

Summary
Many functions of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) have been defined, but relatively little is known about the biology of an alternative mTOR complex, mTORC2. We showed that conditional deletion of rictor, an essential subunit of mTORC2, impaired differentiation into T helper 1 (Th1) and Th2 cells without diversion into FoxP3+ status or substantial effect on Th17 cell differentiation. mTORC2 promoted phosphorylation of protein kinase B (PKB, or Akt) and PKC, Akt activity, and nuclear NF-κB transcription factors in response to T cell activation. Complementation with active Akt restored only T-bet transcription factor expression and Th1 cell differentiation, whereas activated PKC-θ reverted only GATA3 transcription factor and the Th2 cell defect of mTORC2 mutant cells. Collectively, the data uncover vital mTOR – PKC and mTOR–Akt connections in T cell differentiation, and reveal distinct pathways by which mTORC2 regulates development of Th1 and Th2 cell subsets.
Introduction
To meet specific needs for T cell help in immunity, naïve CD4+ T cells can differentiate into functionally distinct subsets of effector and regulatory (Treg) T cells after activation (Glimcher and Murphy, 2000; Zhu and Paul, 2008). This flexibility allows modulation of antigenspecific responses and adaptive immunity against microbes. Among these subsets, T helper 1 (Th1) cells produce cytokines such as IFN-γ after activation and IL-12 and IFN-γ after exposure to signals elicited by intracellular microbes (Glimcher and Murphy, 2000). A Th2 cell subset is induced by different cues and produces a distinct program of cytokines (IL-4, -5, and -13) for allergic and anti-parasitic responses. Several more effector states can develop from naïve CD4+ T cells: IL-17-producing Th17, induced Treg, IL-9-producing Th9, and IL-21-producing follicular helper (Tfh) cells (Locksley, 2009). Although the balance among these CD4 subsets is crucial, much remains unknown as to how signals are integrated to determine T cell fate and function.
T cell activation by antigen is essential for the development of effectors from naïve T cells, and this process is strongly potentiated by engagement of co-stimulatory receptors on the T cells. CD28 dramatically enhances Th1 or Th2 cell responses (Kane et al., 2001; Kuchroo et al., 1995). Similarly, inducible costimulators such as ICOS and OX40 strongly enhance Th2 cell development and Th1 cell responses (Lane, 2000). Thus, costimulation of T cell receptor (TCR) signaling is vital for efficient development of several CD4+ T cell effector states. Furthermore, the precise quantitative and qualitative signaling elicited by the TCR or costimulators can guide the balance of differentiation into the different T helper subsets (Constant and Bottomly, 1997). As such, signaling molecules activated by TCR and costimulation are likely to be of vital importance in identifying means of manipulating the properties of immune responses.
Key molecules activated by costimulation include the mammalian Target of Rapamycin (mTOR), protein kinase B (PKB, also known as Akt), and protein kinase C (PKC)- θ (Huang et al., 2002; Lin et al., 2000). Upon TCR engagement and CD28 ligation, PKC- θ is phosphorylated and enhances T helper responses, in part by promoting nuclear translocation of NF-κB transcription factors (Coudronniere et al., 2000; Wang et al., 2004). In parallel, TCR engagement with costimulation also increases phosphatidylinositol 3-kinase (PI3K) activity. PI3K increases amounts of phosphatidyl inositol (3, 4, 5)-triphosphate (PIP3), which recruits the PI3K-dependent kinase (PDK) 1 and activates Akt via phosphorylation of a conserved residue termed Akt(T308) (Scheid et al., 2002). Among its molecular targets, Akt leads to activation of mTOR (Kane and Weiss, 2003).
The importance of understanding of how specific signaling pathways impact T cell physiology is underscored by the successes and toxicities of immune suppressant drugs such as rapamycin, which targets mTOR. Rapamycin can inhibit proliferation of conventional T lymphocytes without blocking Treg cells (Battaglia et al., 2005; Valmori et al., 2006), and appears to bias the acquisition of CD4+ T cell functions inasmuch as it represses Th1, Th2, and Th17 cell development while enhancing induced Treg cells (Blazar et al., 1998; Kopf et al., 2007). There are at least two independent pools of mTOR in mammalian cells, of which the first is an acutely rapamycin-sensitive assembly termed mTOR complex 1 (mTORC1) (Laplante and Sabatini, 2009). A major role for mTORC1 in T lineage cells is clear from the effects of rapamycin and loss-of-function models for Akt and PDK1 (Hinton et al., 2004; Juntilla et al., 2007). Thus, although mTORC1 can also be activated by Akt- and PI3K-independent mechanisms (Carriere et al., 2008; Fang et al., 2001), an important linear pathway to mTORC1 via Akt is solidly established and the data establish Akt and mTORC1 as key transducers of proliferative and differentiative signals in the T lineage.
After its activation by PDK1, Akt can be further phosphorylated at a C-terminal hydrophobic motif (HM) termed S473 (Scheid et al., 2002). This modification executed by PDK2 activities increased the cell-free in vitro activity of T308-phosphorylated Akt (Chan and Tsichlis, 2001; Scheid et al., 2002). PKC-β, DNA-PK, and integrin-linked kinase can provide PDK2 function in particular cell types (Dong and Liu, 2005). More recently, a second multi-protein complex that includes mTOR, mTORC2, has been identified as a major PDK2 (Sarbassov et al., 2005). In contrast to mTORC1, relatively little is known about the biological roles and functions of mTORC2 (Laplante and Sabatini, 2009). mTORC2 requires unique components - rictor and SIN1 - that are not shared by mTORC1 (Jacinto et al., 2006). Loss of mTORC2 is embryonic lethal, but specific cellular or physiological targets in development are unknown (Guertin et al., 2006; Shiota et al., 2006). Intriguingly, mTORC2 was vital for Akt phosphorylation at T308 as well as S473 in multiple cell lines (Sarbassov et al., 2005) but not in rictor- or SIN1-null mouse embryonic fibroblasts (MEFs) (Guertin et al., 2006; Jacinto et al., 2006). Overall, it remains unclear if mTORC2 impacts Akt activity or its targets in T cells. Apart from Akt, mTORC2 can phosphorylate PKC isoforms or regulate their effects on actin in tumor cells, but the impact of this capacity on physiological functions is not clear (Facchinetti et al., 2008; Ikenoue et al., 2008). Finally, prolonged exposure of tumor cell lines or mice to rapamycin can either reduce or increase activity of mTORC2 (Sarbassov et al., 2006). Thus, the degree to which mTORC2 might contribute to rapamycin effects is unclear.
