

Abstract
Many double-stranded DNA viruses employ ATP-driven motors to translocate their genomes into small, preformed viral capsids against large forces resisting confinement. Here, we show via direct single-molecule measurements that a mutation T194M downstream of the Walker B motif in the phage λ gpA packaging motor causes an 8-fold reduction in translocation velocity without substantially changing processivity or force dependence, whereas the mutation G212S in the putative C (coupling) motif causes a 3-fold reduction in velocity and a 6-fold reduction in processivity. Meanwhile a T194M pseudorevertant (T194V) showed a near restoration of the wild-type dynamics. Structural comparisons and modeling show that these mutations are in a loop-helix-loop region that positions the key residues of the catalytic motifs, Walker B and C, in the ATPase center and is structurally homologous with analogous regions in chromosome transporters and SF2 RNA helicases. Together with recently published studies of SpoIIIE chromosome transporter and Ded1 RNA helicase mutants, these findings suggest the presence of a structurally conserved region that may be a part of the mechanism that determines motor velocity and processivity in several different types of nucleic acid translocases.
Keywords: DNA Viruses, Mutant, Protein Motifs, Single Molecule Biophysics, Virus Assembly
Introduction
Viral DNA packaging motors are remarkable biological nanomachines that are capable of packaging chromosomes to near-crystalline density into viral capsids against enormous internal forces (1, 2). Such motors are present in many double-stranded DNA bacteriophages (3), the medically relevant herpesviruses (4), and possibly in poxviruses and adenoviruses (5). The well characterized genetics and biochemistry of the bacteriophage λ system make it an ideal model system for investigating the structure-function relationships of DNA-packaging machines (6, 7). The “terminase enzyme” is the major active component of the λ packaging motor responsible for DNA translocation. It is composed of multiple heterotrimer units, each containing a large gpA subunit and two small gpNu1 subunits (8). This ATP-powered complex mediates a number of different activities needed to package unit length genomes from concatemeric substrates; these include endonuclease, strand separation, ATP hydrolysis, and DNA translocation catalytic activities. The terminase enzyme initiates packaging by binding to a cos site in a linear DNA concatemer composed of several viral genomes. Terminase introduces staggered nicks at the downstream cosN site, 12-bp apart to form the left mature genome end. The motor then binds to a procapsid and translocates DNA into the capsid interior. Packaging is terminated when the terminase encounters the next downstream cos site, which signals the right end of the genome; terminase again nicks the duplex, releasing the DNA-filled capsid. Recent evidence indicates that gpA1(gpNu1)2 heterotrimers assemble into a tetrameric ring, which we presume is representative of the DNA maturation and packaging motor complex (9).
Our earlier studies of the ATPase center of the λ packaging motor included mutational analysis and ATP binding studies, which indicated that residues Tyr46 and Lys84 of gpA were involved in ATP binding associated with packaging (10, 11). A collection of mutations causing motor defects was mapped to the gene segment encoding the amino terminus of gpA (12). Mutants L180F and G212S could package DNA but never packaged >31 kbp of DNA of the 48.5-kbp genome; we referred to this as a stalled translocation phenotype. Other mutants such as G191S abolished packaging completely, suggesting that these residues are located in important sites for translocation. Among the most interesting mutants isolated was T194M, for which bulk DNase protection assays showed that the full λ genome could be packaged in vivo, but only if the incubation time was extended from 1 to 2.5 h. However, it is unclear from these assays whether the reduced packaging activity of T194M is due to (i) slowed assembly of the motor-DNA and/or motor-DNA-procapsid complexes, (ii) slowed initiation of packaging, (iii) slowed DNA translocation rate, or (iv) increased motor pausing, stalling, or slipping (decreased processivity).
Here, we investigate the effects of mutations on packaging dynamics using a single-molecule optical tweezers assay that measures directly DNA translocation by individual motor complexes in real-time with 0.1-s precision. This technique revealed that the WT6 λ motor-DNA complex can bind the prohead and initiate packaging within seconds, that the average DNA translocation velocity is ∼600 bp/s at the beginning of packaging and slows as packaging proceeds, and that the full-length genome could be packaged in ∼2 min. We further used sequence alignments and packaging measurements of the mutant Y46F to show that gpA contains a “Q motif” that regulates motor-DNA interactions and governs motor force and power generation (13).
Optical tweezer assays also were applied previously to measuring DNA packaging dynamics of the ϕ29 and T4 phage (1, 14–16). In all cases, most strikingly in the T4 system, these measurements of revealed variability in the packaging dynamics of individual complexes, suggesting that the motor may have different conformational states producing different translocation velocities (17). The average DNA translocation rate correlated with the genome sizes of the various phages (1, 17, 18).
