SOO· and SO4˙̄, recognized to play a role in protein oxidation in a pure enzymatic system and in HL-60 cells (clone 15). Therefore, we propose a potential mechanism of EPO-dependent oxidative damage and tissue injury in bisulfite (hydrated sulfur dioxide)-exacerbated eosinophilic inflammatory disorders.
Supplementary Material
Acknowledgments
We thank Mary J. Mason, Dr. Ann Motten, and Jean Corbett for help in the preparation of the manuscript and Jeff Tucker for confocal image analysis.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program, NIEHS.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- EPO
- eosinophil peroxidase
- HSA
- human serum albumin
- ESR
- electron spin resonance
- DMPO
- 5,5-dimethyl-1-pyrroline N-oxide
- HVA
- homovanillic acid
- ELISA
- enzyme-linked immunosorbent assay
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- G/GOx
- glucose/glucose oxidase.
REFERENCES
- 1.Rall D. P. (1974) Environ. Health Perspect. 8, 97–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayon E., Treinin A., Wilf J. (1972) J. Am. Chem. Soc. 94, 47–57 [Google Scholar]
- 3.Neta P., Huie R. E. (1985) Environ. Health Perspect. 64, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunnison A. F. (1981) Food Cosmet. Toxicol. 19, 667–682 [DOI] [PubMed] [Google Scholar]
- 5.Shih V. E., Abroms I. F., Johnson J. L., Carney M., Mandell R., Robb R. M., Cloherty J. P., Rajagopalan K. V. (1977) N. Engl. J. Med. 297, 1022–1028 [DOI] [PubMed] [Google Scholar]
- 6.Lester M. R. (1995) J. Am. Coll. Nutr. 14, 229–232 [DOI] [PubMed] [Google Scholar]
- 7.Cohen H. J., Fridovich I. (1971) J. Biol. Chem. 246, 359–366 [PubMed] [Google Scholar]
- 8.Niknahad H., O'Brien P. J. (2008) Chem. Biol. Interact. 174, 147–154 [DOI] [PubMed] [Google Scholar]
- 9.Mottley C., Mason R. P., Chignell C. F., Sivarajah K., Eling T. E. (1982) J. Biol. Chem. 257, 5050–5055 [PubMed] [Google Scholar]
- 10.Araiso T., Miyoshi K., Yamazaki I. (1976) Biochemistry 15, 3059–3063 [DOI] [PubMed] [Google Scholar]
- 11.Mottley C., Trice T. B., Mason R. P. (1982) Mol. Pharmacol. 22, 732–737 [PubMed] [Google Scholar]
- 12.Furtmüller P. G., Burner U., Regelsberger G., Obinger C. (2000) Biochemistry 39, 15578–15584 [DOI] [PubMed] [Google Scholar]
- 13.Neta P., Huie R. E., Ross A. B. (1988) J. Phys. Chem. Ref. Data 17, 1027–1284 [Google Scholar]
- 14.Reed G. A., Curtis J. F., Mottley C., Eling T. E., Mason R. P. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 7499–7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mottley C., Mason R. P. (1988) Arch. Biochem. Biophys. 267, 681–689 [DOI] [PubMed] [Google Scholar]
- 16.Weiss S. J., Test S. T., Eckmann C. M., Roos D., Regiani S. (1986) Science 234, 200–203 [DOI] [PubMed] [Google Scholar]
- 17.Agosti J. M., Altman L. C., Ayars G. H., Loegering D. A., Gleich G. J., Klebanoff S. J. (1987) J. Allergy Clin. Immunol. 79, 496–504 [DOI] [PubMed] [Google Scholar]
- 18.Slungaard A., Mahoney J. R., Jr. (1991) J. Exp. Med. 173, 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff S. J., Locksley R. M., Jong E. C., Rosen H. (1983) Ciba Found. Symp. 99, 92–112 [DOI] [PubMed] [Google Scholar]
- 20.Wardlaw A. J. (1994) Postgrad. Med. J. 70, 536–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horwitz R. J., Busse W. W. (1995) Clin. Chest Med. 16, 583–602 [PubMed] [Google Scholar]
- 22.Rothenberg M. E. (1998) N. Engl. J. Med. 338, 1592–1600 [DOI] [PubMed] [Google Scholar]
- 23.Slungaard A., Mahoney J. R., Jr. (1991) J. Biol. Chem. 266, 4903–4910 [PubMed] [Google Scholar]
- 24.Wu W., Chen Y., Hazen S. L. (1999) J. Biol. Chem. 274, 25933–25944 [DOI] [PubMed] [Google Scholar]
- 25.Galijasevic S., Proteasa G., Abdulhamid I., Abu-Soud H. M. (2007) Biochemistry 46, 406–415 [DOI] [PubMed] [Google Scholar]
- 26.Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., Lusis A. J., Lee J. J., Lee N. A., Abu-Soud H. M., Ischiropoulos H., Hazen S. L. (2002) J. Biol. Chem. 277, 17415–17427 [DOI] [PubMed] [Google Scholar]
- 27.Mason R. P. (2004) Free Radic. Biol. Med. 36, 1214–1223 [DOI] [PubMed] [Google Scholar]