Because there is no direct evidence of an mTORC2 function in immunity, we analyzed a conditional loss-of-function model using targeted alleles encoding the essential mTORC2 subunit rictor. The principal role revealed for rictor after CD4+ T cell activation was promotion of differentiation into Th1 and Th2 cell subsets. In contrast, mTORC2 inhibited the induction of regulatory T cells under permissive conditions, but the defective cells exhibited no diversion from T helper differentiation into FoxP3+ status or marked abnormality of Th17 cell generation. Biochemical analyses revealed that mTORC2 deficiency impacted both Akt activity and phosphorylation of conserved PKC motifs. Strikingly, constitutively active Akt rescued T-bet expression and Th1 but not Th2 cell differentiation, whereas PKC-θ led to a reciprocal outcome. Thus, each of these mTORC2 targets selectively suppressed distinct defects of rictor-deficient cells, revealing an important branchpoint downstream from this signal-transducing complex in T cells.
Results
Costimulation and T cell activation induce rictor-dependent Akt HM phosphorylation
We first analyzed the relationship of TCR costimulation to Akt phosphorylation and mTORC2 activity using combinations of anti-(α)CD3 and αCD28. Anti-CD28, alone and synergistically with αCD3, increased phosphorylation not only at Akt T308 but also at the Akt S473 (Fig. 1), indicating that costimulation induced PDK2 activity. Costimulation also increased the phosphorylation of p70 ribosomal S6 protein kinase (S6K1) at T389, a site modified by mTORC1. mTORC2 also mediates serum-induced PKC phosphorylation at turn (TM) and hydrophobic (HM) motifs (Guertin et al., 2006; Ikenoue et al., 2008), and we found CD28 stimulation enhanced PKC-θ-mediated PKC HM and TM phosphorylation (Fig. 1).
Figure 1.

Costimulation enhances phosphorylation of Akt HM (S473). Phosphorylated and total pools of the indicated proteins were analyzed by immunoblotting. CD4+ T cells were stimulated (40 min) with 0.5 µg/ml plate-bound αCD3, 2.5 µg/ml of soluble αCD28, or both, as described in the Methods. Bar graphs quantify phosphorylation of Akt, S6K1, and PKC(−θ), with each sample normalized to the level of unphosphorylated protein in one experiment representative of three replicates.
To identify roles of mTORC2 in mature T cells, we used conditional inactivation of rictor. Deletion driven by Cre early in the T lineage (i.e., starting at the DN2 stage) decreased developmental efficiency and led to substantial phenotypic abnormalities of the resultant T cell repertoire (data not shown). Using a codon-optimized Cre cDNA that is directed to start expression after thymocyte progression to the double positive (DP) stage by the distal Lck promoter (“dLck-iCre”) (Zhang et al., 2005), deletion was detected in T cells whereas floxed alleles were almost all intact in the thymus and in Thy1− cells from peripheral lymphoid organs (supplemental Fig. S1a). Thymocytes from dLck-iCre+ Rictorfl/fl (abbreviated as cKO) mice and wild-type controls showed no abnormalities except for very modest decreases in CD8 SP cells and TCRhi HSAlo thymocytes (Fig. S1b–d). Spleen and lymph node CD4+ T cell numbers were normal whereas CD8+ T cells decreased slightly (Fig. 2A) and frequencies of CD44hi, CD62Lhi, and CD25hi cells were similar in cKO and WT mice (supplemental Fig. S1f–i). Thus, mature CD4+ T cells developed normally when rictor deletion was deferred during thymic differentiation, whereas a modest quantitative defect impacted production of CD8+ T cells.
Figure 2.

Impaired Akt phosphorylation and activity in rictor-deficient T cells. (A) T cell numbers from lymphoid organs of 6–8 week-old mice. Shown are mean (±SEM) numbers of cells of the indicated types (spleen and pooled lymph nodes of 8 WT and 8 cKO mice; *p < 0.05) (B) Previously activated CD4+ T cells were stimulated (40 min) with αCD3, αCD28, or both and analyzed by immunoblotting (as in Fig. 1A). Akt S473 and S6K1 T389 phosphorylation were normalized to amounts of unphosphorylated protein in the same sample, and then to amounts in WT CD4+ cells (bar graphs from one experiment representative of two complete replicates, with additional replicates of αCD3 + αCD28 vs control). (C, D) Decreased Akt enzymatic activity in T cells deficient for mTORC2. (C)) Akt activation loop (T308) phosphorylation in T cells lacking rictor (one result representative of two replicates with similar results). (D) A representative result assaying Akt kinase in extracts of WT and cKO CD4+ T cells. Cells were activated and restimulated as in (B); numbers represent quantified signals, normalized to the amounts of Akt in each sample and expressed as arbitrary light units with resting WT cells set as 1. Samples are as in (C). Additional information is in supplemental Fig. S1.
PDK2 function downstream from the TCR and CD28 in mature CD4+ T cells was analyzed by comparison of Akt S473 phosphorylation in WT and cKO cells. This modification was decreased in rictor-deficient T cells after TCR stimulation, costimulation, or both (Fig. 2B). Loss of mTORC2 has been reported either to eliminate the phosphorylation of Akt T308 in some cells (Sarbassov et al., 2005) or to have no impact on this activating modification (Guertin et al., 2006; Jacinto et al., 2006; Shiota et al., 2006). Decreased P-Akt T308, albeit not a complete loss, was observed in cKO T cells (Fig. 2C). Despite the residual T308 phosphorylation, TCR and CD28 engagement stimulated Akt enzymatic activity in mTORC2-deficient T cells far less than WT controls (Fig. 2D). Thus, T cell activation-induced Akt activity relies on mTORC2. In contrast, phosphorylation of S6K1 - which is impacted by Akt but can also be effected through other pathways - was not decreased in cKO cells.