Many molecular motor NTPases have common functional motifs including Walker A motifs, with the consensus sequence GXXXXGK(T/S), that are implicated in binding ATP and Walker B motifs with conserved adjacent Asp and catalytic Glu (DE) residues, which are implicated in arranging Mg2+ and water molecules to hydrolyze ATP (19). Regions downstream from the putative Walker B motif of gpA have not been investigated previously by single molecule measurements and may thus have undetermined functions that relate to the velocity of the motor. For example, the Thr194 residue of gpA is located in a functionally unassigned region between the Walker B motif and the “C motif”.
Sequence alignments suggest that a putative C motif of λ terminase is located 30 residues distal to the Walker B (20, 21). Recent work in a related system, phage T4, has found that mutations of the C motif of the DNA packaging motor result in single turnover hydrolysis with no translocation, suggesting that C motifs of viral packaging motors act as ATP hydrolysis sensors, which couple hydrolysis to translocation of DNA (20). Evidence from crystal structures and bulk chemical assays also strongly suggest the analogous motif III in RNA helicases has important roles in substrate interaction and ATP hydrolysis (22, 23).
In this study, we use single molecule optical tweezers measurements to directly investigate the effect of mutations altering this region of gpA on real-time DNA translocation dynamics. We find that T194M mutants translocate 8–10× slower than WT terminases with no change in processivity, unique for this class of motors. In addition, we find that the mutant G212S has a 3× decreased translocation rate compared with WT but also a large decrease in processivity. We also use genetic screening experiments to identify pseudorevertants of these null mutants that restore the viability of the phages. Structural modeling based on sequence homology and the known structure of phage T4 gp17 indicate that these residues lie in a loop-helix-loop region between the Walker B motif and putative C motif that can position these catalytic motifs in the ATPase center. Comparisons with chromosome transporters and DEAD-box RNA helicases suggest this is a structurally conserved region that may determine velocity and processivity in several different types of nucleic acid translocases.
EXPERIMENTAL PROCEDURES
cos Cleavage
A 34.7-kbp λ biotinylated DNA construct was made as described previously (18). The biotinylated DNA (2 μl DNA (30–40 μg/ml) was incubated with streptavidin-coated microspheres (2.1 μm) for 30 min to create DNA-coated microspheres in a buffer consisting of 20 mm Tris, 10 mm magnesium, 7 mm β-mercaptoethanol at pH 8.0.
Purified complexes of gpNu1 and gpA and crude extracts yielded nearly identical packaging results for mutant- and WT-packaging events (13). 2–2.5 μl of crude cellular extracts containing gpNu1, gpA, and integration host factor were added to the DNA-coated microspheres. Alternatively, in an assay using purified proteins, integration host factor (33 nm final) and 4 μm terminase (267 nm final) were incubated with DNA-coated microspheres for 20 min to 1 h. This reaction afforded the maturation of λ and the binding of terminase to DNA. Protein G microspheres (2.1 nm) were incubated with IgG λ antibodies to create IgG λ microspheres. To make procapsid-bound microspheres, procapsids were incubated with IgG λ microspheres for 30 min.
Initiation of Packaging in an Optical Tweezers Assay
Two microspheres are held in two laser traps, one of which was moved relative to the other using an acousto-optic deflector. Viral packaging of DNA could be initiated by bringing prohead microspheres in contact with DNA/terminase-coated microspheres. The viral DNA packaging assay using optical tweezers was applied to the λ virus using purified terminase and proheads. This induced the assembly of the terminase complex on λ DNA and the maturation of this DNA through its endonuclease and helicase activities. In the λ packaging assay, terminase/DNA-bound beads were brought into contact of prohead-bound microspheres and pulled apart until they reached 5 pN for at least 70 s unless there was instant initiation. Packaging was initiated under saturating ATP conditions (1 mm) in a buffer consisting of 20 mm Tris-HCl and 10 mm magnesium at a pH of 7.5.
Instrumentation
All measurements of DNA packaging were taken using a dual beam trap optical tweezers instrument described previously, under a constant force feedback of ∼5 pN or using a passive mode enabling the measurement of force generation as packaging progressed (24).
Data Analysis
Large slips (>100 bp) and long pauses (>0.2 s) were marked and compiled using custom-made software in Matlab. The tpauses and tslips used in efficiency calculations (Equation 1) were the durations of pausing that end upon resuming of packaging (tpauses) and durations for which the motor slips, resumes packaging, and reaches its previous maximum length translocated before slipping occurs (tslips).
Mutant Purification
Mutations in the A gene, which code for the T194M, T194M/F156Y, T194V, and G212S changes, were introduced into the terminase expression vector pQH101 (25) using the QuikChange II XL site-directed mutagenesis kit (Stratagene). Terminase genes were sequenced to verify that only the desired mutation was present. During sequence analysis, the F156Y change, which resulted from a PCR-introduced mutation, was found in a pQH101 isolate during construction of the T194M expression vector.
