

Abstract
The angiopoietins (Ang-1 and Ang-2) have been identified as agonistic and antagonistic ligands of the endothelial receptor tyrosine kinase Tie2, respectively. Both ligands have been demonstrated to induce translocation of Tie2 to cell-cell junctions. However, only Ang-1 induces Tie2-dependent Akt activation and subsequent survival signaling and endothelial quiescence. Ang-2 interferes negatively with Ang-1/Tie2 signaling, thereby antagonizing the Ang-1/Tie2 axis. Here, we show that both Ang-1 and Ang-2 recruit β3 integrins to Tie2. This co-localization is most prominent in cell-cell junctions. However, only Ang-2 stimulation resulted in complex formation among Tie2, αvβ3 integrin, and focal adhesion kinase as evidenced by co-immunoprecipitation experiments. Focal adhesion kinase was phosphorylated in the FAT domain at Ser910 upon Ang-2 stimulation and the adaptor proteins p130Cas and talin dissociated from αvβ3 integrin. The αvβ3 integrin was internalized, ubiquitinylated, and gated toward lysosomes. Taken together, the experiments define Tie2/αvβ3 integrin association-induced integrin internalization and degradation as mechanistic consequences of endothelial Ang-2 stimulation.
Keywords: Cell Adhesion, Cell Junctions, Endothelium, Integrin, Protein-tyrosine Kinase (Tyrosine Kinase), Tie, Angiopoietin, Vascular Biology
Introduction
Angiogenesis and blood vessel maintenance are orchestrated by the coordinated interplay of multiple vascular receptor tyrosine kinase signaling pathways, including the VEGF/VEGFR,5 the ephrin/Eph, and the angiopoietin/Tie pathways. Among the different vascular receptor tyrosine kinases, angiopoietin/Tie signaling controls later steps of the angiogenic cascade regulating vascular maturation and vessel quiescence.
Genetic experiments have identified angiopoietin-1 (Ang-1) as the nonredundant agonistic Tie2 ligand inducing Tie2 phosphorylation (1–3). Ang-1/Tie2 signaling transduces survival signals, regulates mural cell recruitment, and controls the quiescent phenotype of the endothelium. In contrast, Ang-2 interferes negatively with constitutive Ang-1/Tie2 signaling, acting in a context-dependent manner as antagonistic ligand on resting endothelium (4, 5). Developmentally, transgenic overexpression of Ang-2 phenocopies the properties of Ang-1-deficient mice (6). In the adult, conditional transgenic overexpression of Ang-2 in endothelial cell (EC) silences Tie2 phosphorylation (7), and consequently Ang-2 has been demonstrated to regulate the responsiveness of the quiescent endothelium to exogenous stimuli such as inflammatory mediators (8) or angiogenic signals (9).
Recently, elegant cellular experiments have shown that the downstream signaling consequences of Tie2 activation are dependent on the subcellular localization of Tie2 (10, 11). In contacting, confluent ECs, both angiopoietin ligands are capable of inducing Tie2 translocation to cell-cell junctions. Yet, only Ang-1 induces Akt-Foxo and Akt-eNOS signaling, thereby promoting endothelial quiescence. The molecular consequences of Ang-2 mediated Tie2 translocation have not been fully explored.
Integrins are of major importance in the regulation of endothelial activation and quiescence. Integrins are heterodimers consisting of an α and β subunit, and they connect the extracellular matrix with cytoskeletal proteins. One of the most extensively studied integrins of activated EC is αvβ3 integrin (12). Blocking antibodies against αvβ3 integrin inhibit angiogenesis (13–15). Inhibition of αvβ3 integrin results in reduced adhesion and decreased EC migration (16, 17). Furthermore, the αv integrin antagonist cilengitide induces increased EC permeability (18). Considerable experimental evidence has accumulated in recent years to demonstrate that some integrin-dependent signaling effects may depend on direct interactions with cell surface presented growth factor receptors. For example, αvβ3 integrin has been shown to associate with PDGFRβ and VEGFR-2, thereby enhancing signaling through these vascular receptor tyrosine kinases (19–21).
Based on (i) the angiopoietin-induced translocation of Tie2 receptor, (ii) the antagonizing effects of Ang-2 on quiescent ECs, and (iii) the importance of αvβ3 integrin for activated ECs, we hypothesized that the Ang-2/Tie2 axis may interact directly with αvβ3 integrins. To this end, we examined the effects of Ang-2 stimulation on αvβ3 integrin cell surface trafficking. The experiments revealed that both Ang-1 and Ang-2 stimulation resulted in translocation of Tie2 and β3 integrins to cell-cell junctions. However, only Ang-2 stimulation induced complex formation between Tie2 and αvβ3 integrin. Ang-2 stimulation led to FAK activation, adaptor protein dissociation, and integrin internalization. Internalized αvβ3 integrin was gated toward lysosomal degradation and not to the recycling pathway establishing a critical role of Ang-2-mediated Tie2·FAK·αvβ3 complex formation in integrin turnover.
