

Abstract
Modulation of integrin αvβ5 regulates vascular permeability, angiogenesis, and tumor dissemination. In addition, we previously found a role for p21-activated kinase 4 (PAK4) in selective regulation of integrin αvβ5-mediated cell motility (Zhang, H., Li, Z., Viklund, E. K., and Strömblad, S. (2002) J. Cell Biol. 158, 1287–1297). This report focuses on the molecular mechanisms of this regulation. We here identified a unique PAK4-binding membrane-proximal integrin β5-SERS-motif involved in controlling cell attachment and migration. We also mapped the integrin β5-binding site within PAK4. We found that PAK4 binding to integrin β5 was not sufficient to promote cell migration, but that PAK4 kinase activity was required for PAK4 promotion of cell motility. Importantly, PAK4 specifically phosphorylated the integrin β5 subunit at Ser-759 and Ser-762 within the β5-SERS-motif. Point mutation of these two serine residues abolished the PAK4-induced cell migration, indicating a functional role for these phosphorylations in migration. Our results may give important leads to the functional regulation of integrin αvβ5, with implications for vascular permeability, angiogenesis, and cancer dissemination.
Keywords: Cell Adhesion, Cell Migration, Extracellular Matrix, Integrin, Threonine-Serine Protein Kinase, p21-activated Kinase, Signaling
Introduction
Integrins are heterodimers composed of α and β subunits that contain a large extracellular domain with ligand binding capacity, a transmembrane domain, and a short cytoplasmic domain (2). Integrins mediate cell adhesion and thereby play a central role in cell migration. Integrins also participate in bi-directional signaling processes across the plasma membrane where ligand binding to the extracellular matrix generates intracellular signals (outside-in) and where signaling affecting integrin cytoplasmic tails can lead to regulation of the integrin extracellular binding capacity (inside-out) (3–6).
The membrane-proximal regions of the cytoplasmic domains of the integrin heterodimer interact with each other via a salt bridge. Disruption of this salt bridge can change the integrin affinity state, indicating that the short cytoplasmic domains of integrins are essential for both inside-out and outside-in signaling (3, 7). One important mechanism whereby cellular signaling influences cellular behavior is through the interaction and/or phosphorylation of integrin cytoplasmic tails by intracellular proteins (8–12). Interactions and/or phosphorylation of integrin cytoplasmic tails may also regulate the activation state of integrins, for example, β integrin tyrosine phosphorylation can regulate talin-induced integrin activation (11, 13–16).
Integrin phosphorylation at tyrosine residues has been found in the cytoplasmic domains of α6A, β1, β3, and β4 (17–22). Also, serine/threonine phosphorylation of integrin cytoplasmic domains has been found in α4, β1, β2, β3, and β7 subunits (23–31). However, so far only a few protein kinases have been identified that phosphorylate integrin cytoplasmic domains. c-Src was found to be responsible for tyrosine phosphorylation, whereas protein kinase C and integrin-linked kinase may mediate serine/threonine phosphorylation of integrins (29, 32, 33).
Integrin αvβ5 mediates cell attachment and migration on vitronectin (2, 6, 34, 35). Integrin αvβ5 is induced in keratinocytes during wound healing and facilitates vascular endothelial growth factor-induced vascular permeability (36–38). In addition, growth factor activation of integrin αvβ5-mediated cell motility has been functionally linked to angiogenesis as well as carcinoma cell dissemination (39–41). However, how integrin αvβ5 itself may be controlled and the role of its cytoplasmic domains are both unclear.
Overexpression of p21-activated kinase 4 (PAK4)5 can induce localized actin polymerization and filopodia formation (42) and affect cell adhesion and anchorage-independent growth of rodent fibroblasts (43, 44). We previously found that PAK4 interacts with the integrin β5 cytoplasmic tail and promotes integrin αvβ5-mediated cell migration (1). However, whether PAK4 promotes cell motility through its interaction with integrin αvβ5 and/or its effects on the actin cytoskeleton remains unknown.
In this study, we used site-directed mutagenesis to map the PAK4-binding site within integrin β5 as well as the integrin β5-binding site within PAK4. Importantly, we also identified PAK4-mediated phosphorylation of two serine residues in the integrin β5 cytoplasmic domain that are involved in the regulation of cell motility. These results provide important information regarding the intracellular regulation of αvβ5 activity.
EXPERIMENTAL PROCEDURES
Reagents
Anti-FLAG mouse mAb M2 was acquired from Sigma, anti-integrin αvβ5 (P1F6) mouse mAb from Invitrogen, rabbit anti-HA (Y11) pAb from Santa Cruz Biotechnology, and rabbit anti-human integrin β5 cytoplasmic tail from Chemicon Int. Cell culture media, Lipofectamine Plus, and Lipofectamine 2000 were purchased from Invitrogen. An ECL detection kit, protein G-Sepharose, and glutathione-Sepharose were from Amersham Biosciences. [γ-32P]ATP was from Amersham Biosciences. Vitronectin was purified from human serum as described previously (45). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, myelin basic protein (MBP), and all other chemicals were obtained from Sigma.
Yeast Two-hybrid Assay
Yeast mating test assay was performed using the DupLEX-A Yeast Two-Hybrid System (OriGen Technologies) as previously described (46). The PAK4 C terminus (CT) (amino acids 396–591) was subcloned into the EcoRI and XhoI sites of the pJG4–5 prey vector and transformed into the EGY48 yeast strain, including the pSH18–34 reporter gene (lacZ) plasmid. The integrin β5 cytoplasmic domain (β5-aa-753–799) and various fragments thereof were subcloned into the EcoRI and XhoI sites of the pEG202 bait vector. Point mutations in the integrin β5 tail were generated with the QuikChange site-Directed mutagenesis kit using pEG202-β5-tail as a template and then transformed into the yeast strain RFY206. The correct reading frames and sequences were verified by sequencing. Yeast mating tests were performed by using the RFY206 strain, including different integrin β5-tail constructs with the EGY48 strain, including the PAK4-CT construct.
Mammalian Cell Expression Vectors
FLAG-PAK4-WT, FLAG-PAK4-K350M, FLAG-PAK4-ΔIBD, and FLAG-BAP (bacterial alkaline phosphatase) were previously described (1). The truncated forms of PAK4, N-terminal amino acid 1–322 (NT), kinase domain amino acid 323–591 (KD), and C-terminal amino acid 396–591 (CT), were amplified by PCR using wild-type (WT) human PAK4 cDNA as a template and subcloned into the HindIII/BamHI site of 3 × FLAG-CMVTM-10 expression vector (Sigma-Aldrich). Nine PAK4 point mutations within its integrin-binding domain (IBD) and a PAK4 deletion mutant, PAK4-Δ69–221, were generated using the FLAG-PAK4-WT and QuikChange (Stratagene). To construct Enhanced green fluorescent protein (EGFP)-PAK4-WT and mutants, HindIII/BamHI fragments from FLAG-PAK4 were inserted into the HindIII/BamHI sites of the pEGFP C3 vector (Clontech Laboratories, Inc.). The human integrin β5 full-length cDNA was subcloned into the EcoRI site of the pcDNA3 vector (Invitrogen). Integrin β5 mutants of pCDNA3-β5-ER760,761RE, pcDNA3-β5-ΔSERS, pCDNA3-β5-SS769, 762AA, and pCDNA3-β5-SS759,762EE, were generated by the QuikChange Kit (Stratagene) using pCDNA3-β5-WT as a template. Insertion of CMV-β5 or CMV-β5 mutants into FLAG-PAK4-WT generated FLAG-PAK4-CMV-β5 double cassette constructs.
Cell Culture
African green monkey kidney COS-7 cells and human breast carcinoma MCF-7 cells were grown in Dulbecco's modified Eagle's medium. CS-1 hamster melanoma cells kindly made available by Dr. Caroline Damsky, University of California at San Francisco, were grown in RPMI. All tissue culture media were supplemented with 5% fetal bovine serum and 10 μg/ml Gentamycin (Invitrogen). Cells were maintained in a humidified incubator with 5% CO2. The CS-1-stable clones were grown under the same conditions as parental CS-1 cells, but in the presence of 500 μg/ml G418 (Invitrogen).