Plaque-forming pseudorevertants were isolated by plating 100 λl of lysates of λ-P1 5R:Knr cI857 nin5 (where Knr represents kanamycin-resistant) (26, 27) carrying the T194M or G212S mutations. Lysogens of pseudorevertants served as template for PCR-generated DNA, which was used to sequence the terminase genes. Sequencing assured that the only changes present were the original and second site mutations. To avoid isolation of sibling revertants, 13 independent lysates were examined for the T194M mutant, and 30 for the G212S mutant. All procedures were done using standard phage methods (28).
ATPase Assay
ATPase kinetic activity was determined as described previously (25, 29) using pCTλ as a substrate at 4 nm and ATP at 50 μm. The integration host factor was present in all reactions at 50 μm. Terminase concentration was adjusted between mutants to observe steady-state ATPase activity. Reactions had WT, G212S, and T194M concentrations of 25 nm, 674 nm, and 2.26 μm, respectively. Analysis was performed in the linear range of the reaction. Experiments were performed in triplicate.
Sedimentation Velocity Experiments
Dialyzed protein samples were loaded into Epon charcoal-filled two sector centerpieces, and dialysate was used as a blank. AUC cells were loaded into the precooled rotor and chamber at 7 °C of a Beckman XL-A centrifuge. Experiments were performed at 42,000 rpm, and absorbance was observed at 280 nm at 0.005 cm in continuous scan mode. Determination of a continuous c(s) distribution was performed using SEDFIT. Calculations for buffer density (ρ = 1.03), buffer viscosity (μ = 0.017), and terminase-specific volume (ṽ = 0.7245) were performed with SEDNTERP (30).
RESULTS
Packaging Dynamics of T194M and T194M/F156Y Terminases
Our first measurements employed force clamp feedback control to change the separation between two optical traps as the DNA was translocated into the phage λ capsids under a small constant load (∼5 pN). This enabled us to track double-stranded DNA viral packaging for long distances. The translocation dynamics of several mutant terminases with changes T194M, T194M/F156Y, T194V, and G212S were characterized. Sequence alignments show that these residues are located between the Walker B and ATPase-coupling motifs (C motif) of the large terminase proteins (Fig. 1) (20). Representative packaging events of the mutant terminases are shown in Fig. 2. Notable in each packaging event is the presence of pauses and slips where the motors temporarily stop or temporarily lose grip before resuming packaging. We define the average velocity of translocation (Vmax) as the rate of translocation at saturating ATP conditions (1 mm), excluding the contributions of these slips and pauses (see “Experimental Procedures”).
FIGURE 1.

Conserved features of phage terminases and bacterial translocases. A, sequence alignments (ClustalW) show conservation of ATPase motifs: magenta, Walker A; green, Walker B aspartate; blue, catalytic carboxylate; and red, C motif. Certain features of the slow packaging mutants also are conserved. The λ gpA slow packaging mutants T194M and G212S are located between the Walker B and C motif, as is the SpoIIIe mutant D584A (yellow). The secondary structure of the gpA model (gray) and FtsK structure (green) are shown below the sequence. Numbers in parentheses indicate amino acids not shown. B, the Walker B strand through the C motif strand of E. coli FtsK (green; Protein Data Bank code 2IUS, amino acids 1114–1156), T4 gp17 (violet; Protein Data Bank code 3CPE, amino acids 250–288), and the λ gpA model (gray; amino acids 173–215) were aligned in PyMOL. The gpA slow packaging mutation site Thr194 and the FtsK equivalent of SpoIIIe Asp584, are both in the helices that connect the Walker B and C motif β strands. The gpA mutation site Glu212 is in the C motif β strand, the first residue of the C motif tripeptide, GST.
FIGURE 2.

Representative raw data of packaging events of various terminases using an optical tweezers assay. A schematic depicting the assay is shown in the upper right panel. Beads were held in two optical traps: one bead holds the viral procapsid, and the other holds the DNA/terminase complex. Packaging traces for all mutants shown end with a stall. Packaging was initiated when the beads are brought closer together and the DNA/terminase complex binds to the procapsids. A 5-pN force clamp was utilized to track packaging for long distances.
The T194M single mutant exhibited a Vmax of 82.0 ± 20.7 bp/s, ∼8× slower than WT (Fig. 3). The T194M/F156Y double mutant had similar behavior in translocation dynamics to T194M but with a smaller Vmax of 56.9 ± 8.9 bp/s, a velocity >10× slower than WT. Interestingly, the double mutant also exhibited less variability from complex to complex in translocation velocity than both T194M (S.D. for Vmax of 15% compared with 21% for WT and 25% for T194M). To assess the effects of slipping and pausing on the time it takes for terminase to package DNA, we calculated an efficiency factor as described previously (13).