EXPERIMENTAL PROCEDURES
Materials
Recombinant myc-tagged Ang-1 and myc-tagged Ang-2 were either produced as described (26) or used from ReliaTech or R&D Systems. The following antibodies were used. Mouse anti-αvβ3 (LM609) and rabbit anti-αv were from Chemicon. Rabbit anti-LAMP1 was from Sigma-Aldrich. Mouse anti-Tie2 (05-0584) for immunoprecipitation was from Upstate. Rabbit anti-Tie2 (Sc324) for Western blotting was from Santa Cruz Biotechnology. Mouse anti-Tie2 (Tek9) for immunocytochemistry was from ReliaTech. Rabbit anti-talin-1 was from Cell Signaling. Rabbit anti-FAK ([pY397]), ([pS732]), ([pY910]) was from BIOSOURCE. Rabbit anti-β3, anti-p130Cas, anti-FAK, anti-paxillin, anti-Tie2, mouse anti-Tyr(P)99, anti-Src (C-19), goat anti-actin were from Santa Cruz Biotechnology. Anti-CD31 (M0823) was from R&D Systems. For confocal microcopy, chambered coverglass from Nunc was used.
Cell Culture
Human umbilical vein endothelial cells (HUVECs), endothelial cell growth medium, endothelial cell basal medium, and the corresponding supplements were purchased from Promocell. Cells were cultured at 37 °C, 5% CO2 in the appropriate growth medium containing 10% fetal calf serum (FCS) (Invitrogen). HUVECs were used between passages 2 and 5.
Immunocytochemistry
Coverslips or chambered coverglasses were coated with 5 μg/ml fibronectin for 30 min at 37 °C. Thereafter, HUVECs were seeded at a density of 30,000 cells/24-well dish. For confocal microscopy, HUVECs were seeded at a density of 26,000 cells/4-well dish in chambered coverglass. Cells were allowed to grow for 48 h, starved overnight in EC basal medium and 2% FCS, and stimulated with cytokine for the corresponding time points. HUVECs were washed once with PBS and fixed in 4% PFA for 12 min. Cells were washed again with PBS and blocked with 3% BSA and PBS containing 0.3% Triton X-100 for 30 min followed by incubation with the first antibody. For confocal microscopy, cells were washed again and blocked with 7% goat serum, 1% BSA, and 10% donkey serum in PBS followed by incubation with the first antibody. Anti-FAK (2 μg/ml), anti-FAK ([pS910]) (3 μg/ml), anti-LAMP-1 (2 μg/ml), and anti-β3 antibodies (2 μg/ml) were used as first antibodies. For confocal microscopy, anti-CD31 (1:500), anti-Tie2 (1:800), and anti-β3 (1:100) antibodies were used. Cells were incubated with the antibodies in a 1% BSA and PBS solution containing 0.3% Triton X-100 overnight at 4 °C or for confocal microcopy in 1% BSA and PBS solution overnight at 4 °C. Thereafter, the supernatant was aspirated, and the cells were washed three times with PBS followed by incubation with the corresponding secondary antibody for 30 min at room temperature in the dark. The cells were washed three times with PBS and incubated with 2 μg/ml Hoechst for 10 min (in the dark) to counterstain nuclei. Cells were then washed three times with PBS. Finally, coverslips were mounted with Fluoromount G. For confocal studies, the Leica TCS SP5 of the Core Facility Microscopy, German Cancer Research Center Heidelberg (DKFZ-ZMBH Alliance) was used. For video images, z-stick studies were revised with Amira software.
Western Blot Analysis
Cells were seeded in 6-well plates and allowed to grow to 90% confluence. Prior to stimulation, cells were starved in EC basal medium and 2% FCS overnight. For phosphorylation analyses, the medium was changed to 1 ml of basal medium containing 2 mm sodium orthovanadate prior to stimulation. Thereafter, cells were exposed to the corresponding test compounds at 37 °C for the indicated time points. Following stimulation, cells were washed with ice-cold PBS and 2 mm sodium orthovanadate and lysed in 500 μl of radioimmune precipitation assay buffer for 20 min at 4 °C. To remove cellular debris, lysates were centrifuged for 5 min at 13,000 × g, and the pellet was removed. Thereafter, the lysates were incubated with 40 μl of protein G-Sepharose and 2 μg of the corresponding antibody overnight. The immunoprecipitate was centrifuged for 2 min at 1,000 × g, washed three times with PBS and 2 mm sodium orthovanadate, boiled with 5 × sample buffer, and analyzed by 10% SDS-PAGE and subsequent Western blotting. The membranes were thereafter incubated with 2 μg of the corresponding antibody overnight at 4 °C or for 1 h at room temperature in 1% BSA and PBST. Next, the membrane was washed three times with PBST. The membrane was then incubated with the corresponding horseradish peroxidase-labeled secondary antibody for 45 min at room temperature. Bound antibody was visualized by ECL and exposure of the membrane to film. Thereafter, the antibody was removed from the membrane by incubation with stripping buffer for 12 min at room temperature. To detect total protein levels, the membrane was blocked in blocking buffer followed by incubation with the corresponding antibody in 1% blocking buffer for 1 h. Next, the membranes were washed three times with PBST. To detect bound antibody, the membranes were incubated with 2 μg of horseradish peroxidase-labeled antibody. Bound antibody was visualized by ECL and exposure of the membrane to film.
