

Abstract
Detailed analysis of factors governing high affinity antibody-antigen interactions yields important insight into molecular recognition and facilitates the design of functional antibody libraries. Here we describe comprehensive mutagenesis of the light chain complementarity determining regions (CDRs) of HIV-1 antibody D5 (which binds its target, ‘5-Helix’, with a reported KD of 50 pM). Combinatorial scanning mutagenesis libraries were prepared in which contact residues on the D5 light chain were varied among WT side chain identity or alanine. Selection of these libraries against 5-Helix, and then sequence analysis of the resulting population were used to quantify energetic consequences of mutation from wild-type to alanine (ΔΔGAla-WT) at each contact position. This analysis revealed several hotspot residues (ΔΔGAla-WT ≥ 1 kcal/mol) that formed combining site features critical to the affinity of the interaction. Tolerance of D5 light chain residues to alternative mutations was explored with a second library. We found that light chain residues located at the center and at the periphery of the D5 combining site contribute to shape complementarity and electrostatic characteristics. Thus, the affinity of D5 for 5-Helix arises from extended interactions involving both the heavy and light chains of D5. These results provide significant insight for future antibody engineering efforts.
The specific recognition of an antigen by an antibody is the central feature of humoral immunity, and serves as the basis for countless therapeutic, research, and diagnostic reagents (1, 2). A clear delineation of factors governing high affinity antibody-antigen interactions enhances our understanding of how natural antibodies evolve, and provides information for designing antibody libraries from scratch (3 – 12). Biochemical and biophysical data on protein-targeting antibodies have shown that, similar to other protein-protein interactions, the energetics of intermolecular interfaces between antibodies and their protein antigens are dominated by a small number of residues on either partner (‘hotspots’ for binding) (13 – 20). Residues in the heavy chain CDRs (HCDRs) constitute the majority of the contact surface (the ‘structural paratope’) in many protein-antibody interactions, and most antibody engineering efforts have focused on these regions (3, 10 – 12). However, in some cases both heavy and light chains form extended contacts with the antigen (13 – 16). Mutational analysis on the antibody D1.3, which binds hen egg-white lysozyme (HEL) but can also bind other targets, demonstrated that characteristics of antibody-protein interfaces can vary even within a single antibody that can bind multiple targets (13, 14). Therefore, detailed mutagenesis studies, on a case-by-case basis, of combining site features that contribute most significantly to high affinity binding (‘functional paratopes’) will provide guidelines for development of improved antibody libraries that more accurately mimic physicochemical properties of natural interfaces.
Here we explore light chain requirements for binding in the high affinity HIV-1 antibody D5. Antibody D5 was originally isolated from a phage library constructed from uninfected donors, by screening against 5-Helix, a designed protein that mimics the prehairpin intermediate of HIV-1 gp41 (21). The structure of the D5 antigen-binding fragment (Fab) in complex with 5-Helix, and extensive mutagenesis of heavy chain residues, was previously reported by Luftig et al. (the crystal structure is shown in Figure 1A) (22). Antibody D5 has very high affinity for 5-Helix (the reported KD of the IgG is 50 pM) through a protein-protein interaction involving all six CDRs and > 1000 Å2 of combining site surface area (23, 24). The functional paratope on the D5 heavy chain involves F54 and T56 from HCDR2 which project into a cleft on 5-Helix (Figure 1C). Mutation of these HCDR2 residues to alanine results in significant loss of binding affinity in the IgG (22). The D5 heavy chain is derived from germline sequence VH1-69, and the critical HCDR2 region is identical in sequence to the germline and bears significant sequence and structural homology to other virus neutralizing antibodies derived from the same progenitor (24 – 27) (Figures 1B and 1C). In fact, the HCDR2 regions of influenza antibody CR6261 and HIV-1 antibody 412D are nearly identical in sequence to D5 (Figure 1B) and antigen recognition by these antibodies also involves interaction between F54 of HCDR2 and surface-exposed hydrophobic residues on the antigen (Figure 1C). The similarities among modes of recognition of VH1-69 antibodies suggest that this germline may have unique properties amenable to the design of antibody libraries (28).
Figure 1.

(A) The crystal structure of the D5 Fab bound to 5-Helix reported by Luftig et al. (reference 22). Side chains involved in the interaction are shown in blue (heavy chain), red (light chain), or yellow (5-Helix). The critical HCDR2 is boxed. (B) Sequences of the HCDR2 region of VH1-69, D5, CR6261, and 412D. Residues that differ from the germline are shown in gray. Viral targets for each antibody are indicated in parentheses. (C) Role of HCDR2 in recognition D5 (blue), CR6261 (magenta), and 412D (green). (Antigens shown in gray.) In all three antibodies, F54 contacts surface-exposed hydrophobic residues on the antigen.
Given these similarities among VH1-69 heavy chains, we sought to explore contributions of D5 LCDRs to recognition of 5-Helix. Several groups have shown that critical functional residues at protein-protein interfaces can be rapidly identified using combinatorial mutagenesis coupled to a selection that is dependent on the protein-protein recognition (19, 20, 29 – 31). In this method, ‘combinatorial scanning mutagenesis’ libraries are prepared in which contact positions are allowed to vary among WT or a mutant (in some cases, a 3rd or 4th residue is permitted) and subjected to a selection that is dependent on protein-protein recognition. The output ratio of WT/mutant observed at each position provides an indication of the relative importance of that position to the protein-protein interface. In cases where the selection involves isolation of direct binding clones (i.e., phage display), this WT/mutant ratio can be used to infer energetic information about the consequences of a given WT→mutant mutation by assuming that the WT/mutant ratio observed from the selection is proportional to the ratio of association constants (KA) for WT and a WT→mutant single point mutant (20, 21). This approach is more rapid that traditional site-directed mutagenesis, and allows simultaneous assessment of multiple positions. We performed combinatorial scanning mutagenesis on the LCDR contact residues of D5 by phage display. Our results reveal several unique features that govern affinity in this protein-antibody interaction.
MATERIALS AND METHODS
Expression, Purification, and Refolding of 5-Helix
5-Helix was prepared essentially as described (32). Briefly, an expression plasmid containing 5-Helix was transformed into E. coli BL21(DE3) Rosetta cells. Cells bearing the expression plasmid were grown to an OD600 of ~ 0.6, and expression of 5-Helix induced at 37 °C for 4 hrs by addition of ~1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation and lysed by incubating with ice-cold glacial acetic acid for 1-2 hrs. The lysate was then clarified by ultracentrifugation. Water was added to bring the final acetic acid concentration to < 10 % (v/v) and the mixture lyophilized. The material was resdissolved in water/acetonitrile, and purified by preparative RP-HPLC on a C18 column with water/acetonitrile/trifluoroacetic acid mobile phases. The purified material was lyophilized, and then dissolved in 6 M guanidine HCl (protein concentrations were kept below 0.5 mg/mL). The purified 5-Helix was refolded by step-wise dialysis into 100 mM glycine HCl (pH 3), then phosphate-buffered saline (PBS, pH 7). Precipitated material was removed by centrifugation and the soluble, refolded 5-Helix protein and stored at −80 °C.