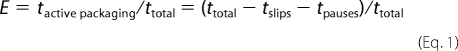 |
This factor quantifies how much packaging is slowed due to pausing and slipping and gives a value for how continuous packaging is in the various terminases. The efficiency factors measured for T194M and T194M/F156Y are near WT (Fig. 3b), which implies that the translocase activity is highly processive. However, these mutants do exhibit increased motor stalling relative to WT, which is not factored in the efficiency factor defined above. Stalls are events where the motor begins translocating but then stops and does not resume again on the time scale of full genome packaging (in <3 min). Stalls in packaging before packaging <50% of the whole genome packaged (instead of the complete dissociation of tethers under a 5-pN load) were found in all packaging measurements of T194M and T194M/F156Y (supplemental Fig. S1). This suggests that stalling is not due to disassembly. Because it was found in bulk biochemical studies that some T194M complexes could eventually package the full-length chromosome after 2.5 h, these mutants may eventually appear to be able to recover from stalling events and complete packaging, or more likely, there only is a very small fraction of these that are able to complete packaging (<4%). Presumably, these are not observed readily in the single molecule experiments due to their low abundance. We note that we expect the in vivo packaging situation to possibly be different from our in vitro packaging measurements because in vivo, the disassembled motor could reassemble or exchange with the terminase subunits. On the other hand, the stalling defects found here along with the 8× slower Vmax and normal pausing and slipping behavior (Fig. 3 and supplemental Fig. S4) compared with WT reconcile the 1.2% measured DNA packaging efficiency found in bulk studies.
FIGURE 3.

Comparison of properties of WT and mutant packaging trajectories measured by optical tweezers assays with a 5-pN force clamp. a, velocity bp/s (Vmax). We define Vmax as the average velocity of translocation with slips and pauses subtracted out of the packaging trajectories, at saturating ATP concentrations (1 mm). Error bars are S.D. b, efficiency (described by Equation 1). Error bars are S.E. c, slips (≥100 bp)/kbp DNA packaged. Error bars are S.E. d, pauses (≥0.2 s)/kbp DNA packaged. Error bars are S.E.
We also carried out experiments where we held the trap positions fixed after initiating packaging to probe the force-generating capability of the mutant complexes. We found a similar relationship of load force versus velocity of translocation (F-V) relationship for T194M when compared with WT (supplemental Fig. S2). This can be contrasted with a much steeper F-V curve measured for the previously studied Q motif mutant, Y46F (13). The functional effects, in terms of velocity, pausing, slipping, force, and power, is distinct from those caused by the K84A and Y46F changes.
Packaging Dynamics of G212S Terminase
The translocation rate of G212S was measured to be ∼3× slower than WT (195 bp/s) with a variability among individual complexes greater than T194M and WT motors (S.D. ∼ 30%). The processivity of this motor also is lower than WT; the slipping frequency of G212S (number of slips per kbp of DNA packaged, number per length = 0.135) is >6× more than WT (0.020) suggesting there are structural defects induced by the mutation (Fig. 3). Despite the decreased processivity, the efficiency of the motor is still 80% of the WT. Similar to T194M and in agreement with bulk biochemical data, G212S displays a stalling phenotype, where no translocation events were measured to exceed 50% of the genome length.
Pseudorevertants of T194M and G212S Terminase
The M194V change was found in plaque-forming pseudorevertants of λ T194M (T194M phages that have additional mutations that partially or fully restore the WT phenotype), and a number of other pseudorevertants have an S230P change, suggesting that the T194V, and T194M/S230P motors must function with near-WT efficiency to sponsor fully infectious virus assembly in vivo. Using single molecule DNA packaging measurements, we found that T194V has a velocity close to WT (420 ± 143 bp/s), implicating the change to Val restored the viability of the virus via the restoration of translocation velocity. The efficiency factor of this motor is similar to WT (0.83 ± 0.05) as expected. (The T194M motor does not show a decrease in efficiency as well.) Stalling statistics of the T194V motor also showed a greater number of events that could reach >50% full genome packaging (supplemental Fig. S1), though the early stalling behavior is still more prevalent than with WT, which would explain why smaller plaque sizes were observed for T194V than WT. A number of second site pseudorevertants for G212S have been isolated and are found in various locations within gpA (Table 1). These have not yet been characterized in detail.
TABLE 1.