Internalization Assay
Internalization assay was performed as described (22). Briefly, HUVECs were serum-starved overnight in 2% FCS, transferred to ice, washed twice in cold PBS, and surface-labeled at 4 °C with 0.2 mg/ml NHS-SS-biotin. Cells were washed in ice-cold PBS and transferred to 37 °C in the absence or in the presence of Ang-2. At the indicated time points, the medium was removed, and the dishes were put on ice. Cells were washed twice with ice-cold PBS, and the remaining biotin on the surface was removed. Toward this end, a solution containing 20 mm sodium Mes in 50 mm Tris (pH 8.6) and 100 mm NaCl was added for 15 min at 4 °C. Sodium Mes was quenched by the addition of 20 mm iodoacetamide for 10 min, and the cells were lysed in radioimmune precipitation assay cell lysis buffer. Lysates were centrifuged for 10 min at 10,000 × g. Supernatants were corrected for equal protein concentration with a BCA kit. Biotinylated integrins were isolated by immunoprecipitation. The immunoprecipitate was separated by SDS-PAGE, transferred to a membrane, and probed for biotin.
Quantification of the Co-localization in Multichannel Two-dimensional and Three-dimensional Images
For quantification of the co-localization in the different image channels, we developed an automatic protocol. The protocol determines the relative co-localization frequency in multichannel two-dimensional images as well as in three-dimensional image stacks and consists of four steps (see supplemental Fig. 1). In the first step, we generated a histogram hc for each image channel gc of an image g. Based on the histograms, we determined which pixels or voxels were taken into account for co-localization analysis. For a robust quantification of the co-localization, we considered only the brightest voxels within the image channels. However, a problem in choosing a threshold was that the intensities in the different channels more or less followed a Poisson distribution so that foreground and background intensities could not be distinguished based on standard histogram-based segmentation methods (e.g. Otsu's method). Instead, we applied a relative threshold Trel, which specified the relative number of the brightest voxels included in the co-localization analysis. In our experiments, we used Trel = 0.1, thus, only the brightest 10% of voxels were considered. Based on Trel and the histograms hc for the different channels, absolute thresholds Tabs,c were determined,
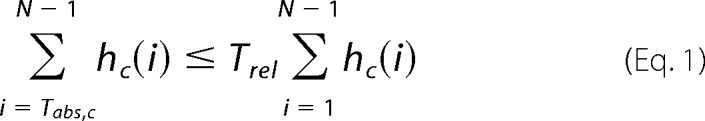 |
where hc(i) denotes the number of voxels with intensity i and N denotes the total number of different intensity levels in hc. Intensity level i = 0 was considered to be the image background, therefore we omitted hc(0) for the computation of Tabs,c. Note, that the histograms hc were different for the different channels, thus different absolute thresholds Tabs,c were obtained for the different channels. For segmentation of the different channels, the absolute thresholds Tabs,c were applied to the original image channels gc. As a result of segmentation, we obtained three binary images bc. Based on the overlap of the segmented images bc, the relative co-localization frequencies were computed.
RESULTS
Ang-1 and Ang-2 Induce Translocation of Tie2 and β3 Integrins to Cell-Cell Junctions
It is well established that both Ang-1 and Ang-2 induce in contacting ECs translocation of their receptor Tie2 to cell-cell junctions (11, 12). Furthermore, the importance of αvβ3 integrin on endothelial stability is well accepted. Therefore, we aimed at examining if angiopoietins induced co-localization of Tie2 and β3 integrins. Immunohistochemical analyses of Tie2, β3 integrins and the endothelial surface marker CD31 were performed (Fig. 1, A–E). Z-stacks of whole cells were analyzed with a newly developed software for co-localization studies (Fig. 1, A and B). The analyses did not reveal increasing co-localization between Tie2 or β3 integrins with the diffusely distributed CD31 on the cell surface upon angiopoietin stimulation in the z-sticks (Fig. 1A, bar 1–6). This indicated that angiopoietins did not induce significant translocation of Tie2 or β3 integrins from intracellular compartments to the cell surface. Therefore, the observed accumulation of Tie2 in the cell-cell junctions upon angiopoietin stimulation (10, 11) seemed to reflect a shift from a diffuse cell surface distribution to an accumulation in cell-cell junctions. Interestingly, angiopoietin stimulation resulted in co-localization of Tie2 and β3 integrins (Fig. 1A, bar 7–9). This accumulation was localized mainly to the cell surface because co-localization studies with all three markers Tie2, β3 integrin, and CD31 detected more co-localization upon angiopoietin stimulation (Fig. 1A, bar 10–12). Taken together, the data imply that angiopoietins induced co-localization of Tie2 and β3 integrins by transposing the two on the cell surface. Interestingly, the accumulation between Tie2 and β3 integrins was found in a pattern resembling cell-cell junctions of neighboring cells (Fig. 1B). Higher magnification confirmed this observation and revealed a pronounced redistribution of Tie2 and β3 integrins to cell-cell junctions (Fig. 1, C–E, and supplemental Fig. 2). Consequently, co-staining analyses with the junctional marker VE-cadherin were performed (Fig. 1F). Ang-1 and Ang-2 stimulation resulted in co-localization of Tie2 and β3 integrins with VE-cadherin, confirming that Tie2-β3 integrin co-localization was mainly restricted to cell-cell junctions.
FIGURE 1.