Cell Transfection
4–8 μg of total DNA was transfected in 10-cm cell culture dishes (80–90% confluence of COS-7 or MCF-7 cells), using Lipofectamine PlusTM (Invitrogen), according to the manufacturer's protocol, and cells were used 30–48 h after transfection. For transfection of CS-1 cells, 4–8 μg of total DNA was transfected in 6-well plates (1 × 106 cells/well) using the Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. CS-1 cells stably transfected with integrin β5 WT or mutant clones were established in medium containing 600 μg/ml G418. Selected cell colonies were transferred to separate culture dishes and were subsequently grown in 500 μg/ml G418 medium. Pools of mixed populations of stable transfectant CS-1 cells expressing comparable levels of each of the wild-type or mutant integrins were established by three times consecutive fluorescence-activated cell sorting with anti-integrin αvβ5 mAb P1F6 performed over a 3-month period.
Pulldown Assay
For in vitro GST pulldown assays, the integrin β1-tail, β5-tail (amino acids 753–799), and β5-tail mutants were individually expressed as GST fusion proteins using the bacterial expression vector pGEM-1λT (Amersham Biosciences). GST fusion proteins were produced and purified using glutathione-Sepharose beads (Amersham Biosciences) according to the manufacturer's protocol. GST pulldown assays were performed as described (1). Briefly, 200 μg of lysates from COS-7 cells overexpressing various hPAK4 constructs were incubated with 5 μg of GST fusion proteins. The result was visualized by immunoblotting, and band intensities were measured using Kodak one-dimensional image analysis or ImageJ 1.43 software (National Institutes of Health).
Kinase Activity Assay and Phosphopeptide Mapping
Various PAK4 constructs were expressed in COS-7 cells and lysed in kinase lysis buffer (50 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 1% Nonidet P-40, 10% glycerol 150 mm NaCl) with addition of fresh protease inhibitors (0.5 μg/ml leupeptin, 1 mm EDTA, 1 μg/ml pepstatin A, 0.2 mm phenylmethylsulfonyl fluoride) and a serine/threonine protein phosphatase inhibitor mixture (Sigma), followed by immunoprecipitation. PAK4 kinase activity was determined in a kinase buffer (50 mm Hepes, pH 7.5, 10 mm MgCl2, 2 mm MnCl2, 0.2 mm dithiothreitol) in the presence of 30 μm cold ATP and 10 μCi of [γ-32P]ATP (3000 Ci/nm, Amersham Biosciences) and in the presence of 5 μg of substrate (MBP, GST, GST-β1 tail, or GST-β5 tail) for 30 min at 30 °C. Incubation was stopped in Laemmli buffer, and samples were heated at 95 °C for 4 min. Phosphorylated proteins were separated by 12.5% SDS-PAGE. The gel was dried and visualized by autoradiography. Phosphorylation sites in GST-β5 were mapped as described previously (47). Briefly, phosphopeptides were resolved by 10% SDS-PAGE and transferred to nitrocellulose membrane. GST or GST-β5 corresponding bands were excised and digested with trypsin as described (48). The first dimension electrophoresis was carried out in a pH 1.9 buffer, and the second dimension separation was performed using TLC in isobutyric acid buffer. The chromatography plates were exposed using Fuji Bas Bio-Imaging Analyzer, and radioactive peptides were scraped off the plate, followed by sequencing and phosphoamino acid analysis. For Edman degradation, phosphopeptides were coupled to Sequelon-AA membranes (Millipore) according to the manufacturer's instructions and sequenced on an Applied Biosystems Gas Phase sequencer. The activity in released phenylthiohydantoin derivatives from each cycle was quantified using the Bio-Imaging Analyzer. For phosphoamino acid analysis, peptides were lyophilized and thereafter hydrolyzed in 6 m HCl for 1 h at 110 °C, followed by TLC as described (49). To determine PAK4 phosphorylation of the integrin β5 subunit in living cells, COS-7 cells transfected with HA-PAK4 underwent a phosphate starvation for 6 h at 40 h post transfection, followed by metabolic labeling with 300 μCi of [γ-32P]ATP for 2 h at 37 °C. Cells were then washed twice with phosphate-free Dulbecco's modified Eagle's medium and lysed in radioimmune precipitation assay buffer. Integrin αvβ5 was immunoprecipitated by mAb P1F6, and the phosphorylated β5 subunit was visualized by autoradiography.
Cell Adhesion Assay
A cell adhesion assay was performed as described (35). Briefly, non-treated 48-well cluster plates (Corning Costar Corp.) were coated with vitronectin (VN) and blocked by 1% heat-denatured bovine serum albumin. 5 × 104 CS-1 cells/well transfected to express integrin β5, integrin β5 mutants, or co-transfected to express integrin β5 and PAK4, were seeded in wells and allowed to attach at 37 °C for 30 min. The attached cells were counted using a microscope (10× objective) after cell staining by crystal violet or, alternatively, quantified using by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Cell Migration Assay
Haptotactic cell migration assays were performed using Transwell chambers (Costar Inc.) with 8.0-μm pore size as described before (1). Briefly, 1 × 105 MCF-7 cells transiently transfected with EGFP, pEGFP-PAK4, or PAK4 mutants were added on top of the Transwell membranes coated with VN on the bottom and allowed to migrate toward VN for 6 h at 37 °C in adhesion buffer (1, 35). The migrated cells were counted using a fluorescence microscope, and adjustment was made for the transfection rate of each population as determined by flow cytometry or by manual counting using fluorescence microscopy. For CS-1 cell migration assay, 1 × 105 CS-1 cells transiently transfected or stable clones with pcDNA3 empty vector, integrin β5, or β5 mutants were added into the Transwell membranes and allowed to migrate toward VN for 24 h at 37 °C. The migrated cells were stained by 0.5% crystal violet and counted using a microscope (10× objective). For CS-1 cells stably expressing integrin β5 or mutant β5 that were co-transfected to express EGFP or EGFP-PAK4, cell migration was quantified as described above for EGFP/EGFP-PAK4 co-transfected MCF-7 cells.
Flow Cytometry Analysis
Cell surface expression levels of integrins were analyzed by measurement of fluorescein isothiocyanate staining intensity by FACScan® flow cytometer using CellQuest software (BD Biosciences) after staining with anti-integrin αvβ5 mAb P1F6 and a fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) essentially as described by Bao and Strömblad (50).
Statistical Analysis
Data were analyzed for statistical significance using an un-paired two-tailed t test.
RESULTS
Identification of an Integrin β5 Cytoplasmic Tail SERS-motif Involved in PAK4 Binding
We previously found that PAK4 binds to the integrin β5-subunit and promotes integrin αvβ5-mediated cell motility (1). We here mapped the PAK4-binding site within the integrin β5-subunit cytoplasmic domain at the amino acid level. Firstly, we analyzed PAK4 interaction in yeast two-hybrid mating tests (a schematic outline is shown in Fig. 1A) of WT β5 and 13 deletion mutants or fragments thereof as indicated in Fig. 1B. The smallest β5 fragment maintaining full binding strength to PAK4 contained a 10-amino acid N-terminal β5-tail sequence (β5–753-762). Any further deletions of this fragment caused loss or partial loss of PAK4 binding. Further, truncation of this 10-amino acid fragment revealed that a β5-SERS motif (β5–759-762) was required for PAK4 binding. Thus, the results suggest that full PAK4 binding requires a 10-aa region of the integrin β5 tail (aa 753–762) and that the membrane-proximal SERS-motif (aa 759–762) within β5 is critical for PAK4 binding. These results were verified in mammalian cells by GST pulldown analyses, where the β5–753-762 fragment pulled down the same amount of PAK4 as the β5-WT cytoplasmic tail (Fig. 1, C and D). However, deletion of the β5-SERS-motif (aa 759–762) abolished PAK4 binding (Fig. 1D). Among integrins, the SERS amino acid sequence only appears in the integrin β5 cytoplasmic tail, although ERS is found at the same position within integrin β6 and ER within integrin β3 (Fig. 1E).
FIGURE 1.

Identification of an integrin β5 SERS-motif in PAK4 binding. A, schematic diagram of the yeast two-hybrid system used in Fig. 1. B, yeast two-hybrid mating test assay. The integrin β5-tail (aa 753–799) and fragments thereof were mated with PAK4-CT. The strength of the interactions judged by the intensity of blue after 48 h is indicated on the far right. C, the schematic diagram shows the GST-integrin β5 tail and the β5 fragments used in the GST pulldown assay in panel D. D, GST pulldown assay of PAK4-WT using GST-integrin β5-tail and fragments thereof (upper panel). Pulldown of PAK4-WT using GST alone served as a negative control. The input lane using a lysate of PAK4 WT-transfected cells marks the size of PAK4-WT. The relative band intensities are indicated below. Coomassie Brilliant Blue gel staining shows the loading of GST proteins (lower panel). E, an alignment of partial amino acids sequences of integrin β cytoplasmic tails with the corresponding regions of the integrin β5 PAK4-binding motif. The SERS-motif and conserved amino acid within are in bold. F, the indicated integrin β5-tail point mutations were introduced within the PAK4-binding region, and the resulting products were mated with PAK4-CT as described in A–B. G, GST pulldown of PAK4-WT using GST, GST-β5-tail, and GST-β5-ER760,761RE mutants. The input lanes show the position of PAK4-WT by direct immunoblot of lysate (upper panel). The relative band intensities are displayed below. Coomassie Brilliant Blue gel staining shows the loading of GST fusion proteins (lower panel). Displayed results are representative of three or more experiments.