Suppressors for lethal mutations of gpA and the locations of the amino acid changes
Modeled residue interactions caused by mutations in the loop-helix-loop region downstream of the Walker B motif and extending into the C motif of the terminase of phage λ also are listed.
| Mutant | Phenotype | Modeled residue interaction | Suppressors |
|---|---|---|---|
| L180S | Stall | Arg211–Pro215 (C motif) | A181S |
| G191S | No packaging | Neighboring residues | None found |
| T194M | Stall, slow (8×) | Phe183 | M194V, S230P |
| G212S | Stall, slow (3×), less processive (6×) | Val74, Ser213 | M61L, E71D, N73S (between Q and Walker A); D185G, S192A (in loop-helix-loop region); and H232Y, H232R, C295S, K297Q, K297N, T323N (near hinge) |
ATPase Activity of Mutant Terminase Holoenzymes
The ATPase activity of the purified terminase holoenzymes also was examined. Terminase possesses three discrete ATPase catalytic activities that are involved putatively in (i) assembly of the motor at cos (gpNu1 ATPase), (ii) modulation of the endonuclease/strand-separation activities of the enzyme, and (iii) fueling motor translocation (packaging ATPase) (6, 31). The packaging ATPase activity can be monitored selectively by using low concentrations of ATP (50 μm) in the reaction mixture because the Km for the packaging ATPase is significantly lower than the other two sites (5 μm versus 500 μm) (29, 32). As can be seen in Fig. 4, the packaging ATPase activities of the G212S and T194M mutant holoenzymes are reduced substantially relative to the WT enzyme (2.5% and 0.8% relative activity, respectively; Table 2). The packaging ATPase activity, which is lower than expected when comparing the less severe reduction in translocation velocity (mutants are 3–10× slower than WT), may be reconciled when taking into account initiation defects (not measured) and the stalling defects previously mentioned (supplemental Fig. S1), which likely terminate hydrolysis before mutants are able to package the full chromosome.
FIGURE 4.

ATP hydrolysis (nmol ADP/nmol terminase/min) for WT (●), G212S (○), and T194M (▲) holoenzymes. ATPase assays were performed as described under “Experimental Procedures.” Each data point represents the average of three separate experiments with S.D. indicated with error bars. Inset, expansion of the data showing slow hydrolysis by the mutant enzymes.
TABLE 2.
Packaging velocities measured by optical tweezer assays and catalytic activities of wild-type and mutant terminase enzymes
NE is not examined.
| Enzyme | Packaging Vmax | ATPase activity |
|---|---|---|
| Wild-type | 580 ± 120 bp/s | 81 ± 8 min−1 |
| G212S | 195 ± 66 bp/s | 2.0 ± 0.2 min−1 |
| T194M | 82.0 ± 20.7 bp/s | 0.6 ± 0.2 min−1 |
| T194M/F156Y | 56.9 ± 8.9 bp/s | NE |
| T194V | 420 ± 143 bp/s | NE |
Structural Features of Mutant Terminase Holoenzymes
We have demonstrated previously that the purified WT terminase holoenzyme partitions into two discrete species, a homogenous protomer composed of one gpA tightly associated with two gpNu1 polypeptides and a heterogenous oligomer predominantly composed of four protomers (the terminase ring species) (8, 9); interconversion between the protomer and homogenous tetrameric ring is facile, but slow. Here, the self-association properties of the purified mutant terminase enzymes were characterized using sedimentation velocity analytical ultracentrifugation as described previously (8, 9). The data presented in supplemental Fig. S3 shows that whereas the G212S mutant exhibits near-WT self-association behavior, the T194M mutant clearly possesses heterogeneous assembly; although the protomer and assembled species are present in the mixture, significant dissociation into smaller components is clearly visible.
DISCUSSION
A Motor Region That Alters DNA Packaging Velocity
Our previous single molecule measurements with ϕ29, λ, and T4 phages found that motor velocity correlated with size of the viral genome. These studies show that the maximum DNA translocation velocity for phage T4 is up to ∼2000 bp/s (170-kbp genome), for λ is ∼800 bp/s (48.5-kbp genome), and for ϕ29 is ∼200 bp/s (19-kbp genome) (supplemental Fig. S4) (1, 17, 18). The velocities of the viral motors appear to be scaled to chromosome size such that full packaging takes a few minutes, regardless of the length of the virus chromosome. It is likely that this relationship evolved to complete DNA packaging within a few minutes of the ∼30 min phage life cycle because of the following: i) packaging must keep pace with the DNA metabolic pathways such as transcription, recombination, and repair, as well as cell lysis; ii) phages should avoid nonspecific endonuclease digestion of their genomes; and iii) potential infection by another phage must be minimized.