Phage Display of the D5 Single Chain Variable Fragment (scFv)
The pCANTAB5E-based vector pAPIII6 was used to display the D5 scFv (33). A synthetic gene (codon optimized for E. coli) was obtained from a commercial supplier (DNA 2.0, Menlo Park, CA) in which the VH and VL domains of D5 were linked by a (GGGS)3 linker (the DNA sequence of the D5 scFv segment and full cloning details are provided in the Supporting Information). This D5 scFv construct was cloned into the phagemid pAPIII6 using HindIII and SalI cloning sites to produce the phagemid pJH3. Cloning of the scFv into pAPIII6 in this way results in display of a chimeric protein containing an N-terminal FLAG epitope (for detection), the scFv, and the C-terminal 188 residues of the minor coat protein pIII all under phoA promoter control (33). To prepare phage particles displaying the D5 scFv (D5Φ), pJH3 was transformed into E. coli XL1-Blue cells, and grown for 3 hours in LB containing 10 μg/mL tetracycline and 50 μg/mL of carbenicillin. The culture was then coinfected with 1011 plaque forming units (pfu) of VCSM13 or K07 helper phage, the media supplemented with 50 μg/mL of kanamycin, and the culture grown at 37 °C overnight. Cells were removed by centrifugation, and the phage particles precipitated by addition of 4 % (w/v) polyethylene glycol (PEG) 8000 and 3% (w/v) NaCl. The precipitated phage were recovered by centrifugation and resuspended in PBS containing 1% (w/v) BSA.
Functional display of the scFv was confirmed using phage enzyme-linked immunosorbent assay (ELISA). High-binding Costar EIA/RIA plates were coated with 0.4 μg of 5-Helix in 100 mM NaHCO3 pH 8.5 at room temperature for 1-2 hours or overnight at 4 °C, then unbound well sites blocked with 1% (w/v) BSA in PBS. The well solution was decanted and the D5Φ incubated for 1-2 hrs. The wells were washed 5 times with PBS containing 0.05% (v/v) Tween (PBS-T), and then an anti-M13/horse radish peroxidase (HRP) conjugate allowed to bind for 1-2 hrs. The wells were washed again with PBS-T and the presence of the anti-M13-HRP detected with the chromogenic substrate tetramethylbenzidine (TMB).
Preparation of D5 scFv Combinatorial Scanning Mutagenesis Libraries
An inactive ‘stop’ clone based on pJH3, in which contact residues of LCDR1-3 regions were replaced with TAA stop codons, was prepared to serve as the template for library synthesis. Kunkel mutagenesis was used as described (34) to replace the stop codons with library DNA using 5′ phosphorylated primers consisting of the reverse complement of the following sequences (degenerate nucleotide mixtures follow standard nomenclature: K = G/T; M = A/C; N = A/T/C/G; R = A/G; S = G/C, W = A/T, Y = C/T): Alanine scanning library: LCDR1, TGC CGC GCG TCT GAA GGT ATT KMT SMT KSG CTG GCT TGG TAC CAG CAG; LCDR2, GCA CCG AAA CTG CTG ATC KMT RMA GCG TCT KCC CTG GCA TCC GGT GCT CCA; LCDR3, GCG ACC TAC TAC TGC CAG CAA KMT KCC RMC KMT SCA SYT ACT TTT GGT GGC GGC ACC. ‘Scan 2′ library: LCDR1, TGC CGC GCG TCT GAA GGT ATT YWT CRT TKG CTT GCT TGG TAC CAG CAG AAA; LCDR2, AAA GCA CCG AAA CTG CTG ATT YWT ARG GCG TCT KCC CTG GCA TCC GGT GCT CCA; LCDR3, GCG ACC TAC TAC TGC CAG CAA YWT KCC MAN YWT SCA MTG ACT TTT GGT GGC GGC ACC.
Typical Kunkel mutagenesis reactions contained 9 μg of single-stranded uridine-enriched template DNA, and 12-fold excess of each library primer. Library DNA was purified and then E. coli XL-1 Blue were transformed with the library DNA to yield 9 × 106 (alanine scan) and 2 × 106 (‘Scan 2’) total clones. These library sizes exceeded the maximum theoretical diversities of 1 × 106 (alanine scan) and 1 × 105 (‘Scan 2’); therefore, all sequences were adequately represented. Large-scale sequencing of resulting library clones indicated that ~60 % (alanine scanning library) or ~40 % (scan 2 library) of the population contained all three library DNA primers (LCDRs 1, 2, and 3) incorporated. The sequence diversity was high among these library clones.
Library Sorting and Analysis
Combinatorial scanning mutagenesis libraries were subjected to two rounds of selection against 5-Helix. For each round, Costar EIA/RIA high-binding plates were coated with 5-Helix (10 μg over 5 wells in 100 mM NaHCO3 pH 8.5) overnight at 4 °C or for 1 hr at room temperature, then blocked with BSA (1% (w/v) in PBS) at room temperature for 1-2 hours. Library phage (> 1010 pfu in PBS-T containing 1% (w/v) BSA) were depleted of non-specific binders by preincubation with BSA-coated wells, and then were incubated with 5-Helix-coated wells at room temperature for 1-2 hrs. The wells were then washed 5 times with PBS-T, then binding phage eluted by incubating with 100 mM glycine HCl pH 2 for 5-10 min. Eluted phage solutions were neutralized with 1 M Tris, pH 8, and then propagated by coinfection of an XL1-Blue culture with VCSM13 or K07 helper phage at 37 °C overnight. Resulting phage mixtures were precipitated with PEG 8000/NaCl, then redissolved in PBS-T containing 1% (w/v) BSA and used for the subsequent round of selection. The display selection (against anti-FLAG antibody M2) was similar except that the libraries were subjected to only one round, and the population was not depleted of non-specific binders prior to this round of selection.
