

Abstract
Toll-like receptors (TLRs) play a central role in host defense by inducing inflammatory and adaptive immune responses following infection. Drugs that target TLRs are of considerable interest as potential inflammatory regulators, vaccine adjuvants, and novel immunotherapeutics. TLR2, in cooperation with either TLR1 or TLR6, mediates responses to a wide variety of microbial products as well as products of host tissue damage. In an effort to understand the structural basis of TLR2 recognition and uncover novel TLR2 agonists, a synthetic chemical library of 24,000 compounds was screened using an IL-8-driven luciferase reporter in cells expressing these human receptors. The screening yielded several novel TLR2-dependent activators that utilize TLR1, TLR6, or both as co-receptors. These novel small molecule compounds are aromatic in nature and structurally unrelated to any known TLR2 agonists. The three most potent compounds do not exhibit synergistic activity, nor do they act as pseudoantagonists toward natural TLR2 activators. Interestingly, two of the compounds exhibit species specificity and are inactive toward murine peritoneal macrophages. Mutational analysis reveals that although the central extracellular region of TLR1 is required for stimulation, there are subtle differences in the mechanism of stimulation mediated by the synthetic compounds in comparison with natural lipoprotein agonists. The three most potent compounds activate cells in the nanomolar range and stimulate cytokine production from human peripheral blood monocytes. Our results confirm the utility of high throughput screens to uncover novel synthetic TLR2 agonists that may be of therapeutic benefit.
Keywords: Inflammation, Innate Immunity, Lipoprotein Receptor, Receptor Structure-Function, Toll-like Receptors (TLR)
Introduction
Cellular innate immune responses drive the immediate release of proinflammatory mediators that enable leukocytes to access the site of infection as well as responses of professional antigen-presenting cells essential for generating effective adaptive immunity (1, 2). The primary triggers of these responses are a family of pattern recognition receptors known as Toll-like receptors (TLRs).2 Humans possess 10 TLR family members, numbered 1 through 10, the subsets of which are expressed in leukocytes and the epithelial cells of mucosal surfaces (3, 4). There are two major types of TLRs, those that reside in intracellular compartments and sense viral and bacterial nucleic acids and those that are expressed on the cell surface and sense outer membrane components of bacteria, fungi, and protozoan organisms (5, 6). TLRs also recognize numerous molecules arising from damage to self-tissues, and this mode of TLR activation appears to play a central role in a number of non-infectious chronic inflammatory conditions (7, 8). Although there are marked differences in signaling and gene induction across various TLRs, all cell surface TLRs engage a core signaling pathway culminating in the activation of NF-κB and the production of proinflammatory chemokines, cytokines, and cell adhesion molecules (9, 10).
TLR2 is a cell surface receptor that senses a remarkable variety of bacterial, fungal, and viral products as well as inflammatory self-components. Among TLR2 agonists, bacterial lipoproteins are by the far the most potent (11, 12). Additional TLR2 agonists comprise a diversity of structures including bacterial and fungal lipids, acylated sugars and proteins, unmodified protein complexes, as well as certain polysaccharides (13, 14). TLR2 needs to form heterodimers with either TLR1 or TLR6 to generate signals, and these TLR2-1 and TLR2-6 complexes discriminate different microbial products (15). For example, triacylated bacterial lipoproteins (mimicked by the lipopeptide Pam3CSK4) and diacylated lipoproteins (including the lipopeptide MALP-2, from mycoplasma) activate cells through TLR2-1 and TLR2-6 heterodimers, respectively (16, 17). We have recently found that similar to TLR1, TLR10 also cooperates with TLR2 in the recognition of triacylated lipopeptides; however, the signaling outcome remains unclear (18). Additional co-receptors are essential for recognition of certain TLR2 agonists and thereby serve to increase the repertoire of agonists for this receptor (13, 14).
Given their therapeutic potential, there is considerable interest in pharmaceuticals that modulate TLR activation. TLR antagonists hold great clinical promise for the treatment of numerous inflammatory conditions and are under investigation for the treatment of viral infections, redirecting allergic helper T cell responses, and as anticancer therapeutics (8, 19, 20). Some TLR agonists also have proven safety and efficacy in humans as vaccine adjuvants and are currently in use in Europe (19, 21). Synthetic lipopeptide agonists for TLR2 exhibit strong adjuvant activity when either mixed with or directly conjugated to various antigens (22, 23). In addition to lipopeptides, a variety of other natural TLR2 agonists exhibit adjuvant activity including zwitterionic polysaccharides from Group B Streptococcus (24), Type IIb heat labile enterotoxin from enteropathogenic Escherichia coli (25), and porin B from pathogenic Neisseriae sp. (26).
Despite their potential clinical utility, no high throughput screens for synthetic compounds that stimulate TLR2 have been reported. In this study, the identification of novel synthetic TLR2 agonists by chemical library screening is presented. The structures of these small agonists are unrelated to any known natural agonists for TLR2. Mutagenesis studies indicate that TLR2-1 recognizes the compounds through mechanisms different from that of microbial lipopeptides. The compounds induce cytokine production in human peripheral blood monocytes, suggesting that they are worthy of further clinical development.