Using yeast two-hybrid mating tests, we then fine-mapped the ten amino acids within the integrin β5 membrane-proximal region involved in PAK4 binding (Fig. 1F). Mutations of β5 aa 756, 757, 758, 759, or 762 did not affect its ability to bind PAK4. However, mutation of β5-EF 753 and 754 to alanine residues partially disrupted PAK4 binding, and reversing the charges by swapping ER 760 and 761 to RE completely abolished PAK4 binding. However, mutation of aa ER 760 and 761 to alanine residues did not influence PAK4 binding. The effect of the ER to RE swap of β5 aa 760 and 761 for binding to PAK4 in mammalian cells was verified by a GST pulldown assay (Fig. 1G). Together, this identified amino acids 753, 754, 760, and 761 in integrin β5 as involved in PAK4 binding and the SERS-motif as critical for PAK4 binding to the integrin β5 subunit.
The Integrin β5 Tail SERS-motif Affects Cell Attachment and Motility
To test the potential role of the integrin β5 SERS-motif in regulation of cell adhesion and migration, full-length integrin β5 (β5-WT) and two mutants, β5-ΔSERS and β5-ER760,761RE, were expressed in CS-1 hamster melanoma cells that do not express endogenous integrin β3 or β5 subunits and therefore are unable to attach to VN (51). The cell surface expression levels of WT β5 and β5-ER760,761RE were similar upon transient transfection when analyzed by FACS using anti-integrin αvβ5 mAb P1F6 (Fig. 2A). Untransfected CS-1 cells did not attach or migrate on VN (data not shown). Interestingly, CS-1 cells transiently transfected with β5-ER760,761RE displayed a significantly lower attachment to VN as compared with cells transfected with β5-WT (Fig. 2B). In parallel, β5-ER760,761RE-transfected CS-1 cells exhibited markedly increased cell motility as compared with WT β5-transfected cells (Fig. 2C) displaying an inverse correlation between cell attachment and motility. Likewise, CS-1 cells stably expressing similar levels of WT β5, β5-ER760,761RE or β5-ΔSERS were analyzed (Fig. 2D). Cells expressing β5-ER760,761RE or β5-ΔSERS displayed a marked reduction in cell attachment combined with a 3- to 6-fold increase in cell motility on VN as compared with WT β5-transfected cells (Fig. 2, E–G). These results indicate that the membrane-proximal SERS motif in the integrin β5 cytoplasmic tail affects the extracellular function of integrin αvβ5.
FIGURE 2.

Mutation or deletion of the integrin β5 tail SERS-motif affects cell attachment and migration. A, the integrin αvβ5 cell surface expression levels of CS-1 cells transiently transfected with integrin β5-WT or β5-ER760,761RE were analyzed by flow cytometry. Non-transfected CS-1 cells served as a control for the FACS settings. The given percentages represent the fraction of cells displaying αvβ5-staining above untransfected CS-1 cells, and M1 shows mean intensity of the cells expressing αvβ5. These cells were then used for cell attachment and migration assays. B and C, bar graphs show quantification of cell attachment (B) or cell migration (C) on VN of CS-1 cells transiently expressing integrin β5-WT or β5-ER760,761RE, where β5-WT-expressing cells are defined as control. D, flow cytometry analysis of integrin αvβ5 cell surface expression in CS-1 cells with or without stable expression of integrin β5 or mutants thereof. M1 is the mean intensity of the cells expressing αvβ5. E and F, cell attachment and (G) motility on VN of CS-1 cells stably expressing integrin β5 or indicated β5 mutants. E, cell attachment onto different VN-coating concentrations and (F) to VN-coating using 2.5 μg/ml. Bars represent mean values ± S.E. (B, C, F, G) or S.D. (E) (n = 3). Statistically discernable differences as determined by t test are indicated (**, p ≤ 0.01). Displayed results are representative of three or more experiments.
PAK4 Regulation of Integrin αvβ5-mediated Cell Attachment and Migration May Involve the Integrin β5-tail SERS-motif
Overexpression of WT PAK4 in MCF-7 cells stimulated integrin αvβ5-mediated cell migration on VN (1). To determine whether the SERS-motif in the integrin β5-tail may play a role in PAK4-mediated regulation of cell motility, we co-transfected WT PAK4 with WT integrin β5 or with non-PAK4-binding integrin β5 SERS-motif mutants in CS-1 cells. These transfected cells were analyzed for the effects on integrin αvβ5-mediated cell attachment and migration. When CS-1 cells were transiently transfected with WT FLAG-PAK4 and WT integrin β5 using a double-cassette vector, cell attachment decreased and cell migration increased as compared with cells transfected with WT β5 alone (Fig. 3, A–D). Consistently, CS-1 cells stably expressing WT β5 were markedly affected by EGFP-PAK4 overexpression resulting in decreased cell attachment and increased cell migration on VN as compared with EGFP control-transfected cells (Fig. 3, E and F). Interestingly, CS-1 cells stably expressing β5-ER760,761RE or β5-ΔSERS displayed similar levels of cell attachment and migration as when WT β5 expressing cells were co-transfected with PAK4. However, no significant changes in cell attachment or migration were observed when either of the mutant β5 expressing cells were transfected with EGFP-PAK4 as compared with control-transfected cells (Fig. 3, E and F). This suggests that the PAK4-binding β5-SERS-motif may be involved in PAK4-mediated regulation of cell attachment and migration.
FIGURE 3.

The PAK4-binding integrin β5 tail SERS-motif regulates cell attachment and migration. A and B, the expression levels of CS-1 cells transiently transfected with integrin β5-WT or co-transfected with integrin β5-WT and FLAG-PAK4-WT analyzed by immunoblotting (A) or flow cytometry (B) with untransfected CS-1 cells as negative control and actin as a loading control. Percentages and M1 values are as shown as in Fig. 2. C, cell attachment and (D) cell motility on VN (10 μg/ml) of CS-1 cells transiently transfected with β5-WT or co-transfected with integrin β5-WT and PAK4-WT. E, cell attachment and (F) cell motility on VN of stable clones of CS-1-β5-WT, CS-1-β5-ER760,761RE, and CS-1-β5-ΔSERS transiently co-expressing EGFP (open bars) or EGFP-PAK4 (solid bars). Parental CS-1 cells transfected with EGFP or EGFP-PAK4 served as background control (not shown). Values obtained with CS-1-β5-WT co-transfected with EGFP empty vector were defined as control. Bars represent mean values ± S.E. (n = 3). Statistically discernable differences as determined by t test are indicated (*, p ≤ 0.05; **, p ≤ 0.01). Displayed results are representative of three or more experiments.
Mapping of the Integrin β5-binding Site within PAK4
To further elucidate the role of the PAK4 to integrin β5 interaction, the β5-binding site in PAK4 was mapped. The PAK4 C-terminal region (PAK4-CT, aa 396–591) directly interacts with integrin β5 in yeast two-hybrid mating tests and the critical IBD was located within aa 505–533 (1). To analyze if this region of PAK4 was also critical for association with integrin αvβ5 in mammalian cells, FLAG-PAK4-WT, a kinase-inactive PAK4 mutant (K350M), FLAG-PAK4-Δ505–533 (ΔIBD), and FLAG-PAK4-Δ69–221 (Fig. 4A) were transiently expressed in COS-7 cells, and a GST pulldown assay was performed (Fig. 4B). Cell extracts of FLAG-PAK4-WT and Δ69–221 were pulled down by GST-β5 tail fusion protein but not K350M or ΔIBD. This showed that the PAK4-IBD was required for integrin β5-association in mammalian cells. Surprisingly, the kinase-inactive PAK4-K350M that contains the IBD did not associate with integrin β5. This is in contrast with our previous results from yeast two-hybrid mating tests where a C-terminal PAK4 fragment with a K350M mutation could still interact with integrin β5 (1). This may be explained by conformation differences between the K350M-mutated full-length PAK4 and a mutated PAK4 C-terminal fragment. However, the fact that eliminating the PAK4 kinase activity also blocked its integrin binding capacity raised the question whether PAK4 kinase activity may be correlated to integrin binding. Therefore, the kinase activity of the PAK4 constructs was tested. FLAG-BAP, PAK4-WT, K350M, ΔIBD, and Δ69–221 were transiently transfected into COS-7 cells, and the FLAG-PAK4 immunoprecipitates were used in an in vitro kinase activity assay. WT PAK4 and PAK4-Δ69–221 displayed autophosphorylation and phosphorylated the substrate MBP, whereas K350M and ΔIBD did not (Fig. 4C). Together, this indicates that the PAK4-IBD is required for integrin binding and that the PAK4 kinase activity correlates with integrin β5 binding in mammalian cells.