To analyze individual clones for 5-Helix binding, 1.5 mL cultures of individual phage from the selected population were grown at 37 °C overnight. The cells were removed by centrifugation, and the culture supernatant applied directly to 5-Helix- or BSA-coated wells. After 1-2 hr incubation, the wells were washed with PBS-T, and the presence of binding phage determined using anti-M13/HRP conjugate and TMB substrate. Clones displaying positive ELISA signal against 5-Helix that was 2-fold or greater over background (BSA) were subjected to DNA sequencing. Analysis for the display selection was similar, except that wells were coated with 50 ng of anti-FLAG antibody M2. For alanine scanning libraries, a WT/Ala ratio was calculated at each position and converted into ΔΔG using the equation ΔΔG = RT ln (WT/Ala) for both the 5-Helix selection and the display selection (19, 20). The ΔΔGAla-WT was then calculated as follows: ΔΔGAla-WT = ΔΔG (5-Helix selection) – ΔΔG (display selection). Analysis to calculate ΔΔGmut-WT for the Scan 2 library was similar.
RESULTS
Expression and Purification of 5-Helix, the D5 Target
5-Helix is engineered to expose a binding groove for the C-heptad repeat (CHR) region of gp41 thereby mimicking the conformational intermediate in the gp41-mediated membrane fusion pathway known as the ‘prehairpin intermediate’ (see Supporting Information) (32, 35). The protein consists of alternating segments corresponding to the N-heptad repeat (NHR) region and the CHR of gp41 linked by flexible protein loops (three total NHR segments and two CHR segments) (32, 35). When folded, 5-Helix adopts a structure similar to the six-helix bundle post-fusion state of gp41 but with one of the CHR helices displaced. Antibody D5 inhibits viral entry by binding the CHR-binding groove and preventing progression from the prehairpin intermediate to the six-helix bundle fusion state. (22, 32, 35) Several versions of 5-Helix proteins have been reported (32, 35, 36). The cocrystal structure of the D5 Fab was solved using a 5-Helix variant designed by Kim and coworkers (22). More recently, Harrison and coworkers designed a similar protein known as ‘gp41-5’ (32); we used the Harrison construct in our studies. The Kim and Harrison designs vary slightly in length of the α-helices and at the linker regions, but the residues that contact D5 are identical among these clones (see Supporting Information). For clarity, we refer to the Harrison construct as ‘5-Helix’ here and throughout the remainder of the text. We expressed, purified, and refolded 5-Helix as previously described (32) (see Supporting Information).
Combinatorial Alanine Scanning Mutagenesis
We expressed the D5 scFv as a pIII fusion (see Supporting Information) and prepared a combinatorial alanine scanning library in which LCDR positions forming van der Waals contacts with 5-Helix were randomized using the scheme of Weiss et al. (19). This library was subjected to two rounds of selection against 5-Helix. Polyclonal phage ELISA analysis of the unselected library (‘R0’) and output populations from rounds 1 and 2 (‘R1’ and ‘R2’, respectively) indicated that the selection against 5-Helix was successful (Figure 2). We tested individual clones from the R2 population for binding by high-throughput phage ELISA and found that over 70% of the clones exhibited a specific binding signal for 5-Helix. We sequenced a sampling of these hits (79 clones) and determined the WT/Ala ratio at each position. The sequence diversity among these clones was high, with only two siblings (clones that had identical amino acid sequence to another clone in the population) in the dataset. To account for display biases, we performed a parallel selection of the alanine scanning libraries against anti-FLAG antibody M2 (a FLAG epitope is included at the N-terminus of the displayed scFv) and then calculated corrected ΔΔGAla-WT values as described in Materials and Methods. Table 1 shows results from this sequence analysis; in cases where additional substitutions other than WT or Ala were permitted (m2 and m3), the ΔΔGm2-WT and ΔΔGm3-WT values are also listed. Figure 3 shows the results of ΔΔGAla-WT scanning mapped onto the crystal structure of the D5 combining site. Based on range of ΔΔGAla-WT values we observed, and on published ΔΔGAla-WT values obtained from combinatorial scanning mutagenesis on the well-characterized human growth hormone-receptor complex (19, 20), the D5 LCDR residues in Figure 3 were color-coded according to the following classifications: red, ΔΔGAla-WT ≥ 1.0 kcal/mol (‘hotspot residues’); orange, 0.4 kcal/mol < ΔΔGAla-WT < 1.0 kcal/mol; green, 0 < ΔΔGAla-WT < 0.4; cyan, ΔΔGAla-WT ≤ 0 kcal/mol.
Figure 2.
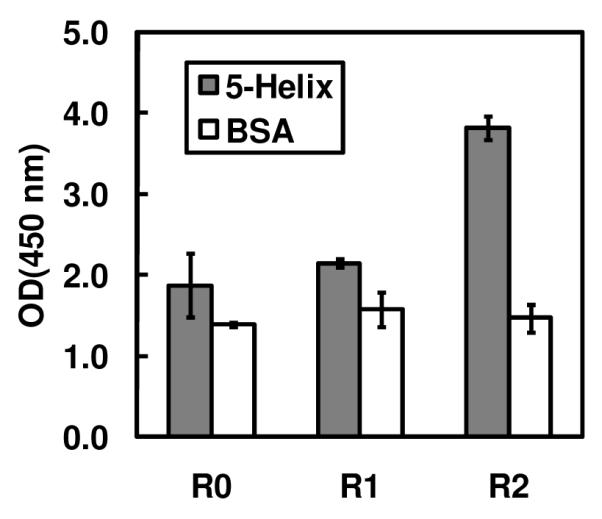
Results from polyclonal phage ELISA of populations from unselected library (R0), and output phage from rounds 1 and 2 of the selection against 5-Helix (R1 and R2, respectively). A specific binding signal was observed in the R2 population. Phage titers for all three populations were ~2 × 1010 infectious units/mL.
Table 1.
Combinatorial alanine scanning results.