EXPERIMENTAL PROCEDURES
Reagents
d-Luciferin and coenzyme A trilithium salt were purchased from Sigma-Aldrich Corp. Luciferin solution and luciferase assay buffer have been described elsewhere (27). The synthetic bacterial lipopeptides Pam3CSK4 and PamOct2CSK4 were obtained from EMC Microcollections. The R isomer of MALP-2 was purchased from Alexis Biochemicals. Chemical compound A (N-methyl-4-nitro-2-[4-(4-nitrophenyl)-1H-imidazolyl]aniline), compound B (methyl 2-[(anilinocarbonothioyl)amino]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate), compound C (ethyl 2-({[(4-methoxyphenyl)amino]carbonothioyl}amino)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate), compound E (N1-(4-chlorobenzyl)-N2-(4-methylphenyl)-N2-(methylsulfonyl)glycinamide), and compound F (N2-(4-bromophenyl)-N1-(4-methoxybenzyl)-N2-(phenylsulfonyl)glycinamide) were individually purchased from Chembridge Corp. Compounds were dissolved and diluted in dimethyl sulfoxide (DMSO).
Plasmid Constructs
A common variant of human TLR1 represented by National Center for Biotechnology Information (NCBI) accession number AAI09095 is referred to as wild type TLR1 in this study and was used as the basis for generating various chimeric variants and mutants. The generation of TLR1 and TLR6 chimeric receptors was described previously (28). The P315L polymorphic variant of TLR1 was generated as described previously based on the technique of overlap extension PCR (29). TLR1 point mutants were generated by random mutagenesis using error-prone PCR (30). Briefly, the central region of TLR1 was excised from a modified FLAG-tagged TLR1 construct with XbaI sites (underlined letters) flanking LRRs 9–12 (28). Error-prone PCR protocol was performed on this fragment using forward 5′-CCAATCTAGAAACAACTTGGAATTCTTTCATTAGGATCC-3′ and reverse 5′-CCAATCTAGATTGTTTAAGGTAAGACTTGATAAGTTTGG-3′ primers. Error-prone fragments were reinserted into TLR1, and the clones were screened by Clone Checker (Invitrogen) and verified by DNA sequencing. Screening of the library revealed several TLR1 variants (V311E, F314D, Q316K, Y320N, E321V, I328N, V399D) with reduced responses to Pam3CSK4 (18, 31).
Cell Culture
All cultured cells were grown at 37 °C in a humidified environment containing 5% CO2. Human colonic epithelial SW620 cells and murine macrophage RAW 264.7 cells were cultured in RPMI 1640 medium containing 10% (v/v) fetal bovine serum and 2 mm l-glutamine. Murine peritoneal macrophages were cultured in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, 2 mm l-glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin. Human peripheral blood monocytes were cultured in RPMI 1640 medium supplemented with 10% autologous plasma and 2 mm l-glutamine.
Chemical Library Screening
The chemical library was composed of ∼24,000 compounds, among which 9,000 were from a private collection of the Department of Chemistry at the University of Illinois, and the rest were from Chembridge Corp. All the compounds were kept in DMSO at 10 mm in a 384-well plate format. The human colonic epithelial cell line SW620 was transiently co-transfected with TLR1, TLR2, and TLR6 along with a firefly luciferase reporter driven by the IL-8 promoter in a 10-cm tissue culture dish. One day after transfection, cells were trypsinized and reseeded into 384-well plates (Corning Corp., Corning, NY) at a density of ∼104 cells/well in a total volume of 20 μl. The next day, chemical compounds were added into each well using a 384-pin transfer apparatus that delivered 0.2–1 μl of each compound at an approximate concentration of 100 μm. Each plate contained wells treated with DMSO alone as a negative control as well as Pam3CSK4 and MALP-2 as TLR2 agonist positive controls. The cells were incubated with individual compounds and controls for at least 6 h followed by cell lysis in a volume of 20 μl. Luciferase values were normalized to DMSO-treated cells to determine relative cell activation levels. Compounds with greater than 2.5-fold activity over DMSO alone were selected for a second round of screening. A total amount of 217 compounds were retested for TLR-dependent activity in SW620 cells transfected with the TLRs against those transfected with the empty expression vector pFLAG-CMV.
Transient Transfection Assays
SW620 cells were co-transfected with various TLR combinations along with an IL-8 promoter-driven firefly luciferase reporter and a Renilla luciferase transfection control reporter. The amount of DNA used for transfection was 20 ng/ml for TLR2, 180 ng/ml for TLR1, TLR6, or TLR1 variants, 150 ng/ml for IL-8-driven luciferase gene, and 50 ng/ml for Renilla control. Transfection was mediated by using FuGENE 6 (Roche Applied Science) at a lipid volume to DNA weight ratio of 4:1. Two days after transfection, cells were stimulated with indicated agonists for at least 6 h, and cell lysates were collected. Luciferase enzyme activities were measured using the Dual-Luciferase reporter assay system (Promega, Madison, WI). The values of firefly luciferase were first divided by those of Renilla luciferase values to normalize the transfection efficiency in different wells. All the values were then normalized to those of unstimulated cells with empty pFLAG-CMV vector and reporters to determine the relative luciferase activity.
Stimulation of Mouse Peritoneal Macrophages
All animal experiments were conducted in accordance with a protocol approved by the University of Illinois Institutional Animal Care and Use Committee. Wild type C57BL6 mice were purchased from The Jackson Laboratory. TLR-deficient mice were kind gifts of Dr. Shizou Akira (Osaka University, Japan). Murine peritoneal macrophages were isolated from the mouse peritoneal cavity. Peritoneal cells were suspended in the medium described above and incubated in tissue culture plates for 2 h. The non-adherent cells were removed by media change. Adherent cells were initially seeded at 2 × 105 cells/well in 96-well cell culture plates. Cells were treated with the indicated compounds, lipopeptides, or DMSO controls for 16 h. The concentration of mouse IL-6 or TNF-α in culture supernatants was determined by ELISA according to manufacturer's instructions (Invitrogen).