What regions of packaging motors have evolved to contribute to the determination of velocity of DNA translocation? Previous sequence alignments show that there are similarities between the ATPase domains of terminases and SF2 DEXD/H box helicases (21). In these studies, it was discovered that adjacent Asp and catalytic Glu residues in the Walker B motif (DE) and the Thr of motif III (C motif) are well conserved in terminases. Furthermore, the sequence 174VAGYDE was identified as a Walker B motif in the large terminase subunit of λ (20, 21). The residue Thr194 is located between the Walker B and C motifs, 15 residues downstream of the Walker B glutamate (Fig. 1). Structural modeling based on the x-ray structure and homology with T4 gp17 shows that the Thr194 residue is part of a conserved long helix that connects the β4 (Walker B) and β5 (C motif) strands of the nucleotide-binding domain (Fig. 1). In addition to our findings of altered motor dynamics with T194M and G212S, we also identified five mutants in genetic screening experiments with changes clustered near T194M that further support the notion that this is a critical functional region for DNA translocation: 1) terminase gpA G191S is null for DNA packaging (12); 2) the change S192A is a suppressor of the lethal change G212S (G212S itself has a slow translocation phenotype); and, farther upstream, there are 3) the lethal L180F, 4) A181S, a suppressor of L180F, and 5) D185G, a suppressor of G212S.
Sequence alignments and structural modeling suggest that the Glu212 residue is directly adjacent to the most critical residues of the putative C motif (Ser213 and Thr214), which has been implicated in the coupling of ATP hydrolysis to DNA translocation in T4 (20). Assuming that the G212S mutation does not affect packaging complex assembly (as suggested by the AUC data), our bulk ATPase and single molecule data indicate that the DNA packaging of the G212S mutant is tightly coupled to hydrolysis. This suggests that this residue may not be required for coupling and may have a different role. The 6× increase in DNA slipping displayed by G212S suggests that residues near the C motif may be in contact with the DNA substrate.
Given the drastically slower phenotypes of T194M and G212S, the near-restoration of velocity from T194V, and the correlation of ATPase activity to translocation speed of the mutant terminases studied, we propose that the region between the Walker B and C motif of gpA is a critical structural region that may be a part of the mechanism that determines motor velocity and processivity. We emphasize that residue Thr194 is a key functional residue in velocity determination because the T194M change dramatically affects motor velocity without affecting force dependence, processivity (slipping), or pausing frequency. This behavior is in sharp contrast to effects we observed previously in response to a lethal mutation Y46F in the Q motif that caused a dramatic decrease in processivity but only a small (∼40%) reduction in velocity, and a mutation adjacent to the Walker A motif (K84A), which also caused only a small (∼40%) reduction in velocity (13).
Taken together with our findings, studies on chromosome transporters and DEAD-box RNA helicases suggest that the region between the Walker B motif and C motif also is a structurally conserved region that may determine velocity and processivity in several different types of nucleic acid translocases. Structural data on MjDEAD RNA helicase suggests that the analogous C motif (also called motif III) is involved in RNA binding (23). Furthermore, mutational analysis by Banroques et al. (22) showed that motif III of the Ded1 helicase is used to create a high affinity single-stranded RNA-binding site and has an important role in ATP hydrolysis. These findings suggest a similar function as our findings that the G212S change causes reduced motor velocity, reduced ATP hydrolysis rate, and increased DNA slipping. Increased slipping is commensurate with decreased motor-substrate interaction.
Also supporting this notion, a mutant of SpoIIIE, a bacterial ring double-stranded DNA translocase responsible for chromosome segregation, recently was isolated with a mutation four residues downstream of its Walker B site (D584A) that exhibits a ∼2.5× reduced in vivo chromosome transport rate (33). Sequence alignments of this region from various DNA translocases show no significant conservation at the amino acid sequence level (Fig. 1). On the other hand, a comparison of the solved crystal structures of the T4 gp17, FtsK translocase (closely homologous with SpoIIIE), and our structural model of λ gpA shows high structural similarity this region. Fig. 1B shows that the nucleotide binding domains of the nucleic acid translocases appear to be extremely well conserved (34). In particular, the loop-(long) helix-loop that connects the β4 (Walker B motif) and β5 (C motif) strands of which the Glu191, Thr194, and Glu212 residues are a part, can be superimposed completely.
The D584A slow mutant of SpoIIIE and FtsK also is part of the same helix. Structural alignments comparing this region with other related nucleic acid translocases including RNA helicases show structural homology as well (supplemental Fig. S5).
Mechanism of Velocity Determination
What is the mechanism by which the loop-helix-loop region influences motor velocity in the viral DNA packaging motor? One possibility proposed by Sisakova et al. (35) is that mutations could modulate a “processivity clamp” in which a tighter grip of the motor on DNA results in a decreased frequency in slipping and a slower translocation rate. However, our data argues against this because the processivity of T194M is similar to WT and 6× less for G212S.