Ang-1 and Ang-2 induce translocation of Tie2 receptor and β3 integrin to cell-cell junctions. A–E, HUVECs were grown to confluence on fibronectin-coated chambered coverglass and stimulated for 15 min with either 200 ng/ml Ang-2 or 200 ng/ml Ang-1. Cells were fixed with 10% PFA followed by incubation with primary antibody against CD31, Tie2, and β3 integrin overnight at 4 °C. After secondary incubation, z-stack pictures (40 pictures/stack) were taken by confocal microscopy (magnification, ×40). A, z-stacks (40 picture/stack) were analyzed with a newly developed software for co-localization analyses. The unstimulated control cells were equalized as 1.0, and stimulated conditions were adjusted. Error bars indicate S.E. Student's t test was not significant (n.s.). B, representatively, the co-localization studies between CD-31·Tie2 and β3 integrin upon Ang-2 stimulation are shown. C–E, enlargements of the interendothelial areas are shown (scale bar, 10 μm). F, HUVECs were grown to confluence on fibronectin-coated chambered coverglass and stimulated for 15 min with either 200 ng/ml Ang-1 or 200 ng/ml Ang-2. Cells were fixed with 10% PFA and incubated with antibodies against VE-cadherin, Tie2, and β3 integrin (scale bar, 25 μm).
Ang-2 Induces Complex Formation among Tie2, αvβ3, and FAK
Based on these co-localization data, we analyzed whether the angiopoietin-induced junctional translocation was just correlatively associated or whether it was a result of a direct interaction between the Tie2 receptor and αvβ3 integrin. Direct and reciprocal immunoprecipitation analyses of Tie2 and the integrin αvβ3 were performed (Fig. 2). Whereas both Ang-1 and Ang-2 induced translocation of Tie2 and β3 integrins into cell-cell junctions (Fig. 1) (9, 10), only Ang-2 induced complex formation between Tie2 and αvβ3 integrin (Fig. 2). Under no experimental conditions was Ang-1 able to mediate complex formation between Tie2 and αvβ3 integrin (Fig. 2 and supplemental Fig. 3). In contrast, Ang-2-induced complex formation was dose-dependent and was already present at low concentrations of Ang-2 (supplemental Fig. 4).
FIGURE 2.

Only Ang-2 induces complex formation between Tie2 receptor and αvβ3 integrin. A–D, HUVECs were grown to confluence and stimulated for different time points with 200 ng/ml Ang-2 (A and B) or 200 ng/ml Ang-1 (C and D). Cell lysates were immunoprecipitated (IP) with αvβ3 integrin antibody or Tie2 antibody and protein G-Sepharose. IgG controls were immunoprecipitated with the corresponding IgG mouse control antibody. The resulting immunoprecipitate was separated by SDS-PAGE and Western blotted. Whole cell lysates (WCL) of HUVECs were used as positive control. Under Ponceau Red control, membranes were cut horizontally at 70 and 35 kDa and probed for β3 integrin, stripped, and reprobed against FAK and Tie2 (B and D) or probed against Tie2, followed by stripping and reprobing against FAK and β3 (A and C). The lower part (<35 kDa) was probed against αv. Densitometric quantiation of the blots was performed with ImageJ. The IgG control result was subtracted from the other values.
Integrin association with growth factor receptors has been shown to enhance signal transduction of both receptors (23), with FAK playing a key role in this process (19, 21, 23–25). We consequently examined the effect of Ang-2-stimulated complex formation between Tie2 and αvβ3 integrin on FAK association and activation. Tie2·αvβ3 complex formation led to rapid recruitment of FAK (Fig. 2, A and B, and supplemental Fig. 4). The adaptor protein Src is commonly involved in FAK activity. However, an increase of Src association with αvβ3 integrin was not observed (data not shown).
Ang-2 Mediates FAK Phosphorylation at Ser910 and Dissociation of p130cas and Talin of αvβ3 Integrin
FAK binding to integrins induces its activation by phosphorylation (26). To assess the effect of Ang-2 on FAK activation, we monitored FAK phosphorylation at Tyr397, Ser732, and Ser910 following Ang-2 stimulation. Western blot analysis showed that FAK was not phosphorylated at Ser732 and Tyr397 following Ang-2 stimulation (Fig. 3A, FAK [pS732] and FAK [pY397] blot). Instead, FAK was time dependently phosphorylated at Ser910 (Fig. 3A, FAK [pS910] blot). Quantitation of FAK phosphorylation revealed strongest Ser910 phosphorylation after 30 min. No changes in the phosphorylation status of Ser732 and Tyr397 were observed. Direct Western blotting for FAK ([pS910]) following αvβ3 integrin immunoprecipitation confirmed the association of activated FAK with the receptor·integrin complex following Ang-2 stimulation (Fig. 3B). Immunohistochemical experiments identified rapid co-localization between FAK ([pS910]) and αvβ3 integrins (Fig. 3C, white arrows). After 20 min of Ang-2 stimulation, FAK ([pS910]) staining revealed a more diffuse pattern (Fig. 3C). In contrast, αvβ3 integrin showed a patchy pattern (Fig. 3C). Previous studies have established that FAK phosphorylation at Ser910 induces dissociation of paxillin (27). Consistently, Ang-2 stimulation led to rapid dissociation of the integrin-associated adaptor proteins talin and p130Cas from the αvβ3 integrin complex (Fig. 3D). Integrin-associated adaptor proteins connect integrins with cytoskeleton proteins, thereby modulating integrin-dependent cell functions.