Stimulation of Human Peripheral Blood Monocytes
Blood was obtained from healthy donors in accordance with a protocol approved by the University of Illinois Institutional Review Board. Human peripheral blood mononuclear cells were isolated from blood of healthy donors by Ficoll gradient centrifugation. CD14+ monocytes were further purified by magnetic negative selection (Miltenyi Biotec, Gladbach, Germany). The purified monocytes were cultured in the medium described above at density of 1 × 105 cells/well in 96-well plates. Cells were treated with indicated compounds, lipopeptides, or DMSO controls for 16 h. The concentration of human TNF-α in culture supernatants was determined by ELISA according to the manufacturer's instructions (Invitrogen).
RESULTS
Identification of Novel TLR2 agonists by Chemical Library Screening
We have previously shown that SW620, a human colonic epithelial cell line lacking endogenous expression of TLR1, TLR2, and TLR6, can be used to reconstitute receptor activity following transient transfection (28). In these transfection assays, a firefly luciferase gene driven by the promoter of the IL-8 provides a robust reporter of TLR2-1 and TLR2-6 activity. To identify novel chemical compounds that activate TLR2 heterodimers, we adapted this assay to a 384-well format and screened a chemical library composed of 24,000 individual synthetic compounds. The primary screen, based on a 2.5-fold enhancement of luciferase activity, yielded 217 initial hits. These 217 compounds were comparatively rescreened against SW620 cells transfected with empty FLAG-CMV vector to exclude compounds that activate the reporter in a TLR-independent fashion. This rescreening identified a total of 16 compounds with reproducible TLR2-dependent activation. To discriminate between TLR2-1- and TLR2-6-mediated activation, cells transfected with different combinations of TLRs were examined for their response to the 10 most potent compounds (Fig. 1A). Most of the compounds were active toward cells co-expressing TLR2 and TLR1, whereas compound F was active toward cells expressing TLR2 and TLR6. Compound E appeared to be an agonist for both TLR2-1 and TLR2-6 receptor pairs. Transfection of TLR2 alone did not render SW620 cells responsive to any of the compounds, supporting the idea that TLR2 requires other TLRs for activity.
FIGURE 1.

Activity and structure of novel chemical agonists for TLR2. A, SW620 cells were co-transfected with various combinations of TLRs and an IL-8-driven luciferase gene and seeded into a 384-well plate. Two days after transfection, cells were stimulated with the indicated compounds at a concentration of ∼100 μm for at least 6 h. Cells were then lysed, and luciferase activities were measured. All values were normalized to those of unstimulated cells with reporter and empty FLAG-CMV vector (CMV). Each bar represents the average of two independent experimental values. B, the chemical structures of the novel TLR2 agonists. Dashed lines show the common structures identified for certain pairs of compounds.
All of the active compounds have low molecular masses ranging from 300 to 500 daltons and are structurally unrelated to any known TLR2 agonists (Fig. 1B). Two of the most active compounds, B and C, share the same core structure consisting of 3-carboxylbenzothiophene linked via a carbonothioylamino bridge to an anilino group. It should be also noted that compounds E and F, which activate cells through TLR2 and TLR6, also contain the same core chemical structure composed of N1-(benzyl)-N2-(phenyl)-N2-(sulfonyl)glycinamide. The independent identification of compounds with similar core structures provides confidence that our screen has identified bona fide TLR2 agonists. The compounds represent the physically smallest agonists for TLR2 reported to date.
Compounds A, B, and C Are Potent and TLR-specific
Compounds A, B, and C were selected for further analyses as they exhibited the highest levels of TLR-dependent stimulation. The activation of TLR2-1 was detected at compound concentrations as low as 30 nm (10 ng/ml), and stimulatory activities comparable with that of Pam3CSK4 were achieved when the concentration reached 3 μm (1 μg/ml) (Fig. 2). The does-response curves of compounds A and B continued to rise over a broad concentration, and saturable levels of reporter activation were not observed even at 30 μm (10 μg/ml). In contrast, despite structural similarity to compound B, compound C appears to reach saturable activation levels at a concentration of 300 nm (100 ng/ml). Notably, none of the three compounds exhibited any activity toward TLR2-6, even at the highest concentration tested.
FIGURE 2.

Compounds A, B, and C are potent TLR2-1 agonists. SW620 cells were co-transfected with vectors encoding the indicated TLRs, an IL-8-driven firefly luciferase gene, and a Renilla luciferase control. Two days after transfection, cells were stimulated with different concentrations of compound A, B, or C for 6 h, and cell lysates were analyzed for Dual-Luciferase activity. After correcting for transfection efficiency using Renilla luciferase, all values were normalized to those of unstimulated cells transfected with empty FLAG-CMV vector (CMV). Error bars represent the S.D. of three independent wells stimulated with the indicated concentration of agonist.