FIGURE 4.
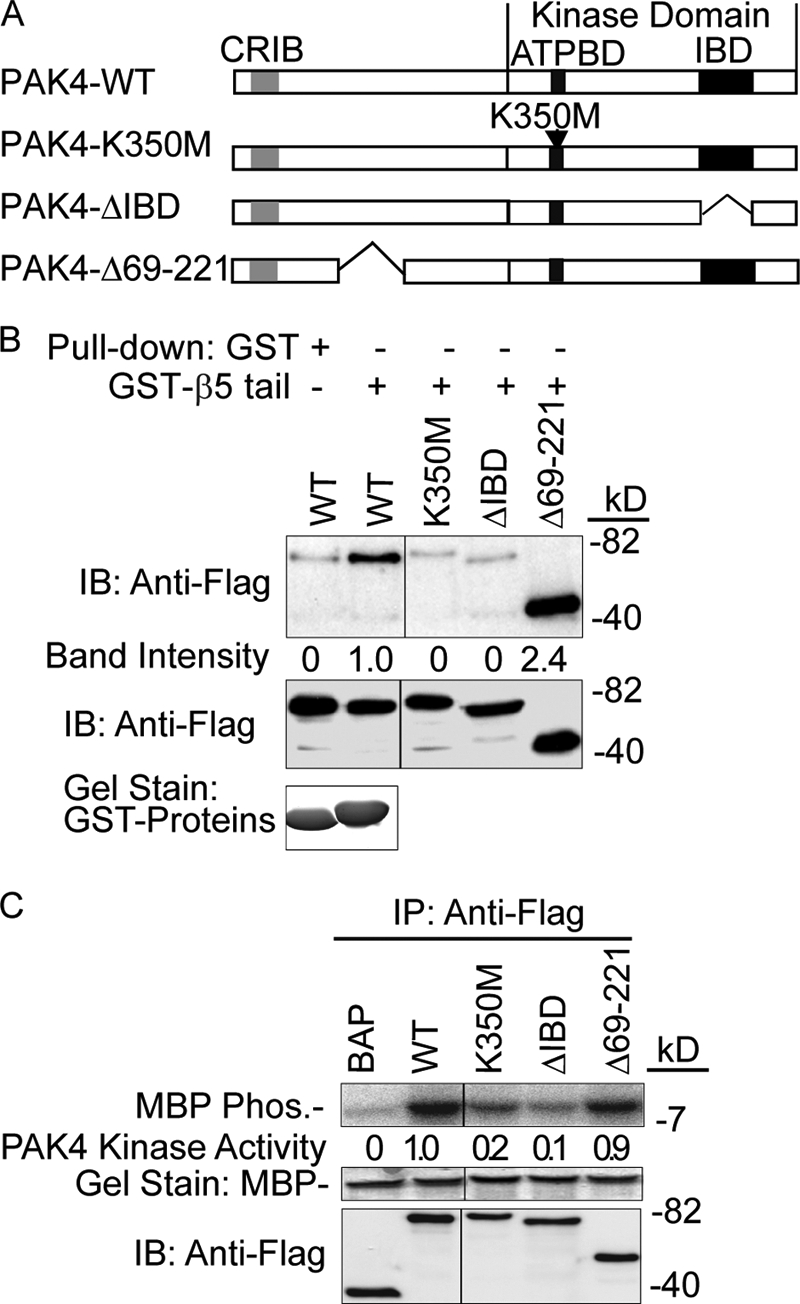
Mapping of the integrin β5 binding region within PAK4 and the role of the IBD for PAK4 kinase activity. A, the schematic diagram shows the domain composition of PAK4 and the PAK4 mutants used in this figure. CRIB denotes the Cdc42- and Rac-binding domain. The ATP-binding domain (ATPBD) and the integrin-binding domain (IBD) are situated within the kinase domain (KD) of PAK4. B, GST pulldown assay of PAK4 mutants. Cell lysates from COS-7 cells transiently expressing FLAG-tagged WT PAK4 (WT), PAK4-ΔIBD, PAK4-K350M, or PAK4-Δ69–221 were pulled down by a GST-β5-WT-tail fusion protein (top panel). PAK4-WT in combination with GST was used as a negative control (top panel). The quantified relative band intensities are shown below. The middle panel shows the used amounts of overexpressed PAK4 analyzed by immunoblotting (IB). Coomassie Brilliant Blue gel staining shows the relative amount of GST fusion proteins (lower panel). C, PAK4 kinase assay. Immunoprecipitates from COS-7 cells transiently transfected with PAK4 constructs were used in an in vitro kinase assay using myelin basic protein (MBP) as a substrate (top panel). The kinase activities of PAK4 mutants were quantified using a PhosphorImager, and numbers relative to WT PAK4 activity for MBP phosphorylation are indicated below. FLAG-BAP was used as a negative control. The middle panel shows Coomassie Brilliant Blue gel staining of MBP loading, and the lower panel shows loading of overexpressed proteins detected by immunoblot (IB).
To further reveal the relationship between the PAK4 integrin binding capacity and its kinase activity, we sought to separate the kinase activity of PAK4 from its integrin binding capacity by introducing nine different point mutations into PAK4-IBD at amino acids conserved among the PAK family (Fig. 5, A and B), because other PAKs can also bind integrins.6 To test the effect of these PAK4 mutations on PAK4 association with integrin β5 in mammalian cells, a GST pulldown assay was performed with lysates from COS-7 cells overexpressing FLAG-tagged bacterial alkaline phosphatase (BAP) used as negative control, PAK4-WT, PAK4-K350M, and the nine PAK4 point mutations introduced in full-length WT PAK4. As shown in Fig. 5C, the mutants K350M; PP513,514AA; PP519,520AA; and A523P displayed no or weak association with the integrin β5-tail in mammalian cells, whereas all the other PAK4 point mutants tested displayed a similar positive β5 association as WT PAK4.
FIGURE 5.

Mapping of the PAK4 integrin-binding site at the amino acid level and function of PAK4-IBD point mutations for PAK4 kinase activity and cell migration. A, the schematic diagram shows the PAK4 point mutations created within the PAK4 IBD. B, alignment of the integrin-binding motif of PAK4 with the corresponding regions of human PAKs, the Drosophila PAK4 homologue mushroom bodies tiny (MBT) gene and yeast STE20. Amino acids in bold show conservation among the PAKs, which was used as the basis for the PAK4-IBD point mutation design. C, GST-β5-tail pulldown of lysates from COS-7 cells transfected with FLAG-tagged full-length PAK4 and point mutants thereof as indicated (top panel). FLAG-BAP was used as a negative control. Immunoblotting shows loading of FLAG-tagged proteins (middle panel). The migration rate of the mutant PP513,534AA protein consistently appeared higher than other mutants. This may be caused by proteolysis as a result of the mutagenesis. The relative band intensity of pulled down PAK4 is indicated in the lower bar graph, and the band intensity was set to 0 for the FLAG-BAP control and100% of control for PAK4-WT. Bars represent mean intensity ± S.E. among three experiments. Statistically discernable differences compared with PAK4-WT according to t test are indicated (**, p ≤ 0.01; ***, p ≤ 0.001). D, FLAG-tagged proteins were analyzed in a kinase assay using MBP as a substrate (top panel). The FLAG-BAP and lysate of non-transfected COS-7 cells were used as negative controls. Coomassie Brilliant Blue gel staining shows the loading of MBP (upper middle panel), and immunoblotting shows the loading of FLAG-tagged proteins (lower middle panel). The relative activities quantified by PhosphorImager are shown in the lower panel (bar graph) for PAK4 MBP substrate phosphorylation activity. The kinase activity was set to 0 for the FLAG-BAP control and 100% of control for PAK4-WT. Bars represent mean values ± S.E. for three distinct experiments. Statistically discernable differences compared with PAK4-WT according to t test are indicated (*, p ≤ 0.05; **, p ≤ 0.01). E, MCF-7 cells transiently transfected with control EGFP, EGFP-PAK4 WT, or EGFP-PAK4 mutants as indicated were analyzed for haptotactic cell motility toward VN. Bar graphs show quantified cell motility relative to the EGFP control (mean value ± S.E. of at least three experiments). Statistically discernable differences compared with EGFP control according to t test are indicated (*, p ≤ 0.05; **, p ≤ 0.01).