FIGURE 3.

Ang-2 stimulation induces FAK phosphorylation at Ser910, recruitment to αvβ3 integrin and dissociation of the adaptor proteins talin, and p130Cas. A, to study the effect of Ang-2 on FAK phosphorylation, HUVECs were stimulated with Ang-2. Total cell lysates were separated by SDS-PAGE, Western blotted, and probed for FAK [pS910], [pS732], and [pT397]. The membranes were stripped and probed for total FAK. FAK was rapidly phosphorylated at Ser910 upon Ang-2 stimulation. Other phosphorylation sites (Tyr397 and Ser732) were not affected. WCL, whole cell lysates. B, HUVECs were stimulated with Ang-2. Lysates were immunoprecipitated (IP) with an antibody against αvβ3, and the immunoprecipitate was subjected to SDS-PAGE followed by Western blotting. Membranes were probed for FAK [pS910], stripped, and reblotted for total β3 integrin subunit. Ser910-phosphorylated FAK interacted rapidly with αvβ3 integrin upon Ang-2 stimulation. C, immunofluorescence staining confirmed the co-localization of FAK [pS910] with αvβ3 integrin after 10 min of Ang-2 treatment (white arrows). After 20 min, FAK910 was distributed more diffusely in the cells whereas αvβ3 integrin showed a punctiform pattern. Cells were unstimulated or stimulated for 10, 20, 30, or 60 min with Ang-2, fixed in 10% PFA, and stained for FAK [pS910], and αvβ3 integrin (scale bar, 25 μm). D, to study the effect of Ang-2 on the association of talin and p130Cas with αvβ3 integrin, HUVECs were stimulated with Ang-2. Cell lysates were immunoprecipitated with an antibody against αvβ3 integrin or IgG control. The resulting immunoprecipitate was separated by SDS-PAGE and Western blotted. The membrane was cut under Ponceau Red control and probed for talin (1st lane) or p130Cas (2nd lane). The membranes were stripped and probed for total β3 integrin subunit. The experiment revealed the rapid dissociation of the integrin-associated adaptor proteins from αvβ3 integrin upon Ang-2 stimulation. Densitometric quantiation of the blots was performed with ImageJ. The IgG control result was subtracted from the other values.
Ang-2 Stimulation Leads to αvβ3 Integrin Ubiquitinylation, Internalization, and Lysosomal Degradation
Focal adhesion complex disassembly is closely coupled to integrin internalization. Immunofluorescence staining of αvβ3 integrin showed a punctiform pattern under Ang-2 treatment, suggesting the presence of αvβ3 integrin in intracellular compartments (Fig. 3C). We consequently examined whether αvβ3 integrin was internalized upon Ang-2 stimulation. The experiments revealed that αvβ3 integrin was rapidly internalized following Ang-2 stimulation (Fig. 4A). The analyses were repeated in the presence of dynasore, an inhibitor of dynamin, which is required for endocytosis. Addition of dynasore strongly reduced Ang-2-induced αvβ3 integrin internalization, indicating that dynamin is required for αvβ3 integrin internalization. Rab5 is a marker for early endosomes. Co-localization studies with Rab5 detected more αvβ3-Rab5 co-localization upon Ang-2 treatment (supplemental Fig. 5), confirming the internalization of αvβ3 integrins. Internalized integrins may be gated to lysosomes for degradation or enter the recycling pathway to be exposed again on the plasma membrane, a process that is most prevalent in migrating cells (28). As shown in Fig. 4B, αvβ3 integrin was rapidly ubiquitinylated upon Ang-2 stimulation. Co-staining of αvβ3 and the lysosome-associated membrane protein-1 LAMP-1 revealed the co-localization of αvβ3 integrin and LAMP-1 upon Ang-2 stimulation (Fig. 4, C and D).
FIGURE 4.

Ang-2 stimulation induces β3 integrin internalization, ubiquitinylation, and gating toward lysosomes. A, serum-starved HUVECs were surface-labeled with NHS-SS-biotin. Cells were then warmed to 37 °C and stimulated with Ang-2 after dynasore pretreatment. Thereafter, biotin was removed from proteins remaining at the cell surface by sodium Mes treatment at 4 °C. The cells were lysed, and protein content was equalized with the BCA detection kit. Equal loading was confirmed by Western blotting against actin (lower lane). Equalized cell lysates were immunoprecipitated (IP) with an antibody against β3 (upper lane). The immunoprecipitate was analyzed by SDS-PAGE followed by Western blotting and probing for peroxidase-conjugated streptavidin to detect internalized biotin-labeled β3 integrin. B, HUVECs were stimulated with Ang-2. Cell lysates were immunoprecipitated with an antibody against αvβ3 integrin. The resulting immunoprecipitate was separated by SDS-PAGE, Western blotted, and probed for ubiquitin sequence. The membrane was stripped and reprobed for total β3 integrin subunit to confirm equal loading. C, cells were stimulated with Ang-2 for 20 min, fixed in PFA, permeabilized with Triton X-100, and stained for total αvβ3 integrin and LAMP-1 (scale bar, 10 μm). In the brightfield (BF) pictures, the cell-cell junction is marked in white. D, HUVECs were stimulated for various time points with Ang-2. Thereafter, cells were fixed with PFA, permeabilized with Triton X-100, and stained for αvβ3 and LAMP-1. Three different experiments were analyzed with the newly developed software for co-localization studies. Error bars, S.E. Student's t test showed significantly more co-localization of LAMP-1 and αvβ3 upon 60 min of Ang-2 stimulation (p < 0.1).