To confirm the specificity of the compounds for TLR1 and TLR2, we treated murine peritoneal macrophages derived from wild type, TLR1, TLR2, and TLR6 knock-out mice with the compounds and measured stimulation of IL-6 production (Fig. 3A). The control stimuli were triacylated lipopeptide PamOct2CSK4 and diacylated lipopeptide MALP-2, which have been identified as strong murine TLR2-1 and TLR2-6 agonists, respectively (16, 32). As expected, macrophages from both TLR2-deficient and TLR6-deficient mice were unresponsive to MALP-2 stimulation, whereas those from TLR1-deficient mice responded well. Conversely, the triacylated lipopeptide PamOct2CSK4 showed reduced activity toward both TLR1-deficient and TLR2-deficient cells but not TLR6-deficient cells. Compound A activated murine macrophages in both a TLR1-dependent and a TLR2-dependent manner, confirming its specificity toward these two receptors. In contrast, compounds B and C did not stimulate cytokine production even from macrophages derived from wild type mice. These results suggest that compounds B and C activate human but not mouse TLRs. To confirm this finding, we examined the activity of the compounds toward RAW 267.4 cells (Fig. 3B). Although compound A was active, compounds B and C failed to induce TNF-α production in this murine-derived macrophage cell line. These results show that compounds B and C exhibit species-specific activity for human but not mouse TLR2-1. Although inactive, neither compound B nor compound C was observed to have antagonistic activity for mouse TLR2 (data not shown).
FIGURE 3.

Compound A, but not B and C, induces IL-6 production in murine macrophages in a TLR1- and TLR2-dependent manner. A, murine peritoneal macrophages prepared from wild type (W.T.), TLR1−/−, TLR2−/−, and TLR6−/− were plated at the density of 1 × 105 cells/well. Cells were then stimulated with 10 μg/ml compound A, B, or C, 100 ng/ml PamOct2CSK4, or 100 ng/ml MALP-2 for 16 h. IL-6 release was measured in the culture supernatant by ELISA. B, RAW cells were plated at a density of 0.8 × 105/well and were stimulated with 5 μg/ml compound A, B, or C for 16 h, and TNF-α release was measured in the culture supernatant by ELISA. Error bars represent the S.D. of values obtained from three independent wells.
Compounds A and B Do Not Antagonize or Synergize with Lipopeptides
Given the distinct structural features between the compounds and the lipopeptide agonists, we next tested the ability of these agonists to exhibit synergy in the activation of TLR2-1. Cells co-expressing TLR2 and TLR1 were stimulated with various concentrations of Pam3CSK4 together with a constant concentration of compound A or compound B (Fig. 4A). An additive response was observed, suggesting a lack of any synergistic interaction. As the concentration of Pam3CSK4 exceeded 10 ng/ml, the more potent stimulatory effect of Pam3CSK4 began to overshadow that of the compounds, suggesting that the former has a higher binding affinity for TLR2-1 when compared with the latter. Given their weaker activity, we next examined whether the compounds could act as antagonists toward Pam3CSK4. To this end, cells were incubated with compound A or compound B for 30 min prior to stimulation with Pam3CSK4 (Fig. 4B). No inhibition was observed, suggesting that the weaker stimulatory activity of the compounds was due to either weaker receptor affinity and/or occupation of unique binding sites on the receptors when compared with the lipopeptides. Although compound A and compound B are structurally distinct small molecule activators, we have observed that they do not exhibit synergism or antagonism with respect to each other (data not shown).
FIGURE 4.

Compounds A and B do not synergize or antagonize Pam3CSK4 activity. SW620 cells were transfected with TLR2, TLR1, IL-8 reporter, and Renilla transfection control. A, 2 days after transfection, cells were stimulated with increasing concentrations of Pam3CSK4 alone or Pam3CSK4 along with either compound A or compound B (1 μg/ml) for 6 h. B, 2 days after transfection, cells were pretreated with compound A or B (1 μg/ml) for 30 min followed by stimulation with increasing concentrations of Pam3CSK4 for 6 h. Luciferase activity was measured as described before. Error bars represent the S.D. of values obtained from three independent wells.
The Central LRRs of TLR1 Are Essential for Activation by the Compounds
We have previously found that the central LRRs of TLR1 and TLR6 are responsible for discriminating between Pam3CSK4 and MALP-2 agonists, respectively, using domain-swapping experiments in which LRRs were exchanged between these two receptors (28). In this approach, all of the chimeric receptors maintain activity toward one of the lipopeptide agonists, indicating that in all cases, the solenoid structure is preserved. To define the region of the extracellular domain in TLR1 responsible for compound recognition, we examined the ability of our compounds to activate the chimeric receptor constructs in conjunction with TLR2 following transient transfection in SW620 cells (Fig. 5). As observed previously, wild type TLR1, in cooperation with TLR2, mediated robust responses to both compounds and Pam3CSK4. An N-terminal exchange construct T6(1–8)/T1, in which LRRs 1 through 8 of TLR1 were replaced with those of TLR6, was activated by the compounds and by Pam3CSK4. However, when the N-terminal replacement of TLR1 with TLR6 was extended to LRR12 (construct T6(1–12)/T1), responses to all the agonists were completely lost. Additionally, the reverse chimera T1(1–8)/T6, in which the first eight LRRs of TLR6 were replaced with those of TLR1, was completely inactive toward all the agonists. Only when the first 12 N-terminal LRRs of TLR1 were replaced with those of TLR6 did the resulting chimera T1(1–12)/T6 exhibit any activity toward the compounds and Pam3CSK4. It is worthy to note that the activation of T1(1–12)/T6 by compounds B and C was barely detectable. Finally, an internal swap chimera T1(6–17)/T6, in which LRRs 6 through 17 of TLR6 replaced those of TLR1, restored activity to all of the agonists. Taken together, these results indicate that similar to Pam3CSK4, the central extracellular domain composed of LRRs 9 through 12 of TLR1 is required for recognition of compound agonists.
FIGURE 5.