The effect of the nine point mutations on PAK4 kinase activity was analyzed using an in vitro kinase assay (Fig. 5D). We observed that the kinase-defective PAK4-K350M as well as three other PAK4 mutants; PP513,514AA; PP519,520AA; and A523P displayed no significant kinase activity. These were the same four PAK4 full-length mutants that also showed no or weak integrin binding capacity. All other mutants displayed equal or stronger PAK4 kinase activity and autophosphorylation compared with WT PAK4. Given that the same four point mutants that lacked kinase activity were also impaired in associating with integrin β5, we were unable to separate the kinase activity of PAK4 and its integrin binding capacity. Based on this, we hypothesize that the PAK4 integrin-binding site might be located in a substrate-binding pocket of PAK4. Alternatively, PAK4 kinase activity may be required for full-length PAK4 integrin binding.
Effects of PAK4 Mutations on Integrin αvβ5-mediated Cell Motility
We previously demonstrated that overexpression of PAK4 induced integrin αvβ5-mediated carcinoma cell migration (1). We now analyzed if the mutations of PAK4-IBD that disrupted integrin binding also affected the capacity of PAK4 to induce integrin αvβ5-mediated cell migration (Fig. 5E). Consistent with our previous findings, overexpression of WT PAK4 in MCF-7 cells induced integrin αvβ5-mediated cell migration on VN ∼2–3 times above control levels. However, the kinase-inactive PAK4-K350M and the PAK4-ΔIBD mutants failed to stimulate αvβ5-mediated cell migration (Fig. 5E). Furthermore, the three point mutations in PAK4-IBD that disrupted the PAK4 integrin binding capacity together with the PAK4 kinase activity did not promote MCF-7 cell migration, whereas WT PAK4 and all other six PAK4-IBD point mutants were able to significantly induce cell motility (Fig. 5E). This suggests that the integrin binding capacity, kinase activity, and the capability to induce cell motility of PAK4 are correlated.
PAK4-mediated Integrin β5 Binding Is Not Sufficient to Promote Cell Migration
PAK4-CT, which lacks the PAK4 ATP-binding pocket, strongly interacted with the integrin β5 cytoplasmic domain in yeast mating tests (1). We compared PAK4-NT (N-terminal aa 1–322) and two C-terminal truncated mutants, PAK4-KD (kinase domain, aa 323–591) and PAK4-CT (C-terminal, aa 396–591) (Fig. 6A), for association with integrin β5 in mammalian cells by a GST pulldown assay (Fig. 6B). Although PAK4-WT, PAK4-KD, and PAK4-CT were pulled down by the GST-β5 tail fusion protein, PAK4-NT was not. Thus, deletion of the NT domain or the ATP-binding domain did not affect the binding capacity of PAK4 to integrin β5 in mammalian cells. Given that PAK4-CT lacks the PAK4 ATP-binding pocket, it is conceivable that it also lacks kinase activity. To test this, we measured the kinase activity of PAK-CT and compared it to that of PAK4-WT, PAK4-NT, and PAK4-KD (Fig. 6C). Although PAK4-WT and PAK4-KD displayed high kinase activities, PAK4-CT and PAK4-NT did not show any significant kinase activity above background. This way, we identified a PAK-CT fragment that lacked kinase activity, but with an intact integrin binding capacity.
FIGURE 6.

Elucidation of the role of PAK4 integrin binding capacity for cell motility. A, schematic diagram shows common structural features of PAK4 and the PAK4 mutations: PAK4-NT (aa 1–322), PAK4-KD (aa 323–591), and PAK4-CT (aa 396–591). B, GST pulldown assay of PAK4 mutants. Cell lysates from COS-7 cells transiently expressing FLAG-tagged WT PAK4 (WT), PAK4-NT, PAK4-KD, or PAK4-CT were pulled down by a GST-β5-tail fusion protein (top panel). PAK4-WT in combination with GST was used as a negative control (top panel). The quantified relative band intensities are shown below. Lower panel shows the loading of overexpressed PAK4 analyzed by immunoblotting (IB). The GST fusion protein relative amounts used are shown in Fig. 4B. C, PAK4 kinase assay. Immunoprecipitates from COS-7 cells transiently transfected with FLAG-tagged PAK4-WT, PAK4-NT, PAK4-KD, or PAK4-CT were used in an in vitro kinase assay using myelin basic protein (MBP) as a substrate (top panel). PAK4-WT in combination with a normal mouse IgG was used as a negative control. The kinase activities of PAK4 mutants were quantified using a PhosphorImager, and the numbers relative to WT PAK4 activity for MBP phosphorylation are indicated below. The middle panel shows Coomassie Brilliant Blue gel staining of MBP loading, and the lower panel shows loading of overexpressed proteins detected by immunoblot (IB). D, cell migration assays of PAK4 mutants. MCF-7 cells were transiently transfected with control EGFP, EGFP-PAK4-WT, EGFP-PAK4-NT, EGFP-PAK4-KD, or EGFP-PAK4-CT mutants. The data represent the mean for three separate experiments ± S.E. Statistically discernable differences compared with EGFP control analyzed by t test are indicated (*, p ≤ 0.05; **, p ≤ 0.01).
To test if PAK4-CT affected cell motility, we overexpressed PAK-WT, PAK4-NT, PAK-KD, and the PAK4-CT and analyzed integrin αvβ5-mediated cell motility of MCF-7 cells. We found that overexpression of PAK4-WT and PAK4-KD consistently enhanced cell motility (Fig. 6D). However, PAK4-CT as well as PAK4-NT failed to induce any substantial cell motility (Fig. 6D). This indicates that PAK4 kinase activity is required for promotion of cell motility, and that PAK4 integrin binding is not sufficient. Taken together, our results indicate that PAK4 kinase activity is critical to promote integrin αvβ5-mediated cell motility.
PAK4 Phosphorylates Integrin β5 Cytoplasmic Domain in Vitro and in Living Cells
Given the role of PAK4 kinase activity in promotion of cell motility, we examined PAK4-mediated phosphorylation of the integrin β5 cytoplasmic tail. Using HA-PAK4 immunoprecipitate prepared from transiently transfected COS-7 cells together with a GST-β5 cytoplasmic domain fusion protein, we found that the β5 cytoplasmic domain was a specific substrate for PAK4 in vitro, whereas GST-β1 integrin was not (Fig. 7A, top panel). Likewise, purified αvβ5 from human placenta could also be phosphorylated by PAK4 on its β5 subunit (Fig. 7B). Importantly, we also found that integrin β5 was phosphorylated in cells transfected with PAK4, as detected by metabolic 32P labeling, but not in mock-transfected cells (Fig. 7C).
FIGURE 7.