| 5-Helix selection (n = 79)A |
Display selection (n = 75)A |
WT/Ala (5-Helix) |
WT/Ala (display) |
ΔΔGAla-WT (kcal/mol) |
ΔΔGm2-WT (kcal/mol) |
ΔΔGm3-WT (kcal/mol) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Res. | WT | Ala | m2B | m3B | WT | Ala | m2B | m3B | |||||
| LCDR1 | |||||||||||||
| Y30 | 57 | 4 | 12 (S) | 6 (D) | 30 | 12 | 19 (S) | 14 (D) | 14.3 | 2.5 | 1.0 | 0.7 | 0.9 |
| H31 | 35 | 11 | 19 (D) | 14 (P) | 16 | 8 | 34 (D) | 17 (P) | 3.2 | 2.0 | 0.3 | 0.8 | 0.6 |
| W32 | 36 | 16 | 14 (S) | 13 (G) | 19 | 5 | 15 (S) | 36 (G) | 2.3 | 3.8 | −0.3 | 0.4 | 1.0 |
| LCDR2 | |||||||||||||
| Y49 | 46 | 4 | 29 (S) | 0 (D) | 29 | 11 | 31 (S) | 4 (D) | 11.5 | 2.6 | 0.9 | 0.3 | > 1.1 |
| K50 | 70C | 1C | 7 (T)C | 0 (E)C | 30 | 14 | 19 (T) | 12 (E) | 70.0 | 2.1 | 2.1 | 1.1 | > 2.0 |
| S53 | 61 | 18 | - | - | 60 | 15 | - | - | 3.4 | 4.0 | −0.1 | - | - |
| LCDR3 | |||||||||||||
| Y91 | 48 | 11 | 14 (S) | 6 (D) | 25 | 9 | 11 (S) | 30 (D) | 4.4 | 2.8 | 0.3 | 0.2 | 1.3 |
| S92 | 42 | 37 | - | - | 26 | 49 | - | - | 1.1 | 0.5 | 0.5 | - | - |
| N93 | 39 | 9 | 6 (D) | 25 (T) | 26 | 9 | 17 (D) | 23 (T) | 4.3 | 2.9 | 0.2 | 0.9 | 0.2 |
| Y94 | 76 | 1 | 1 (S) | 1 (D) | 21 | 22 | 18 (S) | 14 (D) | 76.0 | 1.0 | 2.6 | 2.5 | 2.3 |
| P95 | 74 | 5 | - | - | 43 | 32 | - | - | 14.8 | 1.3 | 1.4 | - | - |
| L96 | 46 | 3 | 17 (V) | 13 (P) | 22 | 18 | 20 (V) | 15 (P) | 15.3 | 1.2 | 1.5 | 0.5 | 0.5 |
Number of times each side chain was observed at a particular position in the selected population. Two siblings were observed in the 5-Helix selection; one was observed in the display selection.
In some cases, a third and forth substitution is permitted (m2 and m3). The occurrence of these side chains is indicated along with identity of the substitution.
In this position, sequencing was obscured in one of the 79 clones (all other positions in this clone were unambiguous).
Figure 3.

Combinatorial alanine scanning results mapped onto the crystal structure of D5. Residues in the VL domain are colored according to ΔΔGAla-WT: red, ≥ 1.0 kcal/mol; orange, 0.4 – 1.0 kcal/mol; green, 0 – 0.4 kcal/mol; cyan, ≤ 0 kcal/mol. Residues previously shown by Luftig et al. to be important for binding in the VH domain are colored blue.
Five of the 12 LCDR residues explored in our libraries had ΔΔGAla-WT ≥ 1.0 kcal/mol (Y30, K50, Y94, P95, L96). The residues Y94, P95, L96 form a cluster of hotspot residues at the interface of the variable domains from heavy (VH) and light (VL) chains involving Y94, P95, and L96 of LCDR3. These residues lie at the ridge of a deep pocket in the D5 combining site into which W571 of 5-Helix protrudes, and are adjacent to the critical HCDR2 residues F54 and T56 on D5 (22). These results further confirm that a critical feature of this high affinity association is an interlocking interaction between opposing hydrophobes on either partner (W571 on 5-Helix, and F54/T56 on D5) (Figure 4). The LCDR hotspot residues (Y94, P95, and L96) line the pocket on the D5 combining site, but are adjacent the critical residues on HCDR2 (F54 and T56).
Figure 4.
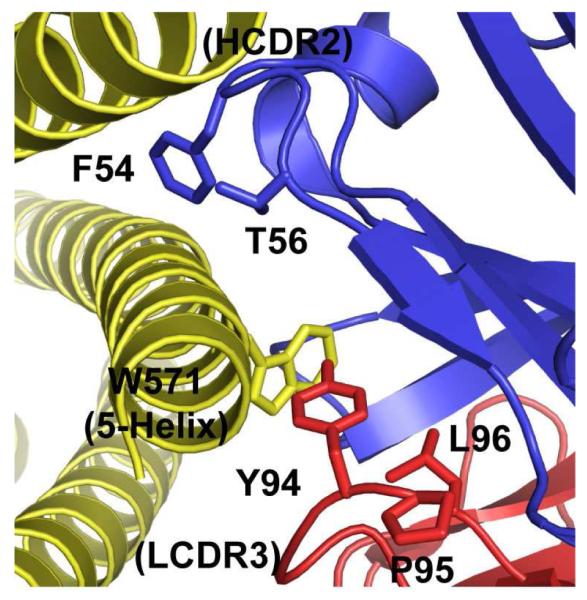
Interlocking interaction between opposing hydrophobes. The pocket on the D5 combining site, into which W571 of 5-Helix inserts, is lined by hotspot residues Y94, P95, and L96. Protruding residues from HCDR2 of D5 (F54 and T56) are shown in blue.
Residue Y30 (ΔΔGAla-WT = 1.0 kcal/mol) is located at the periphery of the combining site and forms the edge of an overall concave binding surface into which two of the α-helices in 5-Helix are nestled (Figure 5A). The identification of Y30 as a hotspot residue is surprising, since hotspot residues tend to cluster at the center of protein-protein interfaces (18 – 20, 37). This position is occupied by a Thr in the five nearest germline progenitors (see Supporting Information), suggesting that Tyr at this position was specifically selected. The combinatorial alanine scanning codon for Tyr also allows substitution with Ser (m2) and Asp (m3); the ΔΔGm2-WT (0.7 kcal/mol) and ΔΔGm3-WT (0.9 kcal/mol) values for these substitutions indicate that substitutions other than Ala also involve an energetic penalty.
Figure 5.

(A) Location of Y30 on the overall concave surface of the D5 combining site. (B) Results from phage ELISA with particles displaying WT D5 or a Y30A point mutant as a function of phage titer. The OD(450nm) at each phage concentration was corrected for background binding by subtracting the binding signal against wells coated with 5-Helix from wells coated with BSA (‘corrected’ OD(450nm)).
Results from combinatorial alanine scanning mutagenesis have been shown to correlate closely with in vitro binding studies with single WT→Ala mutants in several systems (17, 19). However, we sought to confirm the critical role of Y30 in the D5 scFv for binding to 5-Helix since this feature (a hotspot residue located at the periphery of the combining site) is unusual. We prepared a single point mutant phage clone bearing the Y30A scFv and analyzed the binding of this clone by phage ELISA against 5-Helix as a function of phage titer (Figure 5B). Although phage solubility limits prevented saturation of the binding signal, it is clear from the partial binding curve in Figure 5B that the Y30A phage clone is deficient in its ability to bind 5-Helix relative to the clone displaying WT D5 phage clone. The highest phage titer tested in Figure 5B (~7 × 1010 infectious units/mL) corresponds to a phage concentration of ~ 0.1 nM. At this concentration, we observed a significant binding signal for WT D5Φ to 5-Helix, as is consistent with a subnanomolar binding constant (the reported KD for the D5 scFv is 150 pM) (23). However, we observed significantly lower binding signal at these concentrations for the Y30A mutant. The ELISA signals for the WT and Y30A phage clones were within ~20% at higher 5-Helix concentrations (see Supporting Information), indicating that display levels for the Y30A scFv are not grossly perturbed relative to the WT scFv. These results confirm that Y30 plays a critical role in the binding interaction.