The central extracellular region composed of LRRs 9–12 of TLR1 is required for compound-mediated cell activation. SW620 cells were co-transfected with TLR2 and the indicated TLR1 chimeras, an IL-8-driven luciferase gene, and a Renilla transfection control. Cells were stimulated with 5 μg/ml compound A, B, or C or 20 ng/ml Pam3CSK4 for 6 h. Luciferase activity was measured as described before. Error bars represent the S.D. of values obtained from three independent wells. CMV, FLAG-CMV vector. TIR, Toll/IL-1R.
Compound-mediated Activation of TLR2-1 Is Distinct from That of Pam3CSK4
Sequential LRR motifs form spring-like structures in which each LRR contributes a single turn with hydrophobic residues buried in the interior of the solenoid (33, 34). The crystal structure of the human TLR2-TLR1-Pam3CSK4 complex (PDB 2z7x) reveals that the TLR1 and TLR2 solenoids form an m-shaped heterodimer in which the Pam3CSK4 ligand is coordinately bound. In the complex, the two acyl chains of the diacyl glycerol unit of Pam3CSK4 are bound to a hydrophobic pocket of TLR2, whereas the third amide-bound acyl chain of the ligand interacts with a more narrow hydrophobic channel within the central region of TLR1 (Fig. 6A). In addition to sharing ligand binding, the receptors themselves are predicted to make direct contacts in this central region through a number of hydrogen bonds and hydrophobic interactions (35).
FIGURE 6.

Compound-mediated activation of TLR2-1 is distinct from that of Pam3CSK4. A, electrostatic surface representations of the TLR1 and TLR2 heterodimer interface based upon the crystal structure of the TLR2-TLR1-Pam3CSK4 complex (35). The PyMOL software was used to split open the TLR2-TLR1-lipopeptide complex, and each TLR was rotated 90 degrees to display the dimer interfaces of TLR1 and TLR2. Negative and positive charges are shown in red and blue, respectively. The lipopeptide agonist Pam3CSK4 is repeated in stick form in both images with carbons, nitrogens, oxygens, and sulfur colored in green, blue, red, and yellow, respectively. Mutated amino acid residues of TLR1 along with corresponding residues on TLR2 that are presumed to make interactions are indicated. B, SW620 cells were co-transfected with TLR2 and the indicated TLR1 mutants, an IL-8-driven luciferase gene, and a Renilla transfection control. Cells were stimulated with 5 μg/ml compound A, B, or C or 20 ng/ml Pam3CSK4 for 6 h. Luciferase activity was measured as described before. Error bars represent the S.D. from three independent wells. The experiment was repeated three times without significant differences between replicates. A representative experiment is shown. CMV, FLAG-CMV vector.
Given the importance of the central LRRs in the TLR2-TLR1-lipopeptide complex, we generated a random mutagenesis library in which TLR1 variants with single amino acid changes were generated. Through screening of the library, several TLR1 variants were identified that affect responses to Pam3CSK4 (31). To further explore the mechanisms by which TLR recognition of the small compounds and Pam3CSK4 leads to cellular activation, we compared the responses of TLR1 point mutant receptor variants to the different TLR2-1 synthetic agonists. Tyr-320 of TLR1 is predicted to make hydrophobic interactions with residues Leu-324 and Tyr-323 of TLR2. There are also predicted ionic interactions between Glu-321 of TLR1 and Arg-321 of TLR2 (Fig. 6A). The individual substitution of these TLR1 residues results in an overall partial loss of responses to both Pam3CSK4 and the compounds (Fig. 6B). A charged amino acid substitution at Val-339 of TLR1, which engages the center of a hydrophobic patch of TLR2 composed of Phe-322, Phe-349, and Leu-371, resulted in a dramatic and uniform loss of receptor activity to all the agonists. These results suggest that unimpaired receptor dimer interaction is required to mediate efficient responses to both the chemical compounds and the lipopeptides.
Several of the point mutants involve amino acid changes at positions 311–316 of TLR1. These residues form a loop that contributes to the receptor dimer interface and also reside at sites of interaction with the lipopeptide (Fig. 6A). In the TLR2-TLR1-lipopeptide complex, Val-311 of TLR1 appears to make hydrophobic contacts with a hydrophobic patch of TLR2 contributed by Leu-350, Pro-352, and Tyr-376. A TLR1 variant in which Val-311 of the loop is replaced with Glu retained activity toward Pam3CSK4 but exhibited highly attenuated responses to compound A and partial activity to compounds B and C (Fig. 6B). When Phe-314, which appears to orient the protein backbone of the loop through intramolecular contacts with TLR1, is substituted with a charged Lys residue, the responses to either lipopeptide or compound A are almost completely abolished, but those to either compound B or compound C are partially preserved. P315L is a naturally occurring polymorphism of TLR1 that exhibits highly attenuated responses to Pam3CSK4 (29). This proline residue resides in the channel that accommodates the peptide and is predicted to engage TLR2 through hydrophobic interactions (35). Similar to lipopeptides, this receptor variant also displayed a highly attenuated activity to compound A, but responses to compounds B and C were partially retained (Fig. 6B). In addition to the acyl chains, TLR1 also interacts with Pam3CSK4 by forming a hydrogen bond between the side chain of Gln-316 and the amide oxygen of the lipopeptide. The fact that substitution of Gln-316 with lysine abrogated Pam3CSK4 stimulation indicates that this is an important receptor-ligand interaction. Interestingly, this TLR1 variant retained greater than half-maximal responses to all the chemical compounds. Taken together, the results suggest that although the gross configuration of the TLR2-1 heterodimer is similar for the different agonists, there are subtle differences at the receptor interface important for driving cellular activation.