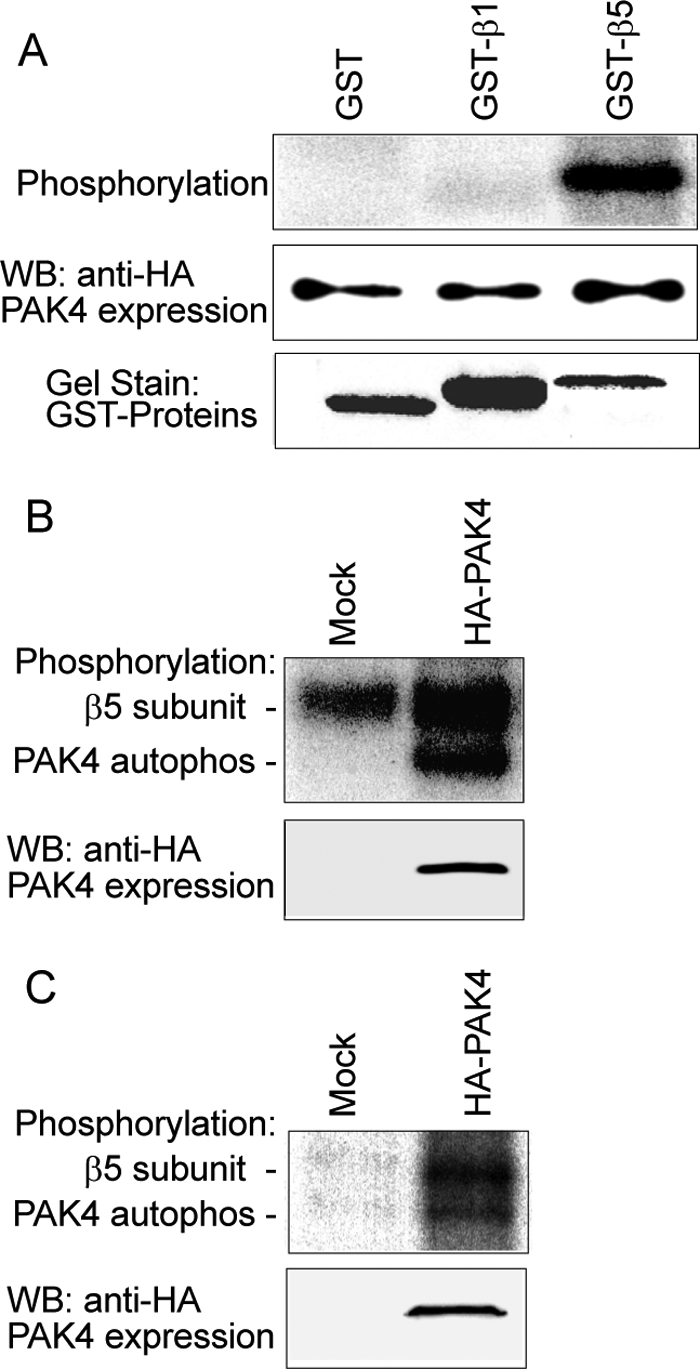
PAK4 phosphorylates the integrin β5 subunit. PAK4 was immunoprecipitated using an anti-HA mAb from COS-7 cells transfected with an HA-PAK4 vector and incubated with integrins in the presence of [γ-32P]ATP. A, PAK4 phosphorylation of integrin β5 cytoplasmic domain analyzed by in vitro phosphorylation using purified GST, GST-β1, or GST-β5 cytoplasmic domain as substrates. PAK4 levels detected by immunoblot (middle panel) and the amounts of GST fusion proteins used are indicated by staining with Coomassie Brilliant Blue (lower panel). B, in the same manner, 5 μg of purified integrin αvβ5 was analyzed for phosphorylation by immunoprecipitated PAK4, separated by 7.5% SDS-PAGE, and visualized by autoradiography. C, PAK4 phosphorylates β5 subunit in living cells. Cells underwent phosphate starvation and then metabolic labeling as described under “Experimental Procedures.” Integrin αvβ5 was immunoprecipitated in cells with or without overexpressed HA-PAK4, exposed to SDS-PAGE and autoradiography (upper panel). The lower panel shows the immunoblot for HA-PAK4 expression.
PAK4 Phosphorylates Integrin β5 at the Membrane Proximal Ser-759 and Ser-762
To determine the location of PAK4 phosphorylation within integrin β5, we employed phospho-peptide mapping. GST-β5 tail and GST control proteins were treated with PAK4 in the presence of [γ-32P]ATP, trypsinized, and subjected to two-dimensional electrophoresis on TLC plates. Two spots (b and c) appeared in the GST-β5 tail sample that were not found in the GST control (Fig. 8A, upper). Following phosphoamino acid analysis, both spots b and c were identified as serines by comparison with the standard marker (Fig. 8A, middle and lower in boxes). Edman sequencing of the spot b and c peptides by 18 cycles of degradation showed that the spot b peptide contained a high level of 32P at the first amino acid, whereas the spot c peptide at the third amino acid contained the most radioactivity. Thus, we identified two distinct PAK4 phosphorylation sites at amino acids Ser-759 and Ser-762 using the GST-β5 cytoplasmic tail as a substrate (Fig. 8, A and B). One additional spot immediately to the left of spot c was also repeatedly observed and was identified as a serine in the third position, consistent with a phosphorylation of Ser-759 in an incompletely cleaved fragment. However, potential additional phosphorylation site(s) cannot be excluded due to the built-in limitations of the two-dimensional-gel electrophoretic separation. Given that PAK4 is a serine/threonine kinase, we then mutated Ser-759 and Ser-762 to Thr-759 and Thr-762, and prepared GST-β5-S759T; GST- β5-S762T, and the double mutant GST-β5-SS759,762TT to further examine the specificity of the PAK4-mediated phosphorylation sites in the β5 tail. In the wild type peptide, spot b contained only phosphorylated serine (Fig. 8C), whereas in the β5-S762T and β5-SS759,762TT mutants, it contained phosphorylated threonine. Further, spot c displayed only serine phosphorylation in WT β5 that was completely reverted to threonine phosphorylation in the β5-S759T and β5-SS759,762TT mutants. This confirms that both the serine residues 759 and 762 of integrin β5 were phosphorylated by PAK4. However, we cannot exclude additional β5 phosphorylation sites, as may be suggested by the observation that spot b displayed serine phosphorylation also upon mutation of GST-β5-S762 to threonine. To further test if the two identified phosphorylation sites were responsible for the observed PAK4 phosphorylation, we generated a mutant with mutation of both Ser-759 and Ser-762 to alanine residues. As shown in Fig. 8D, PAK4 phosphorylated GST-β5-WT significantly above GST control background, but not GST-β5-SS759,762AA, indicating that Ser-759 and Ser-762 were responsible for the observed PAK4 phosphorylation. Amino acid sequence alignment of the human β integrin cytoplasmic domains sharing high homology with integrin β5 indicates that Ser-759 is unique to β5, whereas Ser-762 is conserved between β5, β6, and β8, but with no corresponding residues in β1, β2, β3, or β7 (Fig. 1E). This suggests that phosphorylation of serine residues within the membrane-proximal PAK4-binding SERS-motif is integrin-selective.
FIGURE 8.

Mapping of PAK4 phosphorylation sites within the integrin β5 cytoplasmic domain. A, PAK4-phosphorylated GST-β5 was separated by two-dimensional-gel electrophoresis after trypsin digestion. Spots marked with b and c appeared consistently in the same location in different experiments and did not occur in the GST control. These two spots were further analyzed by phosphopeptide mapping and identified as serines 759 and 762 (middle and bottom panels). The inset shows results of phosphoamino acid analysis. B, arrowheads point out the two PAK4-induced phosphorylation sites within the integrin β5 membrane-proximal region at serine residues 759 and 762. C, phosphoamino acid content of peptides from spots b and c, analyzed from GST-β5-WT, GST-β5-S759T, GST-β5-S762T, and GST-β5-SS759,762TT, and GST fusion proteins were phosphorylated in vitro by PAK4. Migrating positions of phospho amino acid markers are shown in the panel to the right. D, the two phosphorylation sites at Ser-759 and Ser-762 were mutated to alanine residues to further elucidate the identity of the two phosphorylatable residues. PAK4 phosphorylation of GST-β5 was compared with GST and the GST-β5 alanine mutant.
Role of Integrin β5 Ser-759 and Ser-762 in PAK4 Regulation of Cell Motility
Mutations of the integrin β5 to β5-SS759,762AA and β5-SS759,762EE did not influence PAK4 binding in yeast two-hybrid mating tests (Fig. 1F). To determine whether the two serines in the integrin β5 SERS-motif may play a role in PAK4-mediated regulation of cell motility, we made CS-1 cells stably expressing integrin β5-WT, β5-SS759,762AA, or β5-SS759,762EE. The cell surface expression levels of integrin β5 were similar to β5-WT, as determined by FACS analysis using anti-integrin αvβ5 mAb P1F6 (Fig. 9A). We then analyzed the effects on integrin αvβ5-mediated cell migration. When CS-1 cells stably expressing WT integrin β5 were transiently co-transfected with EGFP-PAK4-WT, cell migration increased compared with the same cells co-transfected with EGFP control. CS-1 cells expressing β5-SS759,762AA or β5-SS759,762EE mutants displayed a similar level of cell migration as WT β5-expressing CS-1 cells. However, no significant changes in cell migration were observed when the mutant β5-expressing cells were co-transfected with WT PAK4, in contrast with the WT β5-expressing cells (Fig. 9B). This demonstrates that integrin β5 Ser-759 and Ser-762 are critical for PAK4-induced cell motility and thus that PAK4-mediated phosphorylation of β5 cytoplasmic tail appears to regulate cell motility. This also indicates that a negative charge at positions 759 and 762 in the β5 cytoplasmic tail was not sufficient to promote cell migration.
FIGURE 9.