Rictor promotes a restricted set of helper T cell fate choices
Because rictor was vital for full Akt activity in T cells, we explored its impact on activation, differentiation, and function of T cells. We observed a dramatic decrease in Th2 cell differentiation among naïve CD4+ T cells from cKO mice (Fig. 3A, B). Comparisons of cKO to WT littermates also revealed impaired Th1 cell differentiation (Fig. 3A, B). In contrast, no marked defect in Th17 cell development was observed (Fig. 3A, B), indicating that rictor-containing complexes selectively promoted Th1 and Th2 cell differentiation.
Figure 3.

mTORC2 selectively regulates differentiation of helper T cell subsets and responses. CD4+ T cells cultured for 5 d under Th1, Th2, or Th17 cell conditions, re- stimulated with αCD3 + αCD28, and analyzed by (A) flow cytometry for intracellular cytokines or(B) ELISA with culture supernatants. Shown are profiles for IFN-γ and IL-4 in the CD4+ viable cell gate, or IL-17A and IFN-γ in rictor-deficient CD4+ T cells [one result representative of 3 replicate experiments (A)] and means [±SEM; n=5 (B)]. (C) mTORC2 impedes induction of iTreg cell phenotype. Naïve CD4+ T cells (leftmost panels) from WT and cKO mice were activated (αCD3 + αCD28) and grown in the absence or presence of TGF-β for 3 d. Shown are histograms of FoxP3 expression in the CD4+ gate of freshly isolated naïve T cells or activated T cells, from one representative experiment. (D) Normal Treg cell populations under T helper-inducing conditions. CD4+ T cells activated and cultured under the Th1, Th2, and Th17 cell conditions were analyzed by flow cytometry as in (C). (E–I) Impairment of responses in vivo. Each symbol represents one mouse, solid lines denote mean values; * p<0.05 (two independent experiments). (E, F) Impaired IgG2a, but not IgG3, anti-KLH response in rictor cKO mice. Groups of WT and cKO mice were immunized with KLH in IFA and boosted with KLH in IFA 14 d later. Sera were collected on day 19, 5 d after the boost. Shown are capture ELISA results for IgG2a (E) and IgG3 (F). (G) WT and cKO mice were challenged with L. monocytogenes and analyzed as described in the Methods. Shown are log10-scale results of ELISPOT assays (two independent experiments) performed using the immunodominant class II MHC peptide LLO190–201 as indicated (10, 100 nM); each dot represents one mouse, solid lines denote mean values (panels are arranged vertically). (H, I) Decreased IgG1 and IgE production in rictor cKO mice. As in (E, F), but mice were immunized with low-endotoxin ovalbumin in alum, and ELISA results shown are for Ag-specific IgE (H), and IgG1 (I); ANOVA across the full dilution curves and all indicated differences were significant at p<0.05. Additional information is in supplemental Fig. S2.
Constitutively active Akt can inhibit Treg cell development (Haxhinasto et al., 2008), whereas rapamycin enhances their development at the expense of Th17 cell differentiation (Blazar et al., 1998; Kopf et al., 2007). cKO mice did not perturb the steady-state frequency of Treg cells (supplemental Fig. S2a). However, FoxP3+ progeny developed from activated cKO CD4+ T cells supplemented only with TGF-β at a greater efficiency than controls (Fig. 3C & S2b). In contrast, normal frequencies of the FoxP3+ population were observed despite the mTORC2 defect when activated CD4+ T cells received strong T helper-differentiating signals (Fig. 3D). These results show that mTORC2 inhibits iTreg cell differentiation under conditions already favorable to this process but rictor deficiency neither prevented Th17 cell development nor diverted developing effectors into a FoxP3+ population if IL-4 or Th1 cell-promoting cytokines were present.
To test if rictor influences an immune response in vivo, WT and cKO mice were immunized with keyhole limpet haemocyanin (KLH). The Ag-specific IgG2a response of cKO mice was significantly reduced, whereas IgG2b and IgG3 were not (Fig. 3E, F, & supplemental Fig. S2c). Helper T cell-derived IFN-γ is crucial for isotype switching to IgG2a, suggesting mTORC2 might be important for type 1 helper function. Consistent with this, the frequency of IFN-γ-producing CD4+ T cells was lower in lymphoid samples from cKO mice in an infectious challenge model (Fig. 3G, supplemental Fig. S2d, e). Moreover, total Ig and IgG1 (supplemental Fig. 2f, g) anti-KLH responses also were weaker. To measure IgE concentrations, mice were immunized with ovalbumin in alum. A reduced overall Ag-specific IgG response was observed again and amounts of serum IgE anti-ovalbumin were reduced in the cKO mice (Fig. 3H, I; supplemental Fig. S2h). These results suggested a requirement for mTORC2 in both type 1 and type 2 T cell help to Ab class switching, and the aggregate findings indicate that rictor functions in vivo to mediates a subset of differentiation events and regulates the properties of T-B help.
Rictor promotes proliferation without impacting survival
The best-known functions of Akt are its suppression of apoptosis and acceleration of cell cycling (Manning and Cantley, 2007). TCR-induced proliferation was attenuated in rictor-deficient T cells, at least in part due to a decreased rate of G1-S transitions (Fig. 4A, B). Rictor-deficient CD8+ T cells as well as CD4+ T cells were defective; moreover, IL-4-dependent proliferation of both CD4+ and CD8+ T cells was also attenuated (supplemental Fig. S3a–d). Cytokine receptors that share a common gamma chain signal protection against death by neglect, radiation- or etoposide-induced DNA damage. However, there was no decrease in survival rates of rictor-deficient T cells or increase in their apoptosis in the absence or presence of cytokines (Fig. 4C–E; supplemental Fig. S3e, f). Similarly, we observed no sensitization to apoptosis after re-stimulation of activated CD4+ T cells (supplemental Fig. S3g). Thus, mTORC2 mediates T cell proliferative signaling predominantly via cell cycle regulation rather than suppression of apoptosis.
Figure 4.