Another possibility is the packaging ATPase catalytic site is formed at the interface between gpA subunits, and a small perturbation in the conformation of the assembled motor dramatically affects ATP hydrolysis and translocation rates. Although our AUC data on T194M do suggest a defect in the assembly of gpA protomers, structural evidence on T4 gp17 does not favor a shared ATPase site in the gp17 pentamer (34). It also is possible that only a few complexes assemble correctly at the procapsid during packaging, but they have full WT catalytic activity, and the decreased velocity of the mutants are due to a defect in coupling ATP hydrolysis to translocation. However, the ATPase assays (showing decreased ATPase activity), AUC data (showing WT assembly), and the single molecule data (showing slowed velocity) on G212S suggest that velocity alterations are not caused by defects in assembly or the coupling of ATP hydrolysis to DNA translocation. We also note that because the AUC data only takes into account the assembly of terminase protomers, it does not necessarily measure the state of the complete packaging machine (including the procapsid).
Instead, based on the mutational analysis data, single molecule results (which inherently only measure active packaging complexes), and structural modeling, we favor the interpretation that the velocity of packaging can be modulated by changing intradomain interactions within the ATPase center, which would slightly alter regions responsible for ATP binding and hydrolysis (such as the Walker A and B motifs) and thereby slow down the DNA translocation rate by slowing the ATP hydrolysis rate.
Structural modeling provides insight on how subtle changes in the intradomain interactions could affect the orientation of the Walker B and C motif catalytic residues, resulting in substantial differences in the packaging velocity. For instance, the T194M change could establish an interaction with Phe183, a residue close to the Walker B motif, causing a slight misorientation of the aspartate and catalytic glutamate residues, which in turn would result in a reduced rate of phosphate bond cleavage (Fig. 5). According to our modeling, the T194V change, an amino acid substitution with a similar side chain length to WT) would restore the WT orientation. (The model shows that the distance between the residues would not support interaction as in T194M.) Meanwhile, the G212S change is predicted to interact with residue Val74 (close to the Walker A region). The pseudorevertants appear to compensate for this by slight changes in the orientation of the Walker B and C motif residues.
FIGURE 5.

A model of the gpA ATPase domain (amino acids Phe56–Arg218) using SWISS-MODEL (homology modeling) with T4 gp17 as a template (Protein Data Bank code 3CPE). ATP (black) was docked by alignment with T4 gp17-ATP. Individual models of gpA containing the T194M or G212S mutation (cyan) were constructed in the same way and superimposed with the WT model (Thr194 and Glu212, yellow). The slow packaging mutations are located in the regions connecting the Walker B and C motif catalytic residues, which are oriented into the ATP-binding pocket (blue, Asp178; green, Glu179; and red, Thr214).
Our single molecule packaging dynamics measurements and structural modeling suggest that, in addition to the essential catalytic residues that are responsible for chemical, electrostatic, and base-stacking interactions with ATP, other “nonessential” residues in the nucleotide binding domain are responsible for subtle structural arrangements in the catalytic pocket that may give rise to large variations in translocation speeds. As residue changes that result in the slow phenotypes are mostly found in the region between the Walker B and C motifs (Figs. 1 and 5), and the Walker B motif plays a key role in bond cleavage (19), this region likely is to be critical. However, mutations at secondary sites, some of which are distal to this region, also can compensate for alterations in velocity. Genetic-screening experiments show that residue changes in pseudorevertants for the slow mutant G212S are located in various locations of the N-terminal ATPase domain of gpA (in the vicinity of the Q motif, Walker A motif, the loop-helix-loop region connecting the Walker B and C motif, and downstream of the C motif, Table 1). In addition, packaging measurements of T194M/F156Y mutants show that mutations at secondary sites can subtly alter the packaging dynamics as well; the variability of packaging velocity among different individual complexes decreases, and the velocity is slowed further. These results suggest a possible pathway for the evolution of viral packaging motors having differing DNA translocation velocities; large differences in velocity could be caused by variations in the loop-helix-loop region between the Walker B and C motifs, and further differences could be caused by variations at secondary sites.
Supplementary Material
Acknowledgments
We thank Arielle Yablonovich, Derek Fuller, Al Schweitzer, and Dorian Raymer for technical assistance. We are also indebted to Shelley Grimes and Helene Gaussier for providing reagents for initial studies. We thank Jeff Moffitt and Briana Burton for helpful discussions.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and an additional reference.
- WT
- wild-type
- gp
- gene product
- pN
- piconewtons
- AUC
- analytical ultra centrifugation.