Integrin β5 serines 759 and/or 762 are necessary for PAK4 induced cell migration. A, flow cytometry analysis of integrin αvβ5 cell surface expression in CS-1 cells with or without stable expression of integrin β5-WT, β5-SS759,762AA, or β5-SS759,762EE. M1 shows mean intensity of the cells expressing αvβ5. B, cell migration onto VN of stable mixed clones of β5-WT, β5-SS759,762AA, and β5-SS759,762AA transiently co-expressing EGFP (open bars) or EGFP-PAK4 (solid bars). Bars represent mean values ± S.E. for three experiments. Statistically discernable differences as determined by t test are indicated (*, p ≤ 0.05).
DISCUSSION
Cytoplasmic tails of integrins play key roles in a variety of integrin-mediated events, including adhesion and migration (2). The phosphorylation of integrin cytoplasmic tails has been proposed as a means of regulating integrin functions (10). Our data show that the integrin β5 subunit cytoplasmic tail is a substrate of PAK4. This is the first example of phosphorylation of integrin αvβ5, and how its extracellular functions may be controlled by intracellular phosphorylation. This also adds to the relatively few known substrates of PAK4 (52, 53), thereby extending the knowledge about the immediate signal transduction capacity of PAK4.
The relationship between cell adhesion and migration is complex. Our results showed that, when compared with WT integrin β5, the β5-SERS mutations resulted in a marked induction of integrin αvβ5-mediated cell migration accompanied by a decrease in αvβ5-mediated cell attachment. This could potentially be explained by the fact that cell migration compared with attachment strength can follow an approximate bell-shaped curve (54). The rate of cell migration is a function of matrix concentration, integrin abundance, and the integrin activation state. Change in any one of these properties will affect the rate of cell migration in a manner that is dependent on the original position of the cell on the bell-shaped curve (54, 55). Further, overexpression of PAK4 mimicked the effect on cell motility and attachment of the SERS mutations in the β5-tail. Together, αvβ5 in its normal constitution may mediate cell attachment that is too strong for optimal motility, whereas PAK4 phosphorylation or SERS-motif mutation may lower the attachment strength and resultantly increased cell motility.
The conserved β-integrin membrane-proximal region where PAK4 binds to and phosphorylates β5 is important for integrin oligomerization, inhibition of integrin conformational changes, and tethering of an integrin in the inactive state (56–58). However, although it is possible that PAK4 binding to and phosphorylation of integrin β5 cytoplasmic tail membrane-proximal region may affect the association between integrin αv and β5 subunits and/or binding between αvβ5 and VN, further studies are required to elucidate if the phosphorylation of β5-subunit by PAK4 can affect integrin hinge formation or change the conformation of integrin extracellular domain.
Two other interesting proteins, theta-associated protein 20 (TAP20) and the scaffolding protein, receptor for activated C kinase (RACK1) can also interact with the integrin β5 cytoplasmic tail. TAP20 binds to the integrin β5 tail and reduces integrin αvβ5-mediated cell migration by a protein kinase C signaling pathway (59, 60). RACK1 interacts with a conserved membrane-proximal region of the integrin β subunit cytoplasmic domain and decreased Chinese hamster ovary cell motility in a manner that may also involve its interaction with protein kinase C (59, 61). The RACK1-binding site in integrin β5 is overlapping with that of PAK4. However, further experiments are required to elucidate whether TAP20 and/or RACK1 may be functionally related to PAK4 in the regulation of integrin αvβ5-mediated cell motility.
Phosphorylation of integrin cytoplasmic tails can have both negative and positive roles in integrin regulation, possibly reflecting the importance of dynamically regulated phosphorylation in the integrin function (10). Integrin β5 tail can also be phosphorylated by protein kinase C, but the one or more specific serines to be phosphorylated were not identified (62). In this study, we demonstrate that PAK4 phosphorylation of serines 759/762 in the integrin β5 cytoplasmic tail promoted CS-1 cell migration. Among integrins, the SERS amino acid sequence only appears in the β5 cytoplasmic tail, although ERS is found at the same position within β6, ER within β3, and Ser-762 within β8. Therefore, PAK4 may also be able to phosphorylate also the integrin β6 and β8 cytoplasmic domains.
We demonstrate that phosphorylation of serine residues 759 and 762 in the integrin β5 cytoplasmic tail is necessary for PAK4-mediated promotion of cell migration. Growth factor stimulation can promote αvβ5-mediated cancer cell migration and dissemination as well as induce αvβ5-mediated angiogenesis and vascular permeability (35, 36, 39, 41, 63). Interestingly, PAK4 kinase can be activated by growth factors, such as Hepatocyte growth factor (64). It will therefore be interesting to elucidate the potential role of PAK4 in growth factor stimulation of angiogenesis, vascular permeability, and cancer cell migration and dissemination and determine if PAK4-mediated phosphorylation of integrin β5 serine residues 759 and 762 may play any role. However, PAK4 may also promote cell motility by phosphorylation of additional substrates involved in the control of cell motility, for example by regulation of the actin microfilament system (43).
In summary, we found that a unique membrane-proximal SERS-motif within the integrin β5 cytoplasmic domain can be phosphorylated by PAK4 and that this phosphorylation regulates integrin αvβ5-mediated carcinoma cell motility. This may also contribute to the understanding of intracellular signaling behind vascular permeability, angiogenesis, and carcinoma cell dissemination (36, 39, 63).
Acknowledgments
We thank Drs. Stephen Smith (Karolinska Institutet) and Andrew Paterson (Karolinska Institutet) for critical reading of the manuscript. We are also grateful to Dr. Caroline Damsky (University of California at San Francisco) for providing CS-1 cells, Drs. Audrey Minden (Columbia University) and Errki Ruoslahti (Burnham Institute, La Jolla, CA) for providing human PAK4 and integrin β5 cDNA, respectively, and the Developmental Studies Hybridoma Bank at the University of Iowa for providing anti-actin mAb JLA20.
This work was supported by grants from the Swedish Cancer Society, the Swedish Research Council, the Center for Biosciences at the Karolinska Institutet, and the Swedish Strategic Research Foundation (to S. S.), the Swedish Society of Medicine and Grant NSFC # 30830048 to (H. Z.), and the Swedish Research Council (to L. C.-W.).
H. Zhang and S. Strömblad, unpublished observation.
- PAK4
- p21-activated kinase 4
- mAb
- monoclonal antibody
- MBP
- myelin basic protein
- CT
- C-terminal
- NT
- N-terminal
- aa
- amionj
- BAP
- bacterial alkaline phosphatase
- KD
- kinase domain
- WT
- wild type
- EGFP
- enhanced green fluorescent protein
- CMV
- cytomegalovirus
- GST
- glutathione S-transferase
- VN
- vitronectin
- FACS
- fluorescence-activated cell sorting
- IBD
- integrin-binding domain
- HA
- hemagglutinin
- VN
- vitronectin
- TAP20
- theta-associated protein 20
- RACK1
- scaffolding protein, receptor for activated C kinase.