Decreased proliferation but normal survival of rictor-deficient T cells. (A) Splenocytes of WT and rictor cKO mice assayed for 3H-thymidine incorporation 48 h after activation with αCD3 + αCD28 (representing three replicate experiments). (B) As in (A), except that cells were analyzed by flow cytometry for BrdU incorporation (left panel) or division history using CFSE partitioning (right panel). Shown are histograms of events in the CD4+ and viable lymphoid gates from one experiment representative of three replicates. (C) Mean (±SEM) from one of two replicate experiments measuring viable CD4+ T cell numbers after triplicate cultures (1–3 d) with or without IL-4, expressed as a percentage of the input viable cell counts. (D) Spleen cells were cultured (20 h) in the presence or absence of IL-4 and assayed by TUNEL. Shown are flow cytometry profiles for CD4+ cells; numbers denote % TUNEL positive cells in one experiment representative of 4 replicates. (E) Cells were irradiated, cultured in the presence or absence of IL-4, and assayed by TUNEL. Additional information is in supplemental Fig. S3.
Akt and PKC mediate mTORC2 regulation of T helper differentiation
We transduced constitutively activated kinases targeted by mTORC2 into rictor-deficient T cells to determine the functional impact of Akt and PKC on the defects of T helper differentiation. Th1 cell differentiation of cKO CD4+ T cells was rescued by the constitutively active mutant Myr-Akt (Fig. 5A, B; supplemental Fig. S4a, b). An Akt mutant activated by phospho-mimetic residues at T308 and S473 [Akt(DD)] also suppressed the differentiation defect. In contrast, Akt(DA) - which is refractory to S473 phosphorylation - did not raise the frequency of cKO Th1 cells (supplemental Fig. S4a, b). Akt(DD) also restored normal proliferation to cKO T cells (supplemental Fig. S4d). Strikingly, however, Akt did not rescue the Th2 cell differentiation defect imposed by loss of mTORC2 function (Fig. 5C, D). Thus, mTORC2 modification of Akt promotes Th1 cell differentiation but is not sufficient to explain the defect of Th2 cell differentiation.
Figure 5.
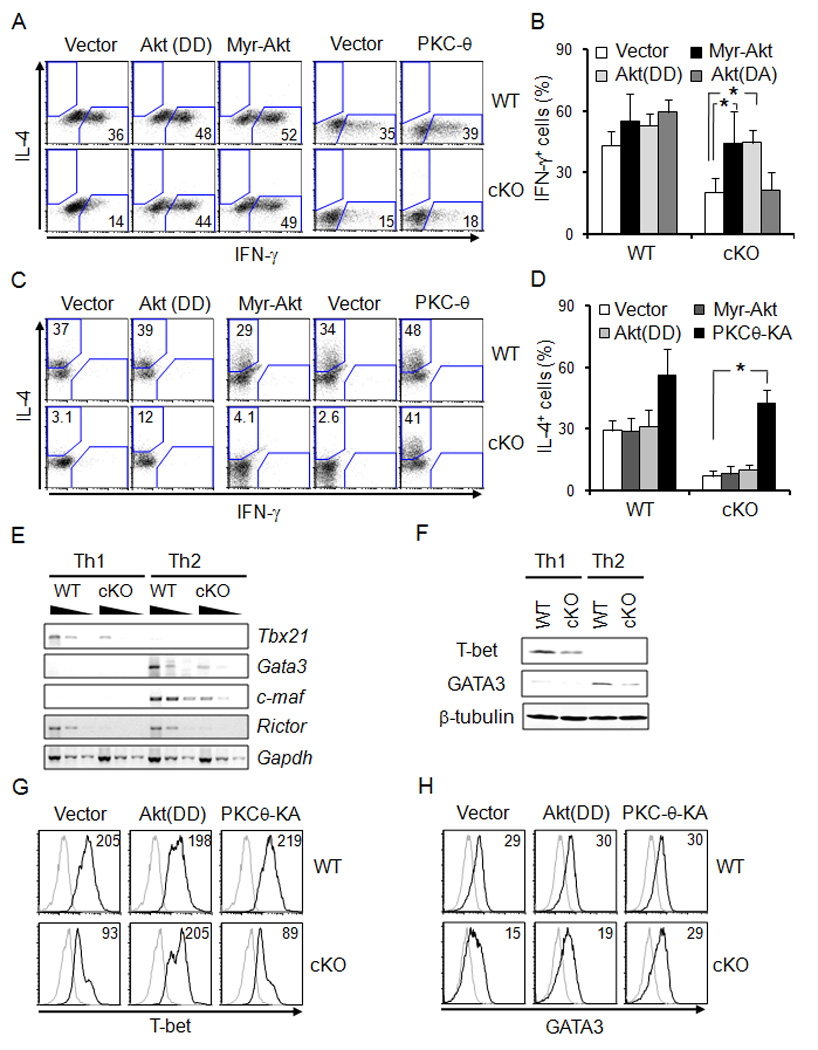
Reciprocal effects of Akt and PKC-θ, downstream from mTORC2, in T helper differentiation. (A–D) CD4+ T cells were activated under non-differentiating conditions, transduced with the indicated constructs, cultured for 5 days under (A, B) Th1 or (C, D) Th2 cell polarizing conditions, restimulated, and analyzed by flow cytometry. Shown are flow data for IFN-γ and IL-4 in the GFP+ CD4+ gate (A, C) (representative result from one of ≥3 replicate experiments). (B, D) Experimental results are summarized as mean (±SEM) % IFN-γ+ or % IL-4+ cells in the in the GFP+ CD4+ gate. (E, F) Impaired T-bet and GATA3 expression in rictor-deficient T cells. CD4+ T cells were cultured (4 d) in Th1 or Th2 cell conditions, and assayed using RT-PCR on serial 5-fold template dilutions to compare levels of mRNAs (E), or immunoblotting (F). (G, H) Selective reversion of decreased T-bet or GATA-3 expression in rictor cKO CD4+ T cells. CD4+ T cells were activated, transduced, switched to differentiating conditions, and analyzed by FACS as in A–D, except that intracellular stains were for T-bet (G) and GATA-3 (H) in the GFP+ CD4+ gate. Shown are representative histograms for the signal of isotype controls (thin line) or α(T-bet or GATA-3, thick line) in one experiment representative of two replicates. Inset numbers represent the net signal as MFI (active Ab – isotype signal) in each sample. Additional information is in supplemental Fig. S4 and Methods.