- 28.Bolscher B. G., Plat H., Wever R. (1984) Biochim. Biophys. Acta 784, 177–186 [DOI] [PubMed] [Google Scholar]
- 29.Duling D. R. (1994) J. Magn. Reson. B 104, 105–110 [DOI] [PubMed] [Google Scholar]
- 30.Holtzman J. L. (1976) Anal. Chem. 48, 229–230 [DOI] [PubMed] [Google Scholar]
- 31.Detweiler C. D., Deterding L. J., Tomer K. B., Chignell C. F., Germolec D., Mason R. P. (2002) Free Radic. Biol. Med. 33, 364–369 [DOI] [PubMed] [Google Scholar]
- 32.Ramirez D. C., Chen Y. R., Mason R. P. (2003) Free Radic. Biol. Med. 34, 830–839 [DOI] [PubMed] [Google Scholar]
- 33.Ehrenshaft M., Mason R. P. (2006) Free Radic. Biol. Med. 41, 422–430 [DOI] [PubMed] [Google Scholar]
- 34.Ranguelova K., Suarez J., Magliozzo R. S., Mason R. P. (2008) Biochemistry 47, 11377–11385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez D. C., Gomez Mejiba S. E., Mason R. P. (2005) Free Radic. Biol. Med. 38, 201–214 [DOI] [PubMed] [Google Scholar]
- 36.Roman R., Dunford H. B. (1973) Can. J. Chem. 51, 588–596 [Google Scholar]
- 37.Furtmüller P. G., Jantschko W., Regelsberger G., Obinger C. (2001) Biochim. Biophys. Acta. 1548, 121–128 [DOI] [PubMed] [Google Scholar]
- 38.Ranguelova K., Mason R. P. (2009) Free Radic. Biol. Med. 47, 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies M. J. (2005) Biochim. Biophys. Acta. 1703, 93–109 [DOI] [PubMed] [Google Scholar]
- 40.Nagy P., Beal J. L., Ashby M. T. (2006) Chem. Res. Toxicol. 19, 587–593 [DOI] [PubMed] [Google Scholar]
- 41.Furtmüller P. G., Burner U., Obinger C. (1998) Biochemistry 37, 17923–17930 [DOI] [PubMed] [Google Scholar]
- 42.Lu T., Galijasevic S., Abdulhamid I., Abu-Soud H. M. (2008) Br. J. Pharmacol. 154, 1308–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischkoff S. A. (1988) Leuk. Res. 12, 679–686 [DOI] [PubMed] [Google Scholar]
- 44.Steele W. V., Appelman E. H. (1982) J. Chem. Thermodynamics 14, 337–344 [Google Scholar]
- 45.Kohler H., Taurog A., Dunford H. B. (1988) Arch. Biochem. Biophys. 264, 438–449 [DOI] [PubMed] [Google Scholar]
- 46.Marquez L. A., Huang J. T., Dunford H. B. (1994) Biochemistry 33, 1447–1454 [DOI] [PubMed] [Google Scholar]
- 47.Arnhold J., Furtmüller P. G., Regelsberger G., Obinger C. (2001) Eur. J. Biochem. 268, 5142–5148 [DOI] [PubMed] [Google Scholar]
- 48.Huie R. E., Neta P. (1984) J. Phys. Chem. 88, 5665–5669 [Google Scholar]
- 49.Mitsuhashi H., Ikeuchi H., Nojima Y. (2001) Clin. Chem. 47, 1872–1873 [PubMed] [Google Scholar]
- 50.Ji A. J., Savon S. R., Jacobsen D. W. (1995) Clin. Chem. 41, 897–903 [PubMed] [Google Scholar]
- 51.Johnson J. L., Waud W. R., Rajagopalan K. V., Duran M., Beemer F. A., Wadman S. K. (1980) Proc. Natl. Acad. Sci. U.S.A. 77, 3715–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwarz G. (2005) Cell Mol. Life Sci. 62, 2792–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acosta R., Granados J., Mourelle M., Perez-Alvarez V., Quezada E. (1989) Ann. Allergy 62, 402–405 [PubMed] [Google Scholar]
- 54.Wilkins J. W., Jr., Greene J. A., Jr., Weller J. M. (1968) Clin. Pharmacol. Ther. 9, 328–332 [DOI] [PubMed] [Google Scholar]
- 55.Cohen H. J., Drew R. T., Johnson J. L., Rajagopalan K. V. (1973) Proc. Natl. Acad. Sci. U.S.A. 70, 3655–3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oshino N., Chance B. (1975) Arch. Biochem. Biophys. 170, 514–528 [DOI] [PubMed] [Google Scholar]
- 57.Stevenson D. D., Simon R. A. (1984) J. Allergy Clin. Immunol. 74, 469–472 [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama E., Yoder R. E., Frank N. R. (1971) Arch. Environ. Health 22, 389–395 [DOI] [PubMed] [Google Scholar]
- 59.Gunnison A. F., Palmes E. D. (1974) AIHAJ 35, 288–291 [DOI] [PubMed] [Google Scholar]
- 60.Fridovich I., Handler P. (1961) J. Biol. Chem. 236, 1836–1840 [PubMed] [Google Scholar]
- 61.Arlandson M., Decker T., Roongta V. A., Bonilla L., Mayo K. H., MacPherson J. C., Hazen S. L., Slungaard A. (2001) J. Biol. Chem. 276, 215–224 [DOI] [PubMed] [Google Scholar]
- 62.Arnhold J., Monzani E., Furtmuller P. G., Zederbauer M., Casella L., Obinger C. (2006) Eur. J. Inorg. Chem. 2006, 3801–3811 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.