DISCUSSION
Angiopoietins induce Tie2 translocation to cell-cell junctions in contacting ECs (Fig. 1) (10, 11). Yet, the net outcome of Ang-1 versus Ang-2-induced Tie2 translocation is different. Whereas Ang-1-induced Tie2 junctional association leads to downstream Tie2 activation along the Akt-Foxo and Akt-eNOS maintenance and survival pathways, Ang-2 stimulation leads to endothelial destabilization (29). Under physiological conditions, angiogenesis is limited to few organs in the adult. In contrasts, tumor growth requires angiogenesis, and Ang-2 levels correlate with tumor burden, indicating the importance during tumor growth (9, 30). Consistently, Ang-2 inhibition leads to vascular normalization in tumors (31). However, the molecular mechanisms underlying the observed Ang-2 effects have not been analyzed, yet.
In the present study we could show that Ang-2 stimulation of contacting EC was found to induce (i) Tie2 and β3 integrin translocation into cell-cell junctions, (ii) complex formation of Tie2 and αvβ3, (iii) FAK recruitment with subsequent phosphorylation at Ser910, (iv) dissociation of the adaptor proteins talin and p130Cas, and (v) integrin internalization and gating toward lysosomal degradation. The observed effect of Ang-2 stimulation on αvβ3 integrin turnover may explain the observed Ang-2-induced endothelial destabilization (29).
The integrin heterodimer αvβ3 has previously been shown to associate with PDGFRβ and VEGFR-2 (19, 21). Similarly, α5β1 integrin has been shown to associate with Tie2 receptor upon Ang-1 and Ang-2 stimulation (32, 33). On the other hand, both Ang-1 and Ang-2 can directly bind to some integrins and thereby signal in the absence of its receptor Tie2 (34–36). The ligand-induced association of αvβ3 with Tie2 as described in this study establishes not just a novel integrin-vascular receptor tyrosine kinase interaction but has paved the way toward studying the cellular consequences of this interaction. Most notably, Tie2·αvβ3 integrin complex formation was selectively induced by Ang-2, but not by Ang-1, suggesting that Tie2·αvβ3 integrin association may be functionally involved in the cellular mechanisms leading to endothelial destabilization (29).
Ang-2-induced Tie2·αvβ3 integrin complex formation was found to involve FAK. FAK recruitment to integrins following growth factor stimulation is usually associated with FAK phosphorylation (32) which in turn is intimately linked to barrier function (37), cell migration, cell survival, and proliferation by regulating the dynamics of focal adhesion complexes (38, 39). FAK phosphorylation occurs primarily at different phosphorylation sites of the kinase domain, including Tyr(P)397 (40). In turn, the Ser910 phosphorylation site located in the C-terminal FAT domain is much less well characterized as a biologically relevant phosphorylation site within FAK (41, 42). The experiments of the present study revealed that Ang-2-mediated Tie2·αvβ3 integrin complex formation and subsequent FAK recruitment lead neither to FAK phosphorylation at Tyr397 nor to Src recruitment, but to FAK phosphorylation at Ser910.
Experiments with PDGF-stimulated fibroblasts have previously shown that phosphorylation of FAK at Ser910 leads to the dissociation of the adaptor protein paxillin from FAK (27). Our experiments correspondingly revealed that talin and p130Cas rapidly dissociate from the αvβ3 integrin complex upon Ang-2 stimulation. These experiments show for the first time that not only paxillin but also p130Cas and talin dissociate from αvβ3 integrin, suggesting a critical hitherto unidentified role of Ser910 phosphorylation in the disassembly of focal adhesion complexes.
The turnover of focal adhesion complexes is known to be involved in vascular remodeling, including angiogenic endothelial cell migration and the regulation of vascular permeability (28, 43). The turnover of focal adhesion complexes is also closely linked to integrin internalization. Upon internalization, integrins are processed in intracellular compartments either to be gated for recycling or for degradation (27, 44). We have examined both pathways. Analyses with the recycling marker Rab11 did not show co-localization of αvβ3 integrin upon Ang-2 stimulation (data not shown). Furthermore, recycling studies were performed as described previously (22), but did not detect αvβ3 integrin recycling upon Ang-2 stimulation. Integrin recycling occurs classically during cell migration. Cellular protrusions adhere through integrins to the extracellular matrix (focal adhesion assembly). Subsequently, contraction via stress fibers allows forward movement. The trailing edges of the cells are released, a process that is initiated by stress fiber-mediated traction forces leading to focal adhesion disassembly. Integrins are internalized and recycled during this cyclic process (28).