Although mTORC2 phosphorylates PKCs at sites not thought of as major regulators of catalytic activity, we hypothesized that a PKC pathway might provide a functional link between mTORC2 and Th2 cell differentiation. Indeed, transduction of a constitutively active PKC-θ mutant (Liu et al., 2001) reverted the Th2 cell defect of cKO CD4+ T cells yet did not impact the frequency of IFN-γ-positive cells (Fig 5A; supplemental Fig. 4a, c). Akt and PKC-θ might collaborate as relays downstream from mTORC2, but co-transduction of these kinases into cKO CD4+ T cells showed only a small additive effect for Th2 cell differentiation (supplemental Fig. S4e). As a mechanism for decreases in Th1 and Th2 cell differentiation when mTORC2 is compromised, amounts of the lineage-determining transcription factors T-bet and GATA-3 were substantially reduced in cKO CD4 T cells (Fig 5E, F). Moreover, Akt restored WT amounts of T-bet expression to cKO T cells developing in Th1 cell conditions, while failing to reverse the deficit of GATA3 in Th2 cell differentiation (Fig 5G, H). Conversely, constitutively active PKC-θ restored GATA3 but not T-bet under Th2 and Th1 cell conditions, respectively (Fig 5G, H).
Because mTORC2 differentially influenced fate choices of mature T cells, we explored relative activities of PDK1, PDK2, and mTORC2 in developing effectors. In comparing nonpolarizing, Th1, Th2, and Th17 cell conditions, there were modest differences in amounts of PKC-θ and its HM-phosphorylated form and in ratios of Akt P-S473 to P-T308 (supplemental Fig. S4f). STAT transcription factors guide choices for CD4+ T cells during T helper development and control the ‘master regulators’ of Th1, Th2, and Th17 cell fates. However, tyrosine phosphorylation of the relevant STAT transcription factors was normal in cKO cells (Fig. 6A; supplemental Fig. S5a). P-STAT induction in cells subjected to inhibition of mTORC1 and 2, or PI3K, was also normal (supplemental Fig. S5b–d). It was intriguing that rapamycin strongly inhibited T cell proliferation despite activated Akt or PKC-θ expression, but also impacted Th1 and Th2 cell frequencies among the hypo-proliferative cells (supplemental Fig. S5e–i). This suggests mTORC1 may also relay signals promoting these states. The results collectively show that mTORC2 drives Th1 cell differentiation via Akt, whereas PKC-θ serves as the relay for the Th2 cell fate, with each of these kinases exerting key effects independent from the STAT pathway.
Figure 6.

mTORC2 exerts differential effects on downstream targets while sparing Stat protein tyrosyl phosphorylation. (A) Normal induction of phosphotyrosyl STAT transcription factors in rictor cKO mice T cells. CD4+ T cells were treated 40 min with the indicated cytokine or left unstimulated, then analyzed by immunoblotting with anti–P-STAT6Y641 or P-STAT3Y705. (B) Naïve, freshly purified CD4+ T cells from WT and cKO mice were stimulated as in Fig. 1 and analyzed by Western blotting (one experiment representative of two replicates). (C) Due to limiting amounts of protein from naïve T cells, purified cells were activated, rested after growth in vitro, restimulated, and analyzed (as for B). The bar graph presents the result of quantitation of the signal for P-FoxO1 (one experiment representative of three replicates). (D) Impairment of the activation-induced increase in expression of galectin-3, a FoxO1-regulated gene. FACS profiles for galectin on freshly isolated and activated (αCD3, αCD28, and 10 ng/ml IL-4 for 2 d) CD4+ T cells in one experiment representative of two replicates, and a bar graph quantitating the results, are shown. Additional information is in supplemental Fig. S5.
Targets downstream from mTORC2 in T cells
The findings raised questions about how rictor affects biochemical targets of mTORC2, Akt, and PKC-θ to regulate the key transcriptional determinants of Th1 or Th2 cell fates. The Akt target FoxO1 represses cell cycling, enhances apoptosis, and has been identified as a regulator of T-bet and galectin-3, proteins that promote Th1 cell responses (Jiang et al., 2009; Ouyang et al., 2009). Consistent with the reductions in Akt activity and T-bet expression (Figs. 2C, D, & 5E, F), P-FoxO1 was decreased in cKO T cells though an analogous phosphorylation of GSK-3β was unaffected (Fig. 6B, C). There also was no significant decrease in phospho-S6K1 (Fig. 6C), a target of mTORC1 regulated indirectly by Akt. GSK-3β(p-S9) and mTORC1 are targeted by other kinases in addition to Akt so the results with GSK-3 and S6K1 may be due to functional redundancy. However, the findings show that mTORC2 relays input signals that are non-redundant for FoxO1 as an Akt target. Because TCR-induced P-FoxO1 as well as T-bet were decreased in cKO T cells, we also measured expression of galectin-3 and found it was reduced in cKO T cells (Fig. 6D).
Unlike the complete deletion in T cells (Kerdiles et al., 2009; Ouyang et al., 2009), decreased P-FoxO1 would only diminish repression by FoxO1, so some FoxO1 target genes might not be impacted by mTORC2 dysfunction. Indeed, mRNA expression was normal for the FoxO1 target IL-7Ra (supplemental Fig. S6a). Recent work on CD8+ T cells reported that PI3K regulates CD62L transcription by a rapamycin-sensitive pathway involving regulation of Klf2 (Sinclair et al., 2008). Although there was no difference in CD62L expression at the surface of freshly isolated CD4+ T cells, we observed marked reductions in the frequency CD62Llo CD4+ (Fig. 7A) and CD8+ (supplemental Fig. S6b) cells after activation of cKO T cells. However, neither Klf2 nor Sell (encoding CD62L) mRNA expression was significantly altered in CD4+ cKO cells (supplemental Fig. S6a). Thus, mTORC2 impacts CD62L down-regulation after T cell activation by a non-transcriptional mechanism. Apart from this identification of a role distinct from the rapamycin-inhibited pathway, the data show that mTORC2 transduces signals controlling mRNA expression for known FoxO1-regulated genes involved in Th1 cell differentiation.
Figure 7.