The basis for conservation of residue K50 (ΔΔGAla-WT = 2.1 kcal/mol) is not clear from the crystal structure – none of the residues within van der Waals radius of the terminal ammonium appear to have polar interactions. However, mutation to Thr (ΔΔGm2-WT = 1.1 kcal/mol) was less perturbing (though still strongly disfavored) and substitution with an acidic residue (Glu) was even more energetically costly than Ala (since no substitutions to Glu were observed in the surviving pool, our analysis provides only a lower ΔΔGm3-WT estimate of 2.0 kcal/mol). The α-helical segments of 5-Helix are collectively negatively charged at neutral pH and therefore K50 may play a role in long-range interactions (32). However, another possibility is that K50 and the adjacent residue, Y49, play a role in maintaining the VH-VL interface to appropriately position residues playing a direct role in the interaction. The Y49 residue falls just short of our categorization as a hotspot residue (ΔΔGAla-WT = 0.9 kcal/mol) but mutation to alanine was nonetheless significantly disfavored. Substitution with Asp at this position was also disfavored (ΔΔGm3-WT > 1.1 kcal/mol). The D5 scFv construct may be particularly sensitive to mutations that alter the VH-VL interface relative to Fab or full-length IgG constructs since minor changes at the VH-VL may have significant effects of scFv folding stability. The D5 scFv is reported to have a KD comparable to that of the IgG (150 pM for the scFv and 50 pM for the IgG) and therefore indirect contributions of residues at the VH-VL interface are nonetheless informative.
Several of the residues assayed in our combinatorial alanine scanning libraries (W32, Y91, N93, Y94) were previously studied by mutagenesis and SPR studies with the full-length D5 IgG by Luftig et al. (22). Our results are in rough agreement with the SPR data for Y94 and N93. Mutation of Y94 to alanine resulted in over 30-fold increase in KD of the IgG, and this matches our data that indicate the ΔΔGAla-WT is high for this position in the scFv (2.6 kcal/mol). Mutation of N93 to alanine had a minor effect on KD for the IgG (4-fold increase); we observed a negligible ΔΔGAla-WT (0.2 kcal/mol) at this position in the scFv. In the SPR studies, substantial effects of KD of the IgG were obtained for the mutants W32A and Y91A (~20-fold increase in each case), however we observed small ΔΔGAla-WT for these positions (−0.3 kcal/mol and 0.3 kcal/mol, respectively). We observed a bias for WT side chain identity at both positions in the display selection (WT/Ala of 2.6 for W32A and 3.1 for Y91A). Therefore, it is possible that mutation of these residues to Ala results in structural effects, and these structural effects contribute to the observed decrease in binding of the IgG. However, another possibility is that discrepancies reflect differences between the IgG (used in the SPR studies) and the scFv (examined here).
Tolerance for Other Substitutions
Mutation of residues from WT to Ala provides information about contribution to binding affinity; however, these data can be biased due to the non-equivalent perturbing nature of WT→Ala mutations (20, 37). To gain further insight into tolerance for mutation at LCDR positions, we generated a second library in which LCDR residues were permitted to vary among WT and a less perturbing mutation (referred to here as the ‘Scan 2’ library). We designed a codon set for this analysis (Table 2) with the goal of examining substitutions that were not as perturbing as WT→Ala mutations, but altered some aspect of the side chain. Specifically, we aimed to include substitutions that contained similar physicochemical features (e.g., charge or hydrogen bonding capacity) but differed either in side chain length or size. We produced the ‘Scan 2’ library and subjected it to two rounds of selection against 5-Helix. We sequenced a sampling of binding clones (60 total clones) and calculated ΔΔGmut-WT using a strategy similar to that used for the combinatorial alanine scanning library. Numerical values for ΔΔGmut-WT are shown in Table 3, and represented graphically in Figure 6 (combinatorial alanine scanning residues are shown for comparison). In some cases, additional substitutions were permitted (which we refer to here as m4 and m5 to prevent confusion with substitutions from the alanine scanning library); the ΔΔGm4-WT and ΔΔGm5-WT values are also listed in Table 3.
Table 2.
Designed ‘Scan 2’ codon set.
| WT residue | Degenerate codonA |
Substitutions |
|---|---|---|
| H | CRT | H/R |
| K | ARG | K/R |
| L | MTG | L/M |
| N | MANB | N/Q(/H/K) |
| P | SCA | P/A |
| S | KCC | S/A |
| W | TKG | W/L |
| Y | YWTB | Y/L(/F/H) |
Degenerate nucleotide mixtures follow standard nomenclature: K = G/T; M = A/C; N = A/T/C/G; R = A/G; S = G/C, W = A/T, Y = C/T.
Due to the degeneracy of the genetic code, additional substitutions are permitted for some residues – these additional substitutions are indicated in parentheses.
Table 3.
Combinatorial ‘Scan 2’ results
| 5-Helix selection (n = 60)A |
Display selection (n = 48)A |
WT/mut (5-Helix) |
WT/mut (display) |
ΔΔGmut-WT (kcal/mol) |
ΔΔGm4-WT (kcal/mol) |
ΔΔGm5-WT (kcal/mol) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Res. | WT | mut | m4B | m5B | WT | mut | m4B | m5B | |||||
| LCDR1 | |||||||||||||
| Y30 | 11 | 12 (L) | 10 (F) | 27 (H) | 4 | 24 (L) | 3 (F) | 17 (H) | 0.9 | 0.2 | 1.0 | −0.1 | 0.3 |
| H31 | 13 | 47 (R) | - | - | 16 | 32 (R) | - | - | 0.3 | 0.5 | −0.4 | - | - |
| W32 | 46 | 14 (L) | - | - | 34 | 14 (L) | - | - | 3.3 | 2.4 | 0.2 | - | - |
| LCDR2 | |||||||||||||
| Y49 | 16 | 13 (L) | 11 (F) | 20 (H) | 5 | 24 (L) | 12 (F) | 7 (H) | 1.2 | 0.2 | 1.1 | 0.7 | 2.4 |
| K50 | 23 | 37 (R) | - | - | 18 | 30 (R) | - | - | 0.6 | 0.6 | 0.0 | - | - |
| S53 | 16C | 43 (A)C | - | - | 11 | 37 (A) | - | - | 0.4 | 0.3 | 0.1 | - | - |
| LCDR3 | |||||||||||||
| Y91 | 10 | 28 (L) | 15 (F) | 7 (H) | 8C | 16 (L)C | 8 (F)C | 15 (H)C | 0.4 | 0.5 | −0.2 | −0.2 | 0.6 |
| S92 | 12 | 48 (A) | - | - | 17 | 31 (A) | - | - | 0.3 | 0.5 | −0.5 | - | - |
| N93 | 12 | 19 (Q) | 20 (H) | 9 (K) | 13 | 12 (Q) | 17 (H) | 6 (K) | 0.6 | 1.1 | −0.3 | −0.1 | −0.3 |
| Y94 | 23 | 11 (L) | 23 (F) | 3 (H) | 7 | 20 (L) | 10 (F) | 11 (H) | 2.1 | 0.4 | 1.1 | 0.2 | 1.5 |
| P95 | 49 | 11 (L) | - | - | 23 | 25 (A) | - | - | 4.5 | 0.9 | 0.9 | - | - |
| L96 | 43 | 17 (M) | - | - | 31 | 17 (M) | - | - | 2.5 | 1.8 | 0.2 | - | - |
Number of times each side chain was observed at a particular position in the selected population. One sibling was observed in the 5-Helix selection; none was observed in the display selection.