Contrasting the recycling pathway, internalized integrins can be ubiquitinylated and lysosomally gated toward degradation. The experiments of this study have shown that Ang-2 stimulation induces internalization of the integrin subunit β3 associated with rapid ubiquitinylation. This gates the internalized integrin toward lysosomal degradation as evidenced by co-localization with the endosomal marker LAMP-1.
Taken together, the present study has unraveled the novel molecular mechanisms of Ang-2-dependent regulation of αvβ3 integrin turnover. Both the agonistic ligand Ang-1 and the antagonistic ligand Ang-2 induce transjunctional Tie2 receptor clustering. Yet, whereas Ang-1 transjunctionally clustered Tie2 receptor stimulates downstream signaling supporting endothelial survival, Ang-2-induced junctional clustering of Tie2 leads to recruitment of and clustering with αvβ3 integrin. This complex leads to FAK activation at Ser910 with subsequent focal adhesion dissociation and integrin internalization and degradation. The experiments establish a plausible model for Ang-2-mediated endothelial destabilization (29). Future work will need to unravel the exogenous cytokine-dependent contextuality of Ang-2 effects as a mediator of endothelial destabilization associated vessel regression or as facilitator of EC activating cellular responses.
Supplementary Material
Acknowledgments
We thank Dr. Felix Bestvater and Manuela Brom from the Microscopy Core Facility, German Cancer Research Center Heidelberg (DKFZ-ZMBH Alliance), for help with confocal microscopy.
This work was supported by grants from the German Research Council (DFG, SFB-TR23, Vascular Differentiation and Remodeling, Project A3).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5.
- VEGF and VEGFR
- vascular endothelial growth factor and its receptor
- Ang
- angiopoietin
- BSA
- bovine serum albumin
- EC
- endothelial cell
- FAK
- focal adhesion kinase
- FCS
- fetal calf serum
- HUVEC
- human umbilical vein endothelial cell
- Mes
- 4-morpholineethanesulfonic acid
- PDGF and PDGFR
- platelet-derived growth factor and its receptor
- PFA
- paraformaldehyde.
REFERENCES
- 1.Dumont D. J., Gradwohl G., Fong G. H., Puri M. C., Gertsenstein M., Auerbach A., Breitman M. L. (1994) Genes Dev. 8, 1897–1909 [DOI] [PubMed] [Google Scholar]
- 2.Sato T. N., Tozawa Y., Deutsch U., Wolburg-Buchholz K., Fujiwara Y., Gendron-Maguire M., Gridley T., Wolburg H., Risau W., Qin Y. (1995) Nature 376, 70–74 [DOI] [PubMed] [Google Scholar]
- 3.Suri C., Jones P. F., Patan S., Bartunkova S., Maisonpierre P. C., Davis S., Sato T. N., Yancopoulos G. D. (1996) Cell 87, 1171–1180 [DOI] [PubMed] [Google Scholar]
- 4.Augustin H. G., Koh G. Y., Thurston G., Alitalo K. (2009) Nat. Rev. Mol. Cell Biol. 10, 165–177 [DOI] [PubMed] [Google Scholar]
- 5.Thomas M., Augustin H. G. (2009) Angiogenesis 12, 125–137 [DOI] [PubMed] [Google Scholar]
- 6.Maisonpierre P. C., Suri C., Jones P. F., Bartunkova S., Wiegand S. J., Radziejewski C., Compton D., McClain J., Aldrich T. H., Papadopoulos N., Daly T. J., Davis S., Sato T. N., Yancopoulos G. D. (1997) Science 277, 55–60 [DOI] [PubMed] [Google Scholar]
- 7.Reiss Y., Droste J., Heil M., Tribulova S., Schmidt M. H., Schaper W., Dumont D. J., Plate K. H. (2007) Circ. Res. 101, 88–96 [DOI] [PubMed] [Google Scholar]
- 8.Fiedler U., Reiss Y., Scharpfenecker M., Grunow V., Koidl S., Thurston G., Gale N. W., Witzenrath M., Rosseau S., Suttorp N., Sobke A., Herrmann M., Preissner K. T., Vajkoczy P., Augustin H. G. (2006) Nat. Med. 12, 235–239 [DOI] [PubMed] [Google Scholar]
- 9.Nasarre P., Thomas M., Kruse K., Helfrich I., Wolter V., Deppermann C., Schadendorf D., Thurston G., Fiedler U., Augustin H. G. (2009) Cancer Res. 69, 1324–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukuhara S., Sako K., Minami T., Noda K., Kim H. Z., Kodama T., Shibuya M., Takakura N., Koh G. Y., Mochizuki N. (2008) Nat. Cell Biol. 10, 513–526 [DOI] [PubMed] [Google Scholar]
- 11.Saharinen P., Eklund L., Miettinen J., Wirkkala R., Anisimov A., Winderlich M., Nottebaum A., Vestweber D., Deutsch U., Koh G. Y., Olsen B. R., Alitalo K. (2008) Nat. Cell Biol. 10, 527–537 [DOI] [PubMed] [Google Scholar]