The Compounds Stimulate TNF-α Production from Human Peripheral Monocytes
We next evaluated the ability of compounds A, B, and C to activate human monocytes isolated from peripheral blood of healthy donors (Fig. 7). We found that the compounds dose dependently activated TNF-α production in human monocytes. Although all three compounds induced TNF-α release at micromolar concentrations, compounds B and C exhibited weaker activity than compound A. The fact that the compounds are able to activate primary human cells suggests that they may have clinical utility, perhaps as novel vaccine adjuvants.
FIGURE 7.
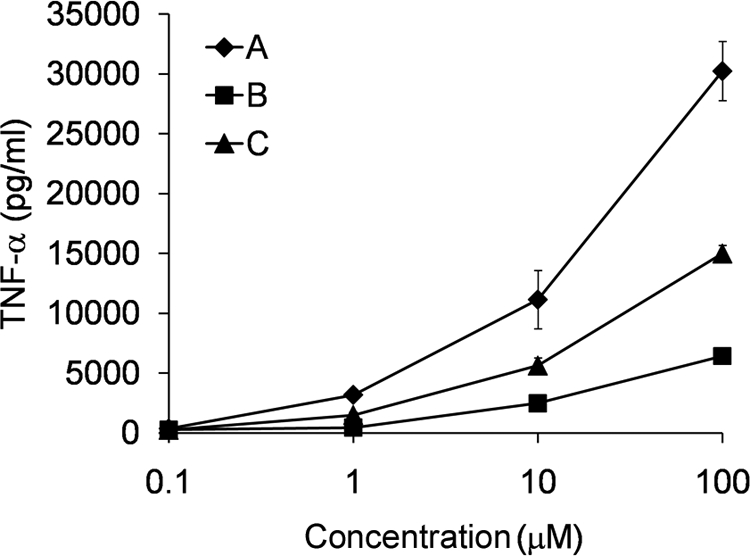
Compounds A, B, and C induce TNF-α production from human monocytes. Human peripheral blood monocytes were cultured at a density of 1 × 105 cells/well with increasing concentrations of compound A, B, or C for 18 h. TNF-α release was measured in the culture supernatant by ELISA. Error bars represent the S.D. of values obtained from three independent wells.
DISCUSSION
In this study, we have adapted a cell-based TLR2 activity assay to a format that permits screening of small chemical libraries for novel TLR2 agonists. The assay utilizes an SW620 epithelial cell line expressing TLR1, TLR2, and TLR6 as well as a luciferase reporter driven by the promoter of the IL-8. This assay remains sensitive and robust, exhibiting a 30–50-fold increase in luciferase activity in response to TLR2 lipopeptide agonists despite adaptation to a 384-well format in which measurements are derived from as few as 5,000 cells/well. An initial screen of 24,000 compounds provided 217 hits, and a secondary screen, which permits as assessment of TLR2 dependence, revealed five compounds with greater than 5-fold activity. All five compounds share five or six atom-ring structures including phenol, thiophene, and imidazole groups, which are absent in TLR2 natural agonists. Compounds B and C share a core (anilinocarbonothioyl) amino-benzothiophene structure. Similarly, compounds E and F are both sulfonylglycinamides with terminal phenyl groups. These compounds are the smallest TLR2 agonists identified to date and only slightly larger than the imidazoquinoline agonists for TLR7 and TLR8. The independent identification of structurally similar compounds from a diverse chemical library serves to validate the screening assay and demonstrates that it presents a robust method of identifying novel TLR2 agonists.
TLR activation involves agonist-induced receptor dimerization. This event brings together two receptor signaling domains that serve as platforms for the recruitment of adaptor molecules required to initiate signaling (9, 36). The structure of the TLR2-TLR1-Pam3CSK4 complex reveals coordinate binding of the lipopeptide by both receptors in which two acyl chains are embedded within a hydrophobic pocket of TLR2 with the third amide-linked acyl chain bound within a hydrophobic channel of TLR1 (35). Additionally, hydrogen bonds between the glycerol and peptide backbone of Pam3CSK4 and the TLRs, as well as direct contacts between the receptors themselves at the receptor dimer interface, contribute to stable heterodimer formation. As diacylated lipopeptide agonists of TLR2-6 lack the third amide-linked acyl chain, the TLR2-TLR6-Pam2CSK4 complex relies more heavily on these latter interactions. Indeed, the crystal structure reveals that the hydrophobic channel of TLR6 cannot accommodate long acyl chains due to blockage by two phenylalanine residues (37).
Assessment of both domain exchange and point mutants revealed that similar to lipopeptides, activation by our compounds requires the central LRRs of TLR1. Strikingly, TLR1 residues Tyr-320, Glu-321, and Val-339, which are farther removed from the Pam3CSK4 binding site and appear to interact with TLR2 at the receptor dimer interface, make similar contributions to receptor activity independent of the agonist used. Taken together, these results suggest that the orientation and interactions between TLR1 and TLR2 in the activated heterodimer complex are similar irrespective of the agonist.