In some cases, a third and forth substitution is permitted (m4 and m5). The occurrence of these side chains are indicated along with identity of the substitution.
In this position, sequencing was obscured in one of the 60 clones; all other positions were unambiguous.
Figure 6.

ΔΔGAla-WT and ΔΔGmut-WT values obtained from combinatorial alanine scanning and ‘Scan 2’libraries. The positions are labeled with WT residue identity preceding the position number, and the substitution of the ‘Scan 2’ library following the number. A ‘cut-off’ of 1 kcal/mol (dashed line) was used to identify hotspot residues.
Substitutions from the ‘Scan 2’ library were tolerated to a higher degree at positions Y94, P95, and L96 though a strong preference for WT (ΔΔGmut-WT ≥ 0.9 kcal/mol) was still observed at positions Y94 and P95. These results further confirm that ridge lining the pocket into which W571 of 5-Helix protrudes is a critical feature for this interaction. The combinatorial codon we employed for the ‘Scan 2’ library permitted addition substitutions of Phe or His at position Y94; Phe at this position was minimally perturbing (ΔΔGm4-WT = 0.2 kcal/mol) but His was poorly tolerated (ΔΔGm5-WT = 1.5 kcal/mol). The residue P95 likely plays a role in positioning the aromatic group at Y94 and therefore P95 cannot be functionally replaced by other residues. Residue L96 lines the bottom of the combining site pocket; we found that mutation to Met was well tolerated (ΔΔGmut-WT = 0.2 kcal/mol), therefore this position is more malleable to substitution by other aliphatic groups.
We observed a high ΔΔGmut-WT for replacement of the peripheral Y30 residue with Leu (1.0 kcal/mol). However, substitution with Phe (ΔΔGm4-WT = −0.1 kcal/mol) or His (ΔΔGm5-WT = 0.3 kcal/mol) was tolerated to a higher degree. The residue K50 could be replaced by Arg without penalty, further implicating the charge contribution to binding for this position. The adjacent residue, Y49, could not be replaced with Leu (ΔΔGmut-WT = 1.2 kcal/mol) or His (2.4 kcal/mol) and even the conservative Tyr→Phe mutation was found to be energetically costly (0.7 kcal/mol). Together with data from the neighboring K50 residue, these observations suggest that residues at the interface of VH and VL domains play indirect roles in supporting this high affinity interaction.
All other residues that bore modest ΔΔGAla-WT from the alanine scanning library also did not have high ΔΔGmut-WT from the ‘Scan 2’ library. This result is consistent with studies on the human growth hormone-receptor interaction that have shown that alanine scanning and other mutagenesis schemes are in rough agreement for identification of those residues that play major and minor roles in the intermolecular interaction (20). Notably, the substitutions in the ‘Scan 2’ library for three of the LCDR residues were alanine (S53, S92, and P95) and, in two of these three positions (S92 and P95), the ΔΔGAla-WT varied considerably among the ‘Scan 2’ library and the alanine scanning library (−0.5 kcal/mol vs. 0.5 kcal/mol for S92, and 0.9 kcal/mol vs. 1.4 kcal/mol for P95). These results indicate that context-dependent results may affect analysis of residue contributions when using combinatorial scanning mutagenesis. It is possible that other mutations were less deleterious in the ‘Scan 2’ library and therefore WT→Ala at positions S92 and P95 was more highly tolerated due to compensatory effects not present in the alanine scanning library. Combinatorial alanine scanning is based on the predicate that ΔΔGAla-WT values are independent from other positions (20); proof that this predicate is true would come from obtaining equal ΔΔGAla-WT values from different libraries. The discrepancy in ΔΔGAla-WT values for S92 and P95 suggest that this predicate may not hold true for all positions or all cases. A comprehensive analysis of Scan 2 binding clones containing WT→Ala mutations at S92 and P95, to identify coupled compensatory mutations elsewhere, might provide some insight into this question. However, our sampling size (n = 60 clones) was too small to identify significant mutational couplings. We note that Pal et al. observed similar behavior with the human growth hormone-receptor interaction (20).
DISCUSSION
Light Chain Contributions to High-Affinity Binding in D5
Our analysis demonstrates that light chain residues contribute substantially to the high affinity interaction between the D5 scFv and 5-Helix. In particular, a cluster of hotspot residues at the VH-VL interface (Y94, P95, L96) form the ridge of a pocket on the D5 combining site into which a hydrophobe (W571) from 5-Helix protrudes. These results, in conjunction with previous studies on the D5 IgG by Luftig et al. (22), indicate that the critical feature for this intermolecular association is an interlocking interaction between the opposing hydrophobes of W571 on 5-Helix and D5 HCDR2 residues F54 and T56. Furthermore, our results suggest that Y30, a residue with high ΔΔGAla-WT at the periphery of the combining site on LCDR1, also plays a role in this high affinity interaction. Interestingly, Y30 contacts one of the CHR segments of 5-Helix. The CHR region is likely not relevant to inhibiting HIV-1 membrane fusion since the NHR core trimer of the prehairpin intermediate is thought to consist only of NHR segments. Recently, D5 variants were isolated by phage display affinity maturation against a mimic of the prehairpin intermediate lacking the CHR (‘IZN23’) (23). The D5 variants selected against this target did not maintain identity at position Y30, which supports the hypothesis that this residue is important for recognition of 5-Helix.