REFERENCES
- 1.Smith D. E., Tans S. J., Smith S. B., Grimes S., Anderson D. L., Bustamante C. (2001) Nature 413, 748–752 [DOI] [PubMed] [Google Scholar]
- 2.Catalano C. (ed) (2005) Viral Genome Packaging Machines: Genetics, Structure, and Mechanism, Landes Bioscience, Georgetown, TX [Google Scholar]
- 3.Campbell A. M. (2007) in Fields Virology (Knipe D. M., Howley P. M. eds.) 5th Ed., pp. 769–794, Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 4.Roizman B., Knipe D. M., Whitley R. J. (2007) in Fields Virology (Knipe D. M., Howley P. M. eds.) 5th Ed., pp. 2501–2602, Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 5.Berk A. J. (2007) in Fields Virology (Knipe D. M., Howley P. M. eds.) 5th Ed., pp. 2355–2394, Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 6.Feiss M., Catalano C. (2005) in Viral Genome Packaging Machines: Genetics, Structure, and Mechanism (Catalano C. ed.) pp. 5–33, Landes Bioscience, Georgetown, TX [Google Scholar]
- 7.Rao V. B., Feiss M. (2008) Annu. Rev. Genet. 42, 647–681 [DOI] [PubMed] [Google Scholar]
- 8.Maluf N. K., Yang Q., Catalano C. E. (2005) J. Mol. Biol. 347, 523–542 [DOI] [PubMed] [Google Scholar]
- 9.Maluf N. K., Gaussier H., Bogner E., Feiss M., Catalano C. E. (2006) Biochemistry 45, 15259–15268 [DOI] [PubMed] [Google Scholar]
- 10.Dhar A., Feiss M. (2005) J. Mol. Biol. 354, 738–738 [DOI] [PubMed] [Google Scholar]
- 11.Hang J. Q., Tack B. F., Feiss M. (2000) J. Mol. Biol. 302, 777–795 [DOI] [PubMed] [Google Scholar]
- 12.Duffy C., Feiss M. (2002) J. Mol. Biol. 316, 547–561 [DOI] [PubMed] [Google Scholar]
- 13.Tsay J. M., Sippy J., Feiss M., Smith D. E. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14355–14360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chemla Y. R., Aathavan K., Michaelis J., Grimes S., Jardine P. J., Anderson D. L., Bustamante C. (2005) Cell 122, 683–692 [DOI] [PubMed] [Google Scholar]
- 15.Fuller D. N., Rickgauer J. P., Jardine P. J., Grimes S., Anderson D. L., Smith D. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 11245–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moffitt J. R., Chemla Y. R., Aathavan K., Grimes S., Jardine P. J., Anderson D. L., Bustamante C. (2009) Nature 457, 446–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuller D. N., Raymer D. M., Kottadiel V. I., Rao V. B., Smith D. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16868–16873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuller D. N., Raymer D. M., Rickgauer J. P., Robertson R. M., Catalano C. E., Anderson D. L., Grimes S., Smith D. E. (2007) J. Mol. Biol. 373, 1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) EMBO J. 8, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Draper B., Rao V. B. (2007) J. Mol. Biol. 369, 79–94 [DOI] [PubMed] [Google Scholar]
- 21.Mitchell M. S., Matsuzaki S., Imai S., Rao V. B. (2002) Nucleic Acids Res. 30, 4009–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banroques J., Doère M., Dreyfus M., Linder P., Tanner N. K. (2010) J. Mol. Biol. 396, 949–966 [DOI] [PubMed] [Google Scholar]
- 23.Story R. M., Li H., Abelson J. N. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3624–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rickgauer J. R., Fuller D. N., GrimeS S., Jardine P. J., Anderson D. L., Smith D. E. (2007) Biophys. J. 48a–48a [Google Scholar]
- 25.Hang Q., Woods L., Feiss M., Catalano C. E. (1999) J. Biol. Chem. 274, 15305–15314 [DOI] [PubMed] [Google Scholar]
- 26.Pal S. K., Chattoraj D. K. (1988) J. Bacteriol. 172, 2819–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sternberg N., Austin S. (1983) J. Bacteriol. 153, 800–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arber W., Enquist L., Hohn B., Murray N. E., Murray K. (1983) in Lambda II (Hendrix R. W., Roberts J. W., Stahl F. W., Weisberg R. A. eds.) pp. 433–366, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 29.Tomka M. A., Catalano C. E. (1993) J. Biol. Chem. 268, 3056–3065 [PubMed] [Google Scholar]
- 30.Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ortega M. E., Gaussier H., Catalano C. E. (2007) J. Mol. Biol. 373, 851–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang Y., Catalano C. E., Feiss M. (1996) Biochemistry 35, 2796–2803 [DOI] [PubMed] [Google Scholar]
- 33.Burton B. M., Marquis K. A., Sullivan N. L., Rapoport T. A., Rudner D. Z. (2007) Cell 131, 1301–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S., Kondabagil K., Draper B., Alam T. I., Bowman V. D., Zhang Z., Hegde S., Fokine A., Rossmann M. G., Rao V. B. (2008) Cell 135, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sisáková E., Weiserová M., Dekker C., Seidel R., Szczelkun M. D. (2008) J. Mol. Biol. 384, 1273–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.