- 12.Garmy-Susini B., Varner J. A. (2008) Lymphat. Res. Biol. 6, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks P. C., Montgomery A. M., Rosenfeld M., Reisfeld R. A., Hu T., Klier G., Cheresh D. A. (1994) Cell 79, 1157–1164 [DOI] [PubMed] [Google Scholar]
- 14.Drake C. J., Cheresh D. A., Little C. D. (1995) J. Cell Sci. 108, 2655–2661 [DOI] [PubMed] [Google Scholar]
- 15.Storgard C. M., Stupack D. G., Jonczyk A., Goodman S. L., Fox R. I., Cheresh D. A. (1999) J. Clin. Invest. 103, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheu J. R., Yen M. H., Kan Y. C., Hung W. C., Chang P. T., Luk H. N. (1997) Biochim. Biophys. Acta 1336, 445–454 [DOI] [PubMed] [Google Scholar]
- 17.Penta K., Varner J. A., Liaw L., Hidai C., Schatzman R., Quertermous T. (1999) J. Biol. Chem. 274, 11101–11109 [DOI] [PubMed] [Google Scholar]
- 18.Alghisi G. C., Ponsonnet L., Rüegg C. (2009) PLoS One 4, e4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borges E., Jan Y., Ruoslahti E. (2000) J. Biol. Chem. 275, 39867–39873 [DOI] [PubMed] [Google Scholar]
- 20.Masson-Gadais B., Houle F., Laferrière J., Huot J. (2003) Cell Stress Chaperones 8, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soldi R., Mitola S., Strasly M., Defilippi P., Tarone G., Bussolino F. (1999) EMBO J. 18, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts M., Barry S., Woods A., van der Sluijs P., Norman J. (2001) Curr. Biol. 11, 1392–1402 [DOI] [PubMed] [Google Scholar]
- 23.Serini G., Napione L., Arese M., Bussolino F. (2008) Cardiovasc. Res. 78, 213–222 [DOI] [PubMed] [Google Scholar]
- 24.Geiger B., Spatz J. P., Bershadsky A. D. (2009) Nat. Rev. Mol. Cell Biol. 10, 21–33 [DOI] [PubMed] [Google Scholar]
- 25.Abedi H., Zachary I. (1997) J. Biol. Chem. 272, 15442–15451 [DOI] [PubMed] [Google Scholar]
- 26.Avraham H. K., Lee T. H., Koh Y., Kim T. A., Jiang S., Sussman M., Samarel A. M., Avraham S. (2003) J. Biol. Chem. 278, 36661–36668 [DOI] [PubMed] [Google Scholar]
- 27.Pellinen T., Ivaska J. (2006) J. Cell Sci. 119, 3723–3731 [DOI] [PubMed] [Google Scholar]
- 28.Lamalice L., Le Boeuf F., Huot J. (2007) Circ. Res. 100, 782–794 [DOI] [PubMed] [Google Scholar]
- 29.Scharpfenecker M., Fiedler U., Reiss Y., Augustin H. G. (2005) J. Cell Sci. 118, 771–780 [DOI] [PubMed] [Google Scholar]
- 30.Helfrich I., Edler L., Sucker A., Thomas M., Christian S., Schadendorf D., Augustin H. G. (2009) Clin. Cancer Res. 15, 1384–1392 [DOI] [PubMed] [Google Scholar]
- 31.Hashizume H., Falcón B. L., Kuroda T., Baluk P., Coxon A., Yu D., Bready J. V., Oliner J. D., McDonald D. M. (2009) Cancer Res. 70, 2213–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cascone I., Napione L., Maniero F., Serini G., Bussolino F. (2005) J. Cell Biol. 170, 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imanishi Y., Hu B., Jarzynka M. J., Guo P., Elishaev E., Bar-Joseph I., Cheng S.-Y. (2007) Cancer Res. 67, 4254–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlson T. R., Feng Y., Maisonpierre P. C., Mrksich M., Morla A. O. (2001) J. Biol. Chem. 276, 26516–26525 [DOI] [PubMed] [Google Scholar]
- 35.Hu B., Jarzynka M. J., Guo P., Imanishi Y., Schlaepfer D. D., Cheng S. Y. (2006) Cancer Res. 66, 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bezuidenhout L., Zilla P., Davies N. (2009) Inflammation 32, 393–401 [DOI] [PubMed] [Google Scholar]
- 37.Wu M. H. (2005) J. Physiol. 569, 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamadi A., Bouali M., Dontenwill M., Stoeckel H., Takeda K., Rondé P. (2005) J. Cell Sci. 118, 4415–4425 [DOI] [PubMed] [Google Scholar]
- 39.Kumar C. C. (1998) Oncogene 17, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 40.Hood J. D., Cheresh D. A. (2002) Nat. Rev. Cancer 2, 91–100 [DOI] [PubMed] [Google Scholar]
- 41.Schlaepfer D. D., Mitra S. K. (2004) Curr. Opin. Genet. Dev. 14, 92–101 [DOI] [PubMed] [Google Scholar]
- 42.Hunger-Glaser I., Fan R. S., Perez-Salazar E., Rozengurt E. (2004) J. Cell. Physiol. 200, 213–222 [DOI] [PubMed] [Google Scholar]
- 43.Gao B., Curtis T. M., Blumenstock F. A., Minnear F. L., Saba T. M. (2000) J. Cell Sci. 113, 247–257 [DOI] [PubMed] [Google Scholar]
- 44.Marmor M. D., Yarden Y. (2004) Oncogene 23, 2057–2070 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.