Despite these similarities, the lipopeptide and chemical agonists differ in their ability to activate TLR1 variants that possess individually altered residues located in the loop of LRR11. These residues line the lipopeptide binding pocket, and many are presumed to interact with TLR2 at the receptor dimer interface. The stronger inhibitory effect of the Gln-316 mutation on Pam3CSK4 activation versus the compounds is supported by the crystal structure, which predicts a hydrogen bond between this residue and the peptide backbone of the lipopeptide. However the differential effects of individual TLR1 mutations at Val-311, Phe-314, or Pro-315 are harder to explain and suggest that the contribution of these residues, either to a ligand binding pocket or to interactions at the receptor dimer interface, is subtly different dependent upon the agonist. As expected, however, the effect of TLR1 mutations on the activity of the structurally related compound B and C agonists is similar. Interestingly, compounds B and C were inactive toward murine cells and thus exhibit species specificity for human but not mouse TLR2-1. To our knowledge, the only other species-specific TLR2 agonist reported to date is a trilauroylated lipopeptide, which stimulates mouse but not human TLR2 (38).
The activity of our point mutants, and the fact that the bulky phenyl groups of our compounds are too large to be accommodated by the hydrophobic channel of TLR1, suggests that our compounds bind directly within the interface of the TLR2-1 heterodimer. It is important to note that even the most active compounds identified in our screen are ∼3 orders of magnitude less potent in the TLR2 activation assay than the lipopeptide control agonists (Fig. 2). This is perhaps not surprising given that none of the screened chemical libraries contained compounds with long acyl chains, which are known to contribute to ligand binding and heterodimer formation. Similarly, a wide variety of naturally occurring agonists that lack acyl chains are far less potent TLR2 activators than bacterial lipopeptides and lipoproteins (13, 14). Because TLR2 mediates responses to such a wide range of agonists, additional ligand docking analyses, mutagenesis studies, and crystal structures will be required to fully understand the full scope of interactions that can ultimately drive heterodimer formation and activation of TLR2 complexes.
The incorporation of TLR agonists in vaccine development represents a promising mechanism to boost immune responses to infectious agents and tumor antigens. However, inflammatory toxicity associated with strong TLR agonists limits their broad application (39, 40). The TLR2-dependent adjuvants under development include lipopeptides, zwitterionic polysaccharides, and larger bacteria-derived proteins (22–26). Although we have not tested compounds A, B, and C for adjuvanticity, we have observed that these synthetic TLR2 agonists weakly induce TNF-α production from human monocytes (Fig. 7). We have also found that even at high concentrations, the compounds fail to activate TLR2 as potently as lipopeptides (Fig. 2). This may be of clinical benefit as such weak or partial agonists often avoid the toxicity associated with strong inflammation. For example, the lipopolysaccharide analogue monophosphoryl lipid A is a weak or partial TLR4 agonist and an effective adjuvant in hepatitis B vaccine formulations approved in Europe and Argentina. The small size and defined chemistry of the compounds also favors direct conjugation to antigen, an approach with lipopeptide that elicits robust antibody responses to tumor antigens in mice (41).
In conclusion, the high throughput screening assay developed in this study has uncovered novel synthetic small molecule TLR2 agonists. Next generation analogues of the compounds may exhibit pharmacologic and clinically favorable characteristics and will enable a closer examination of the structure-function relationship between agonists and TLR2 complexes.
Acknowledgments
We gratefully acknowledge the High Throughput Screening Facility and the Division of Animal Resources at the University of Illinois for assistance with these studies.
This work was supported, in whole or in part, by National Institutes of Health Grant AI052344 (to R. I. T.).
- TLR
- Toll-like receptor
- IL
- interleukin
- Pam3CSK4
- N-palmitoyl-(S)-[2,3-bis(palmitoyloxy)-propyl]-(R)-cysteinyl-(lysyl)3-lysine
- MALP-2
- S-[2,3-bis(palmityloxy)-propyl]-(R)-cysteinyl-GNNDESNISFKEK (macrophage-activating lipopeptide-2)
- PamOct2CSK4
- N-palmitoyl-(S)-[2,3-bis(octonoyloxy)-propyl]-(R)-cysteinyl-(lysyl)3-lysine
- TNF
- tumor necrosis factor
- LRR
- leucine-rich repeat
- DMSO
- dimethyl sulfoxide
- ELISA
- enzyme-linked immunosorbent assay.