REFERENCES
- 1.Zhang H., Li Z., Viklund E. K., Strömblad S. (2002) J. Cell Biol. 158, 1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes R. O. (2002) Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 3.Liddington R. C., Ginsberg M. H. (2002) J. Cell Biol. 158, 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim M., Carman C. V., Springer T. A. (2003) Science 301, 1720–1725 [DOI] [PubMed] [Google Scholar]
- 5.Miranti C. K., Brugge J. S. (2002) Nat. Cell Biol. 4, E83–E90 [DOI] [PubMed] [Google Scholar]
- 6.Lock J. G., Wehrle-Haller B., Strömblad S. (2008) Semin. Cancer Biol. 18, 65–76 [DOI] [PubMed] [Google Scholar]
- 7.Giancotti F. G., Ruoslahti E. (1999) Science 285, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 8.Liu S., Calderwood D. A., Ginsberg M. H. (2000) J. Cell Sci. 113, 3563–3571 [DOI] [PubMed] [Google Scholar]
- 9.Phillips D. R., Prasad K. S., Manganello J., Bao M., Nannizzi-Alaimo L. (2001) Curr. Opin. Cell Biol. 13, 546–554 [DOI] [PubMed] [Google Scholar]
- 10.Fagerholm S. C., Hilden T. J., Gahmberg C. G. (2004) Trends Biochem. Sci. 29, 504–512 [DOI] [PubMed] [Google Scholar]
- 11.Anthis N. J., Haling J. R., Oxley C. L., Memo M., Wegener K. L., Lim C. J., Ginsberg M. H., Campbell I. D. (2009) J. Biol. Chem. 284, 36700–36710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legate K. R., Fässler R. (2009) J. Cell Sci. 122, 187–198 [DOI] [PubMed] [Google Scholar]
- 13.Calderwood D. A., Zent R., Grant R., Rees D. J., Hynes R. O., Ginsberg M. H. (1999) J. Biol. Chem. 274, 28071–28074 [DOI] [PubMed] [Google Scholar]
- 14.Tadokoro S., Shattil S. J., Eto K., Tai V., Liddington R. C., de Pereda J. M., Ginsberg M. H., Calderwood D. A. (2003) Science 302, 103–106 [DOI] [PubMed] [Google Scholar]
- 15.Millon-Frémillon A., Bouvard D., Grichine A., Manet-Dupé S., Block M. R., Albiges-Rizo C. (2008) J. Cell Biol. 180, 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wegener K. L., Partridge A. W., Han J., Pickford A. R., Liddington R. C., Ginsberg M. H., Campbell I. D. (2007) Cell 128, 171–182 [DOI] [PubMed] [Google Scholar]
- 17.Gimond C., de Melker A., Aumailley M., Sonnenberg A. (1995) Exp. Cell Res. 216, 232–235 [DOI] [PubMed] [Google Scholar]
- 18.Sakai T., Zhang Q., Fässler R., Mosher D. F. (1998) J. Cell Biol. 141, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowan K. J., Law D. A., Phillips D. R. (2000) J. Biol. Chem. 275, 36423–36429 [DOI] [PubMed] [Google Scholar]
- 20.Boettiger D., Lynch L., Blystone S., Huber F. (2001) J. Biol. Chem. 276, 31684–31690 [DOI] [PubMed] [Google Scholar]
- 21.Datta A., Huber F., Boettiger D. (2002) J. Biol. Chem. 277, 3943–3949 [DOI] [PubMed] [Google Scholar]
- 22.Dans M., Gagnoux-Palacios L., Blaikie P., Klein S., Mariotti A., Giancotti F. G. (2001) J. Biol. Chem. 276, 1494–1502 [DOI] [PubMed] [Google Scholar]
- 23.Dahl S. C., Grabel L. B. (1989) J. Cell Biol. 108, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J., Liu S., Rose D. M., Schlaepfer D. D., McDonald H., Ginsberg M. H. (2001) J. Biol. Chem. 276, 40903–40909 [DOI] [PubMed] [Google Scholar]
- 25.Reszka A. A., Hayashi Y., Horwitz A. F. (1992) J. Cell Biol. 117, 1321–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barreuther M. F., Grabel L. B. (1996) Exp. Cell Res. 222, 10–15 [DOI] [PubMed] [Google Scholar]
- 27.Valmu L., Fagerholm S., Suila H., Gahmberg C. G. (1999) Eur. J. Immunol. 29, 2107–2118 [DOI] [PubMed] [Google Scholar]
- 28.Valmu L., Hilden T. J., van Willigen G., Gahmberg C. G. (1999) Biochem. J. 339, 119–125 [PMC free article] [PubMed] [Google Scholar]
- 29.Fagerholm S., Morrice N., Gahmberg C. G., Cohen P. (2002) J. Biol. Chem. 277, 1728–1738 [DOI] [PubMed] [Google Scholar]
- 30.Kirk R. I., Sanderson M. R., Lerea K. M. (2000) J. Biol. Chem. 275, 30901–30906 [DOI] [PubMed] [Google Scholar]
- 31.Hilden T. J., Valmu L., Kärkkäinen S., Gahmberg C. G. (2003) J. Immunol. 170, 4170–4177 [DOI] [PubMed] [Google Scholar]
- 32.Sakai T., Jove R., Fässler R., Mosher D. F. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3808–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novak A., Hsu S. C., Leung-Hagesteijn C., Radeva G., Papkoff J., Montesano R., Roskelley C., Grosschedl R., Dedhar S. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4374–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wayner E. A., Orlando R. A., Cheresh D. A. (1991) J. Cell Biol. 113, 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yebra M., Parry G. C., Strömblad S., Mackman N., Rosenberg S., Mueller B. M., Cheresh D. A. (1996) J. Biol. Chem. 271, 29393–29399 [DOI] [PubMed] [Google Scholar]
- 36.Eliceiri B. P., Puente X. S., Hood J. D., Stupack D. G., Schlaepfer D. D., Huang X. Z., Sheppard D., Cheresh D. A. (2002) J. Cell Biol. 157, 149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larjava H., Salo T., Haapasalmi K., Kramer R. H., Heino J. (1993) J. Clin. Invest. 92, 1425–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheppard D. (2004) Curr. Opin. Cell Biol. 16, 552–557 [DOI] [PubMed] [Google Scholar]
- 39.Brooks P. C., Klemke R. L., Schon S., Lewis J. M., Schwartz M. A., Cheresh D. A. (1997) J. Clin. Invest. 99, 1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedlander M., Brooks P. C., Shaffer R. W., Kincaid C. M., Varner J. A., Cheresh D. A. (1995) Science 270, 1500–1502 [DOI] [PubMed] [Google Scholar]
- 41.Strömblad S., Cheresh D. A. (1996) Chem. Biol. 3, 881–885 [DOI] [PubMed] [Google Scholar]
- 42.Abo A., Qu J., Cammarano M. S., Dan C., Fritsch A., Baud V., Belisle B., Minden A. (1998) EMBO J. 17, 6527–6540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Callow M. G., Clairvoyant F., Zhu S., Schryver B., Whyte D. B., Bischoff J. R., Jallal B., Smeal T. (2002) J. Biol. Chem. 277, 550–558 [DOI] [PubMed] [Google Scholar]
- 44.Qu J., Cammarano M. S., Shi Q., Ha K. C., de Lanerolle P., Minden A. (2001) Mol. Cell. Biol. 21, 3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yatohgo T., Izumi M., Kashiwagi H., Hayashi M. (1988) Cell Struct. Funct. 13, 281–292 [DOI] [PubMed] [Google Scholar]
- 46.Zhang H., Berg J. S., Li Z., Wang Y., Lång P., Sousa A. D., Bhaskar A., Cheney R. E., Strömblad S. (2004) Nat. Cell Biol. 6, 523–531 [DOI] [PubMed] [Google Scholar]
- 47.Boyle W. J., van der Geer P., Hunter T. (1991) Methods Enzymol. 201, 110–149 [DOI] [PubMed] [Google Scholar]
- 48.Ito N., Wernstedt C., Engström U., Claesson-Welsh L. (1998) J. Biol. Chem. 273, 23410–23418 [DOI] [PubMed] [Google Scholar]
- 49.Aebersold R. H., Leavitt J., Saavedra R. A., Hood L. E., Kent S. B. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 6970–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bao W., Strömblad S. (2004) J. Cell Biol. 167, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas L., Chan P. W., Chang S., Damsky C. (1993) J. Cell Sci. 105, 191–201 [DOI] [PubMed] [Google Scholar]
- 52.Callow M. G., Zozulya S., Gishizky M. L., Jallal B., Smeal T. (2005) J. Cell Sci. 118, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 53.Dan C., Kelly A., Bernard O., Minden A. (2001) J. Biol. Chem. 276, 32115–32121 [DOI] [PubMed] [Google Scholar]
- 54.Palecek S. P., Loftus J. C., Ginsberg M. H., Lauffenburger D. A., Horwitz A. F. (1997) Nature 385, 537–540 [DOI] [PubMed] [Google Scholar]
- 55.Holly S. P., Larson M. K., Parise L. V. (2000) Exp. Cell Res. 261, 69–74 [DOI] [PubMed] [Google Scholar]
- 56.Zage P. E., Marcantonio E. E. (1998) Cell Adhes. Commun. 5, 335–347 [DOI] [PubMed] [Google Scholar]
- 57.Bodeau A. L., Berrier A. L., Mastrangelo A. M., Martinez R., LaFlamme S. E. (2001) J. Cell Sci. 114, 2795–2807 [DOI] [PubMed] [Google Scholar]
- 58.Wegener K. L., Campbell I. D. (2008) Mol. Membr. Biol. 25, 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liliental J., Chang D. D. (1998) J. Biol. Chem. 273, 2379–2383 [DOI] [PubMed] [Google Scholar]
- 60.Tang S., Gao Y., Ware J. A. (1999) J. Cell Biol. 147, 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buensuceso C. S., Woodside D., Huff J. L., Plopper G. E., O'Toole T. E. (2001) J. Cell Sci. 114, 1691–1698 [DOI] [PubMed] [Google Scholar]
- 62.Freed E., Gailit J., van der Geer P., Ruoslahti E., Hunter T. (1989) EMBO J. 8, 2955–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewis J. M., Cheresh D. A., Schwartz M. A. (1996) J. Cell Biol. 134, 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wells C. M., Abo A., Ridley A. J. (2002) J. Cell Sci. 115, 3947–3956 [DOI] [PubMed] [Google Scholar]