Comparison to Other Protein Antibodies
Extensive site-directed mutagenesis studies have been performed on the anti-HEL antibody D1.3 as a model for antibody-protein recognition (13, 14). The interface of D1.3 for HEL binding has properties similar to that of the human growth hormone-receptor interaction, in that a small cluster of hotspot residues (four total residues across VH and VL with ΔΔG > 1 kcal/mol) account for the majority of the energy of the association (13). However, the interface involving an anti-D1.3 antibody (E5.2) that binds at the D1.3 combining site and contacts the same residues as HEL was found to be more evenly distributed among D1.3 combining site residues (14). In the D1.3-E5.2 interaction, six of nine residues examined in VL, and all eight residues examined in the VH had ΔΔG values > 1 kcal/mol for single WT→Ala mutations. From our studies with D5, the ΔΔGAla-WT values ranged from −0.3 kcal/mol to 2.6 kcal/mol, consistent with the range of values obtained from combinatorial alanine scanning studies of the human growth hormone-receptor interaction and the interaction between Fab2C4 and its target ErbB2 (17, 19, 20). However, the distribution of hotspot residues differs in that not all critical residues are clustered together at the center of the protein-protein interface. Instead, critical residues contributing to the high affinity association of D5 and 5-Helix appear to be distributed across the extended interface consisting of all six CDRs.
Unique Recognition Properties of VH1-69 Antibodies
Several groups have noted that the VH1-69 germline segment is frequently selected among virus neutralizing antibodies (24 – 27, 38), perhaps a surprising observation since antibodies derived from this germline account for less than 2% of productive rearrangements in normal (uninfected) circulating blood (39). Antibody D5 represents a prototypical VH1-69 antibody since it was isolated from a library constructed from uninfected donors and therefore contains relatively few mutations from the originating germline segments (21). In particular, the critical heavy chain paratope on D5 – involving residues on HCDR2 – is identical in sequence to the VH1-69 germline. The similarity among modes of recognition in D5, CR6261, and 412D involving HCDR2 (Figure 1C) suggest that unique properties of the hydrophobic VH1-69 HCDR2 make it particularly amenable intermolecular association with surface-exposed hydrophobic patches on antigens. As an example, the structural paratope for CR6261 is dominated by HCDR1 and HCDR2 (HCDR3 plays only a minor role) (25); this feature is notable since most other protein-antibody interactions involve major contributions from HCDR3 (3). We suggest that HCDR2 of VH1-69 antibodies form a ‘hydrophobic anchor point’ and other CDR contacts contribute to optimize specificity and affinity. This unique recognition property could potentially be exploited to generate novel synthetic antibody libraries based on VH1-69 scaffolds. Here we have shown that LCDR residue contribute substantially to interaction between D5 and 5-Helix. Studies to determine if the specificity of D5 can be altered by light chain engineering alone are currently underway.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the reviewers of this manuscript for their insightful comments. We acknowledge Mark Sullivan (University of Rochester) for providing the pAPIII6 phagemid; Stephen Harrison (Harvard Medical School) for the 5-Helix epression plasmid; Sachdev Sidhu (University of Toronto), Greg Weiss (University of California, Irvine), and Sam Gellman (University of Wisconsin) for helpful discussions.
ABBREVIATIONS
- HIV-1
human immunodeficiency virus, type 1
- CDR
complementarity determining region
- HCDR
heavy chain CDR
- LCDR
light chain CDR
- HEL
hen egg-white lysozyme
- Fab
antigen binding fragment
- scFv
single chain variable fragment
- IgG
immunoglobulin G
- SPR
surface plasmon resonance
- OD
optical density
- IPTG
isopropyl-β-D-thiogalactopyranoside
- RP-HPLC
reverse-phase high performance liquid chromatography
- PBS
phosphate-buffered saline
- pfu
plaque forming units
- PEG
polyethylene glycol
- BSA
bovine serum albumin
- ELISA
enzyme-linked immunosorbent assay
- HRP
horseradish peroxidase
- TMB
tetramethylbenzidine
- CHR
C-heptad repeat
- NHR
N-heptad repeat
Footnotes
This work was funded by Einstein start-up funds, a pilot project grant from the Einstein-Montefiore Center for AIDS Research, and the Arnold and Mabel Beckman Foundation Young Investigators Program. J. S. H. is supported by a National Institutes of Health Biophysics Training Grant (T32-GM008572).
SUPPORTING INFORMATION AVAILABLE Expression, purification, and characterization of 5-Helix protein; details for cloning and phage display of the D5 scFv; comparison of the D5 VL domain to germline segments; phage ELISA for WT D5 and Y30A phage at varying 5-Helix concentrations. This material is available free of charge at http://pubs.acs.org.
REFERENCES
- 1.Dimitrov DS, Marks JD. Therapeutic antibodies: current state and future trends – is a paradigm change coming soon? Methods Mol. Biol. 2009;525:1–27. doi: 10.1007/978-1-59745-554-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maynard J, Georgiou G. Antibody engineering. Annu. Rev. Biomed. Eng. 2000;2:339–376. doi: 10.1146/annurev.bioeng.2.1.339. [DOI] [PubMed] [Google Scholar]
- 3.MacCallum RM, Martin CA, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J. Mol. Biol. 1996;262:732–745. doi: 10.1006/jmbi.1996.0548. [DOI] [PubMed] [Google Scholar]
- 4.Almagro JC. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: implications for the rational design of antibody repertoires. J. Mol. Recognit. 2004;17:132–143. doi: 10.1002/jmr.659. [DOI] [PubMed] [Google Scholar]
- 5.Wilson IA, Stanfield RL. Antibody-antigen interactions: new structures and conformational changes. Curr. Opin. Struct. Biol. 1994;4:857–867. doi: 10.1016/0959-440x(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 6.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nat. Chem. Biol. 2006;2:682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 7.Kossiakoff AA, Koide S. Understanding mechanisms governing protein-protein interactions from synthetic binding interfaces. Curr. Opin. Struct. Biol. 2008;18:499–506. doi: 10.1016/j.sbi.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Lerner RA. Manufacturing immunity to disease in a test tube: the magic bullet realized. Angew. Chem. Int. Ed. Engl. 2006;45:8106–8125. doi: 10.1002/anie.200603381. [DOI] [PubMed] [Google Scholar]
- 9.Winter G. Synthetic human antibodies and a strategy for protein engineering. FEBS Lett. 1998;430:92–94. doi: 10.1016/s0014-5793(98)00628-0. [DOI] [PubMed] [Google Scholar]