mTORC2 regulation of NF-κB activity via PKC. (A) Regulation of CD62L expression on T cells. Shown are FACS profiles of CD62L and CD44 expression on freshly isolated or activated (as in Fig. 4) CD4+ T cells (one experiment representative of four replicates). Inset numbers: frequencies (%) in each quadrant. (B) Rictor dependence of nuclear NF-κB subunits and the NF-κB-regulated protein Bcl-3 after TCR-CD28 costimulation. CD4+ T cells were activated and a portion of each was re-stimulated with αCD3 and αCD28 as in Fig. 6. After separation of nuclear (N) and cytosolic (C) fractions and resolution by SDS-PAGE, immunoblots were probed for the indicated species (one experiment representative of 2–3 replicates). Inset numbers: relative signals for each protein in the nuclei after normalization to lamin-A, with the nuclei of resting, previously activated WT cells set at 1. (C, D) PKC-θ reverses a defect mTORC2-deficient T cells in TCR-CD28 costimulatory induction of NF-κB transcriptional activity. (C) Activated CD4+ T cells were transfected with the RE/AP-luciferase reporter construct along with a constitutively active Renilla luciferase, and restimulated with αCD3 and αCD28. Relative activity: firefly luciferase measured 6 h after restimulation and normalized for transfection efficiency (one experiment representative of three replicates). (D) As in (C) except that cells also were co-transfected with empty vector or vector encoding the kinase-active PKC-θ mutant and the data are pooled from two independent replicates. (E) Decreased expression of Bcl-3. As in (A) except that the overall level of Bcl-3 was analyzed using whole-cell extracts. (F) mTORC2 promotes TCR-CD28-induction of ICAM-1 avidity. Using lymph node T cells ± stimulation with αCD3 and αCD28, activation- and avidity-dependent binding of Fc-ICAM-1-αFc complexes to CD4+ T cells was measured by flow cytometry. The frequencies (%) of ICAM-1-binding cells within the CD4+ gate are indicated by inset numbers, with results from two independent experiments summarized (right). Additional information is in supplemental Fig. S6.
mTORC2 phosphorylates PKC-α and regulates 3T3 and HeLa cell cytoskeletons (Jacinto et al., 2004; Sarbassov et al., 2004), but no role in T cells is known. Phosphorylation of PKC HM and PKC-θ TM - direct targets of mTORC2 - also decreased in cKO T cells (Fig. 6B, C). PKC-θ is essential for coupling TCR and CD28 costimulation to nuclear amounts of NF-κB and Th2 cell differentiation (Cannons et al., 2004; Marsland et al., 2004). Importantly, TCR-stimulated NF-κB amounts in nuclei of rictor-deficient CD4+ T cells were substantially decreased (Fig. 7B). Moreover, TCR induction of a Carma1-dependent NF-κB-driven promoter was decreased in CD4+ cKO cells (Fig. 7C), whereas transfection of cKO CD4+ T cells with the PKC-θ construct restored promoter activity to the amount of WT cells transfected with empty vector (Fig 7D). These data indicate that PKC-dependent NF-κB activity downstream from the TCR depends on mTORC2. Of note, in addition to decreased NF-κB transcription factors in the nuclei of cKO CD4+ T cells, Bcl-3 was also diminished (Fig. 7E). This atypical IκB-like protein converts NF-κB1-p50 homodimers to transcriptionally active complexes, so that GATA3 and Th2 cell differentiation are decreased in Bcl-3-deficient T cells as well as NF-κB1-p50-null cells (Corn et al., 2005). Consistent with this, decreased GATA-3 expression in mTORC2-deficient developing Th2 cells were restored by activated PKC-θ (Fig. 5F, H). PKC-θ also promotes increased binding of the integrin LFA-1 to ICAM-1 (Letschka et al., 2008). Consistent with a functional impact of mTORC2 on the PKC pathway in T cells, the activation-induced increase in LFA-1 binding to ICAM-1 was impaired in cKO T cells (Fig. 7F). Taken together with the data on Akt-targeted FoxO1 and reversion results, the findings place Akt and PKC functions downstream from mTORC2 in T cells. Further, the data provide evidence of a functional dichotomy in helper T cell differentiation, such that Akt mediates mTORC2 regulation of Th1 cell development and PKC that in the Th2 cell direction.
Discussion
We have shown that mTORC2 selectively promotes subsets of helper T cell differentiation and restrains acquisition of FoxP3 expression, but a defect of mTORC2 does not divert differentiating populations into a Treg cell-like fate. Thus, Ag-specific antibody isotypes driven by type 1 and type 2 help decreased in cKO mice, and proliferation-independent analyses revealed blocks to Th1 and Th2 but not Th17 cell differentiation. Dissection of mTORC2 effects on differentiation, survival, and cell cycling provided evidence of an unexpected hierarchy in the roles of this complex. T cell survival was unaffected and proliferation decreased only modestly despite decreased Akt activity, yet the effects on differentiation were substantial but selective. TCR-CD28-induced increases in phosphorylation of both Akt S473, a direct target of mTORC2, and the T308 residue, which is the most critical determinant of Akt activation by PDK1, were attenuated in cKO T cells. In addition, the defect of mTORC2 also weakened functional signaling to PKC. This biochemical bifurcation downstream from mTORC2 was reflected in function. Thus, active Akt sufficed to restore expression of Tbx21, a FoxO1-regulated gene, but not Gata3, and a normal efficiency of Th1 but not Th2 cell differentiation. In contrast, PKC-θ yielded the reciprocal result. We conclude that the dominant function of mTORC2 under physiological conditions in T cells is to guide differentiation mediated dichotomously by the mTORC2 targets Akt and PKC.