REFERENCES
- 1.Fearon D. T. (1997) Nature 388, 323–324 [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R., Janeway C. A., Jr. (1997) Cell 91, 295–298 [DOI] [PubMed] [Google Scholar]
- 3.Muzio M., Bosisio D., Polentarutti N., D'amico G., Stoppacciaro A., Mancinelli R., van't Veer C., Penton-Rol G., Ruco L. P., Allavena P., Mantovani A. (2000) J. Immunol. 164, 5998–6004 [DOI] [PubMed] [Google Scholar]
- 4.Zarember K. A., Godowski P. J. (2002) J. Immunol. 168, 554–561 [DOI] [PubMed] [Google Scholar]
- 5.Akira S., Uematsu S., Takeuchi O. (2006) Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 6.Iwasaki A., Medzhitov R. (2004) Nat. Immunol. 5, 987–995 [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z., Schluesener H. J. (2006) Cell. Mol. Life Sci. 63, 2901–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Neill L. A., Bryant C. E., Doyle S. L. (2009) Pharmacol. Rev. 61, 177–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Neill L. A., Bowie A. G. (2007) Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 10.Kawai T., Akira S. (2007) Trends. Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 11.Aliprantis A. O., Yang R. B., Mark M. R., Suggett S., Devaux B., Radolf J. D., Klimpel G. R., Godowski P., Zychlinsky A. (1999) Science 285, 736–739 [DOI] [PubMed] [Google Scholar]
- 12.Brightbill H. D., Libraty D. H., Krutzik S. R., Yang R. B., Belisle J. T., Bleharski J. R., Maitland M., Norgard M. V., Plevy S. E., Smale S. T., Brennan P. J., Bloom B. R., Godowski P. J., Modlin R. L. (1999) Science 285, 732–736 [DOI] [PubMed] [Google Scholar]
- 13.Miyake K. (2007) Semin. Immunol. 19, 3–10 [DOI] [PubMed] [Google Scholar]
- 14.Tapping R. I. (2009) Semin. Immunol. 21, 175–184 [DOI] [PubMed] [Google Scholar]
- 15.Ozinsky A., Underhill D. M., Fontenot J. D., Hajjar A. M., Smith K. D., Wilson C. B., Schroeder L., Aderem A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi O., Kawai T., Mühlradt P. F., Morr M., Radolf J. D., Zychlinsky A., Takeda K., Akira S. (2001) Int. Immunol. 13, 933–940 [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi O., Sato S., Horiuchi T., Hoshino K., Takeda K., Dong Z., Modlin R. L., Akira S. (2002) J. Immunol. 169, 10–14 [DOI] [PubMed] [Google Scholar]
- 18.Guan Y., Ranoa D. R., Jiang S., Mutha S. K., Li X., Baudry J., Tapping R. I. (2010) J. Immunol. 184, 5094–5103 [DOI] [PubMed] [Google Scholar]
- 19.Kanzler H., Barrat F. J., Hessel E. M., Coffman R. L. (2007) Nat. Med. 13, 552–559 [DOI] [PubMed] [Google Scholar]
- 20.Makkouk A., Abdelnoor A. M. (2009) Immunopharmacol. Immunotoxicol. 31, 331–338 [DOI] [PubMed] [Google Scholar]
- 21.Casella C. R., Mitchell T. C. (2008) Cell. Mol. Life Sci. 65, 3231–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eriksson E. M., Jackson D. C. (2007) Curr. Protein Pept. Sci. 8, 412–417 [DOI] [PubMed] [Google Scholar]
- 23.Moyle P. M., Toth I. (2008) Curr. Med. Chem. 15, 506–516 [DOI] [PubMed] [Google Scholar]
- 24.Gallorini S., Berti F., Mancuso G., Cozzi R., Tortoli M., Volpini G., Telford J. L., Beninati C., Maione D., Wack A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 17481–17486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang S., Hosur K. B., Nawar H. F., Russell M. W., Connell T. D., Hajishengallis G. (2009) Vaccine 27, 4302–4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X., Wetzler L. M., Massari P. (2008) Vaccine 26, 786–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pazzagli M., Devine J. H., Peterson D. O., Baldwin T. O. (1992) Anal. Biochem. 204, 315–323 [DOI] [PubMed] [Google Scholar]
- 28.Omueti K. O., Beyer J. M., Johnson C. M., Lyle E. A., Tapping R. I. (2005) J. Biol. Chem. 280, 36616–36625 [DOI] [PubMed] [Google Scholar]
- 29.Omueti K. O., Mazur D. J., Thompson K. S., Lyle E. A., Tapping R. I. (2007) J. Immunol. 178, 6387–6394 [DOI] [PubMed] [Google Scholar]
- 30.Cirino P. C., Mayer K. M., Umeno D. (2003) Methods Mol. Biol. 231, 3–9 [DOI] [PubMed] [Google Scholar]
- 31.Liang S., Hosur K. B., Lu S., Nawar H. F., Weber B. R., Tapping R. I., Connell T. D., Hajishengallis G. (2009) J. Immunol. 182, 2978–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buwitt-Beckmann U., Heine H., Wiesmüller K. H., Jung G., Brock R., Akira S., Ulmer A. J. (2006) J. Biol. Chem. 281, 9049–9057 [DOI] [PubMed] [Google Scholar]
- 33.Bella J., Hindle K. L., McEwan P. A., Lovell S. C. (2008) Cell. Mol. Life Sci. 65, 2307–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin M. S., Lee J. O. (2008) Immunity 29, 182–191 [DOI] [PubMed] [Google Scholar]
- 35.Jin M. S., Kim S. E., Heo J. Y., Lee M. E., Kim H. M., Paik S. G., Lee H., Lee J. O. (2007) Cell 130, 1071–1082 [DOI] [PubMed] [Google Scholar]
- 36.Monie T. P., Moncrieffe M. C., Gay N. J. (2009) Immunol. Rev. 227, 161–175 [DOI] [PubMed] [Google Scholar]
- 37.Kang J. Y., Nan X., Jin M. S., Youn S. J., Ryu Y. H., Mah S., Han S. H., Lee H., Paik S. G., Lee J. O. (2009) Immunity 31, 873–884 [DOI] [PubMed] [Google Scholar]
- 38.Grabiec A., Meng G., Fichte S., Bessler W., Wagner H., Kirschning C. J. (2004) J. Biol. Chem. 279, 48004–48012 [DOI] [PubMed] [Google Scholar]
- 39.Hauguel T. M., Hackett C. J. (2008) Front. Biosci. 13, 2806–2813 [DOI] [PubMed] [Google Scholar]
- 40.Lahiri A., Das P., Chakravortty D. (2008) Vaccine 26, 6777–6783 [DOI] [PubMed] [Google Scholar]
- 41.Ingale S., Wolfert M. A., Gaekwad J., Buskas T., Boons G. J. (2007) Nat. Chem. Biol. 3, 663–667 [DOI] [PMC free article] [PubMed] [Google Scholar]