- 10.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J. Mol. Biol. 2005;348:1153–1162. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 11.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Koissiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Cobaugh CW, Almagro JC, Pogson M, Iverson B, Georgiou G. Synthetic antibody libraries focused towards peptide ligands. J. Mol. Biol. 2008;378:622–633. doi: 10.1016/j.jmb.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dall’Acqua W, Goldman ER, Lin W, Teng C, Tsuchiya D, Li H, Ysern X, Braden BC, Li Y, Smith-Gill SJ, Mariuzza RA. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–7991. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- 14.Dall’Acqua W, Goldman ER, Eisenstein D, Mariuzza RA. A mutational analysis of the binding of two different proteins to the same antibody. Biochemistry. 1996;35:9667–9676. doi: 10.1021/bi960819i. [DOI] [PubMed] [Google Scholar]
- 15.Pons J, Rajpal A, Kirsch J. Energetic analysis of an antigen/antibody interface: alanine scanning mutagenesis and double mutant cycles on the HyHEL-10/lysozyme interaction. Protein Sci. 1999;8:958–968. doi: 10.1110/ps.8.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Urrutia M, Smith-Gill SJ, Mariuzza RA. Dissection of binding interactions in the complex between the anti-lysozyme antibody HyHEL-63 and its antigen. Biochemistry. 2003;42:11–22. doi: 10.1021/bi020589+. [DOI] [PubMed] [Google Scholar]
- 17.Vajdos FF, Adams CW, Breece TN, Presta LG, de Vos AM, Sidhu SS. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J. Mol. Biol. 2002;320:415–428. doi: 10.1016/S0022-2836(02)00264-4. [DOI] [PubMed] [Google Scholar]
- 18.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 19.Weiss GA, Watanabe CK, Zhong A, Goddard A, Sidhu SS. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA. 2000;97:8950–8954. doi: 10.1073/pnas.160252097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal G, Fong S-Y, Kossiakoff AA, Sidhu SS. Alternative views of functional protein binding epitopes obtained by combinatorial shotgun scanning mutagenesis. Protein Sci. 2005;14:2405–2413. doi: 10.1110/ps.051519805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller MD, Geleziunas R, Bianchi E, Lennard S, Hrin R, Zhang H, Lu M, An Z, Ingallinella P, Finotto M, Mattu M, Finnefrock AC, Bramhill D, Cook J, Eckert DM, Hampton R, Patel M, Jarantow S, Joyce J, Ciliberto G, Cortese R, Lu P, Strohl W, McElhaugh M, Lane S, Lloyd C, Lowe D, Osbourn J, Vauhan T, Emini E, Barbato G, Kim PS, Hazuda DJ, Shiver JW, Pessi A. A human monoconal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc. Natl. Acad. Sci. USA. 2005;102:14759–14764. doi: 10.1073/pnas.0506927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luftig MA, Mattu M, DiGiovine P, Geleziunas R, Hrin R, Barbato G, Bianchi E, Miller MD, Pessi A, Carfi A. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat. Struct. Mol. Biol. 2006;13:740–747. doi: 10.1038/nsmb1127. [DOI] [PubMed] [Google Scholar]
- 23.Montgomery DL, Wang Y-J, Hrin R, Luftig M, Su B, Miller MD, Wang F, Hatyko P, Huang L, Vitelli S, Condra J, Liu X, Hampton R, Carfi A, Pessi A, Bianchi E, Joyce J, Lloyd C, Geleziunas R, Bramhill D, King VM, Finnerfrock AC, Strohl W, An Z. Affinity maturation and characterization of a human monoclonal antibody against HIV-1 gp41. Monoclonal antibodies. 2009;1:462–474. doi: 10.4161/mabs.1.5.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang CC, Venturi M, Majeed S, Moore MJ, Phogat S, Zhang MY, Dimitrov DS, Hendrickson WA, Robinson J, Sodroski J, Wyatt R, Choe H, Farzan M, Kwong PD. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc. Natl. Acad. Sci. USA. 2004;101:2706–2711. doi: 10.1073/pnas.0308527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, Wyatt R, Bewley CR, Kwong PD. Structures of the CCR5 N-terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong PD, Wilson IA. HIV-1 and influenza antibodies: seeing antigens in new ways. Nat. Immunol. 2009;10:573–578. doi: 10.1038/ni.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, Wheeler JC, Sweet RW, Lu J, Luo J, Tornetta M, Whitaker B, Reddy R, Brittingham R, Borozdina L, Chen Q, Amegadzie B, Knight DM, Almagro JC, Tsui P. De novo selection of high-affinity antibodies from synthetic Fab libraries displayed on phage as pIX fusion proteins. J. Mol. Biol. 2010;397:385–396. doi: 10.1016/j.jmb.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 29.Lai JR, Fischbach MA, Liu DR, Walsh CT. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc. Natl. Acad. Sci. USA. 2006;103:5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai JR, Fischbach MA, Liu DR, Walsh CT. Localized protein interaction surfaces on the EntB carrier protein revealed by combinatorial mutagenesis and selection. J. Am. Chem. Soc. 2006;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z, Lai JR, Walsh CT. Interdomain communication between the thiolation and thioesterase domains of EntF explored by combinatorial mutagenesis and selection. Chem. Biol. 2006;13:869–870. doi: 10.1016/j.chembiol.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Frey G, Rits-Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. USA. 2006;103:13938–13943. doi: 10.1073/pnas.0601036103. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haidaris CG, Malone J, Sherrill LA, Bliss JM, Gaspari AA, Insel RA, Sullivan MA. Recombinant Human Antibody Single Chain Variable Fragments Reactive with Candida Albicans Surface Antigents. J. Immunol. Methods. 2001;257:185–202. doi: 10.1016/s0022-1759(01)00463-x. [DOI] [PubMed] [Google Scholar]
- 34.Sidhu SS, Weiss GA. Chapter 2: Constructing phage display libraries by oligonucleotide-directed mutagenesis. In: Clackson T, Lowman HB, editors. Phage display: a practical approach. Oxford University Press; New York, NY: pp. 27–41. [Google Scholar]
- 35.Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 36.Champagne K, Shishido A, Root MJ. Interactions of HIV-1 inhibitory peptide T20 with the gp41 N-HR coiled coil. J. Biol. Chem. 2009;284:3619–3627. doi: 10.1074/jbc.M809269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 38.Kashyap AK, Steel J, Orner AF, Dillon MA, Swale RE, Wall KM, Perry KJ, Faynboym A, Ilhan M, Horowitz M, Horowitx L, Palese P, Bhatt RR, Lerner RA. Combinatorial antibody libraries from survivors of the rurkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl. Acad. Sci. USA. 2008;105:5986–5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brezinschek H-P, Brezinschek RI, Dorner T, Lipsky PE. Similar characteristics of the CDR3 of VH1-69/DP-10 rearrangements in normal human peripheral blood and chronic lymphocytic leukaemia B cells. Br. J. Haematology. 1998;102:516–521. doi: 10.1046/j.1365-2141.1998.00787.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.