TOR was identified as the key target of a clinically important immune suppressant drug rapamycin. Attention focused on the capacity of this agent to inhibit proliferation and promote apoptosis, but rapamycin was later shown to block Th1 cell development, inhibit Th2 and Th17 cell differentiation, and enhance the population of Treg cells in Th17 cultures (Blazar et al., 1998; Kopf et al., 2007; Valmori et al., 2006). Loss-of-function for mTOR in T cells has been reported to mimic the effects of rapamycin on T helper proliferation and differentiation, and to divert CD4+ T cells under helper-differentiating conditions into FoxP3+ cells (Delgoffe et al., 2009). Signals that can reverse these abnormalities are not known. The lack of mTOR was reported to cause potentially meaningful decreases of IL-4-induced STAT6 and IL-6-induced STAT3. It is intriguing to compare these findings to those in the rictor-deficient state. Rictor cKO T cells exhibited defects primarily of Th1 and Th2 cell differentiation and were not diverted into FoxP3+ status under any condition promoting T helper effector differentiation, although the prevalence of iTreg cell increased in response to TGF-β under otherwise neutral conditions. Moreover, no defect of STAT phosphorylation was detectable in either rictor-deficient T cells or those inhibited for PI3K or mTOR. Collectively, these data provide direct evidence that a rictor-deficient state leads to attenuation of Th1 and Th2 cell differentiation without any diversion into iTreg cell or apparent abnormality of STAT protein induction by tyrosine phosphorylation. Instead, mTORC2 targets Akt and PKC, and the activated kinases suffice to restore normal differentiation to rictor cKO T cells.
Comparison of these results for mTORC2 and mTOR deficiencies might suggest that the differences should be attributable to mTORC1. This model is made more attractive because, unlike the observation with splenocytes of rapamycin-treated mice, prolonged rapamycin treatment of CD4+ T cells eliminated Akt(HM) phosphorylation. Thus, mTORC1 might promote Th17 cell differentiation, repression of FoxP3 and the Treg cell state, and perhaps the capacity of cytokines to induce tyrosine phosphorylation of STAT transcription factors. However, rapamycin blocks mTOR activity without inhibiting IL-4-induced STAT6 phosphorylation, and even substantial reductions in P-STAT6 only minimally impair Th2 cell differentiation (Mora et al., 2003; Wang et al., 1997). Moreover, almost all signaling proteins ultimately prove to serve in several heterologous complexes and have multiple signal inputs. Although an alternative signaling complex (McDonald et al., 2008) including rictor might contribute to results reported with mTOR cKO T cells when mTOR is depleted, the molecular epistasis results with active Akt and PKC-θ show that targets of mTORC2 suffice to revert the main defects in T helper differentiation if mTORC1 is active. Because Akt can liberate Rheb, a signal-transducing GTPase and activator of mTORC1, from repression by a Tsc1/Tsc2 complex (Inoki et al., 2002), the attenuated Akt activation observed in rictor cKO T cells might suggest a complex circuit involving mTORC1 in the data. Consistent with rictor or SIN1-null MEFs, however, we observed normal activity of mTORC1 at least at the target kinase S6K1. Moreover, T cell-specific Rheb depletion did not phenocopy loss of mTOR in terms of repressing FoxP3 (other components of the Rheb-deficient phenotype are unknown) (Delgoffe et al., 2009). Analysis of mTORC2 inactivation along with the Rheb data suggests intriguing possibilities. These include that the threshold for mTORC1 to repress FoxP3 is very low, so that PI3K and Rheb-independent mTORC1 activation contributes to differences (Gwinn et al., 2008; Ha et al., 2006). Alternatively, Rheb may function in a signaling pathway that impacts FoxP3 in an mTORC1-independent manner, or each complex (mTORC1, mTORC2) may independently mediate FoxP3 repression. Thus, while our data directly show that mTORC2 function is vital for Th1 and Th2 differentiation, mTORC1 is likely independently to contribute to these processes.
Notwithstanding these issues pertaining to mTORC1, our data underscore new features of mTOR in T cells. These include mTORC2 as the predominant PDK2 activity driving costimulation-enhanced HM phosphorylation and Akt activity, and the impact of this pathway on T-bet expression and Th1 cell differentiation. Intriguingly, measurements comparing WT, heterozygous (Cre+, Rictorfl/+), and homozygous cKO T cells found that Th2 cell differentiation was strongly reduced at the intermediate expression of mTORC2 whereas Th1 cell differentiation of heterozygous cells was almost normal (data not shown). Thus, mTORC2 may function as a scalar regulator of the balance among T helper subsets so that when Akt activity is less affected, Th1 cell differentiation is preserved. The findings also provide evidence of an Akt-independent, PKC-dependent role for rictor in T helper differentiation.
The cytoplasmic tail of CD28 both stimulates PI3K activity by collaboration of two separable transducer motifs and enhances recruitment of both PDK1 and PKC-θ into the immunological synapse (Andres et al., 2004). The requirements for rictor in T helper differentiation is similar to one aspect of signaling downstream from CD28 in that Myr-Akt reversed only the impairment of Th1, but not Th2 cell, differentiation observed when CD28 was absent (Kane et al., 2001). In terms of Th2 cell differentiation, both CD28 and PKC-θ are vital determinants of the efficiency of Th2 cell differentiation (Andres et al., 2004). The finding that rictor-deficient cells have reduced TCR-induced NF-κB activity, GATA3, and Th2 cell differentiation reversible by constitutively active PKC-θ suggests mTORC2 is a critical link connecting costimulation to PKC-θ and PI3K. Further, our data suggest that this role connects to a well-established pathway in which PKC-θ activates NF-κB after TCR-CD28 stimulation by mobilizing a Carma1, Bcl-10, MALT-1 signaling complex.
Expression of c-Maf, a potent regulator of Il4 transcription in Th2 cell responses, was also rictor-dependent. Vav1 couples the TCR pathway to Maf (encoding c-Maf) (Tanaka et al., 2005), but PI3K-PKB regulation is normal in Vav-deficient T cells (Wood et al., 2006). IL-6 collaborates with this TCR signal to induce c-Maf via STAT3 (Yang et al., 2005). However, rictor cKO cells had normal STAT3 induction and were reverted by PKC-θ. Thus, mTORC2 likely relays signals essential for c-Maf induction by a pathway that collaborates with but is parallel to these previously established mechanisms. More generally, the data indicate that loss of mTORC2 impairs a subset of biological targets of mTOR in T cells and acts via protein kinases B or C without an impact on cytokine-induced Stat protein tyrosine phosphorylation. This selectivity suggests the possibility of alternate therapeutic windows for agents that would impair mTORC2 function, for disease states in which complete mTOR inhibition might add toxicities but not benefits.
Experimental Procedures
Mice, antibodies and reagents
Rictorfl/fl mice were bred to a B6 background. For T lineage-specific deletion, Rictorfl/fl mice were crossed with transgenic mice expressing codon-optimizedCre recombinase (iCre) under control of the distal promoter of Lck gene (Zhang et al., 2005). Mice were maintained using ventilated micro-isolator cages in specified pathogen-free conditions and used in accordance with IACUC regulations of Vanderbilt University. Sources of chemical and biological reagents (cytokines, antibodies) detailed in Supplemental Experimental Procedures.
Cell culture and retroviral transduction
CD4+ cells were purified by positive selection using αCD4-conjugated magnetic beads (Miltyeni Biotec). Lymphoid cells were cultured, proliferation assays were performed, and virions were generated after transfection of ΦNX ecotropic packaging cells with constructs of Akt(DD) and Akt(DA) mutants (T308D, S473D or A), Myr-Akt, and PKC-θ in MSCV-IRES-GFP and -Thy1.1 (MiG RV, MiT RV, respectively) as described (Corn et al., 2003). Activated CD4 T cells were ‘spinfected’ and then cultured in Th1 or Th2 conditions. For in vitro T helper differentiation, lymph node cells were activated with plate-bound αCD3 plus soluble αCD28 and cultured with IL-2. Subset-specific culture additions were: anti-IL-4 and IL-12 (Th1 culture), αIL-12, αIFN-γ, and mIL-4 (Th2 culture), or TGF-β, IL6, IL-23, and αIL-4, αIL-12, and αIFN-γ (Th17 conditions). After 5 d, cells were re-stimulated with plate-bound αCD3 and soluble αCD28 (0.5 µg/ml each), stained for CD4, intracellular IFN-γ and IL-4 as described (Corn et al., 2003), or restimulated to yield culture supernatants and measure cytokine production by ELISA. Details are tabulated in Supplemental Experimental Procedures.
Transfection and luciferase assays
Activated T cells (4 × 106 /sample) were rested in IL-2-free medium for 5 h, and co-transfected with 20 µg of RE/AP reporter DNA, 5 µg of TK-pRL (Promega), and, where indicated, 10 µg of MSCV or MSCV-PKC- θ -KA using the Mouse T cells Nucleofector kit (Amaxa). Transfectants were returned to culture 4 h, restimulated (αCD3 and αCD28, 1 µg/ml each) for 6 h. Luciferase assays were performed using the Dual-luciferase kit (Promega). NF-κB-driven firefly luciferase activity was determined using a luminometer and normalized to Renilla luciferase activity.
Immunoblotting and Akt In vitro kinase assay
Proteins in cell extracts were prepared and analyzed as described (Mora et al., 2003) using appropriate IR dye-conjugated second Abs and laser excitation/quantitative fluorescence detection [Odyssey Infrared Imaging System (Li-Cor)]. To analyze cytosolic and nuclear proteins, previously activated T cells were washed and fractionated into cytosolic and nuclear portions as described (Sun et al., 2000). To measure Akt activity, αAkt immunoprecipitates of lysate proteins were used for in vitro kinase assays of GSK-3α/β (S21/S9) phosphorylation following kit instructions (Cell Signaling Technology). For each sample, Akt kinase activity determined by Western blotting was normalized to the Akt in immune complexes.
Flow cytometry, cell division and TUNEL assays
Cells were stained with Ab, or stained, fixed, permeabilized, and then further processed for detection of intracellular epitopes, all as described (Corn et al., 2003). To measure divisions, cells were fluorescein-labeled using CFSE (1 µM, 5 min), activated as described for TdR incorporation, cultured 48 h, stained with anti-CD4 and anti-CD8, and analyzed by FACS. S phase entry rates were analyzed 24 hr after activation by plate-bound αCD3 (1 µg/ml), soluble αCD28 (1 µg/ml), labeling with BrdU (6 h), staining CD4 and CD8, and processing for DNAse I digestion, αBrdU staining, and flow cytometry analysis as described (Corn et al, 2003). To assay apoptosis, cells cultured in the presence or absence of 10 ng/ml IL-4 for 20 h, with or without irradiation (2 Gy) in a 137Cs irradiator, were stained for surface markers, subjected to TUNEL assays, and analyzed by flow cytometry in comparison to control reactions lacking TdT.
Immunizations, ELISA, and ELISPOT assays
Mice were injected intraperitoneally (i.p.) with 20 µg KLH in incomplete Freund’s adjuvant (IFA) and boosted (20 µg KLH in IFA) 2 wk later. Sera collected 5 d after the boost (19 d after primary immunization) were analyzed by isotype-specific capture ELISA. For IgG1 and IgE responses, mice were immunized twice with 20 µg low-endotoxin ovalbumin (Hyglos GmbH, Germany) in alum (Imject, Pierce) at a 1 wk interval. Sera collected 19 d after the first immunization were analyzed for ovalbumin-specific IgG, IgG1, and IgE by capture ELISA. Alternatively, mice were immunized with Listeria monocytogenes (5 × 104 colony-forming units i.v.), and analyzed for IFN- γ expression by FACS and ELISPOT assays 6 d post-infection. ELISPOT assays from two independent experiments with n=8 (WT) and 6 (cKO) mice were performed by stimulating portions of the same experimental samples with 10 nM and 100 nM of the class II MHC-restricted LLO190–201 peptide.
Ligand-complex-based adhesion assay
Avidity-dependent assays of ICAM-1 binding were performed as described (Konstandin et al., 2007) with minor modifications. ICAM-1-human Fc (Fc-ICAM-1, R&D Systems) was incubated with biotinylated anti-human IgG Fc (eBioscience) in PBS at 4°C. Freshly isolated LN cells were incubate d with Fc-ICAM-1-αIgG Fc complexes in the presence or absence of 10 µg/ml anti-CD3 and 2 µg/ml anti-CD28 for 20 min, washed with PBS, fixed with 4% paraformaldehyde, and stained with streptavidin-PerCP and CD4-APC.
Supplementary Material
Acknowledgements
This work was supported by NIH R01AI068149 and Vanderbilt Institutional Bridge Funds; additional acknowledgements are in included in Supplemental Information.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andres P, Howland K, Nirula A, Kane L, Barron L, Dresnek D, Sadra A, Imboden J, Weiss A, Abbas AK. Distinct regions in the CD28 cytoplasmic domain are required for T helper type 2 differentiation. Nat Immunol. 2004;5:435–442. doi: 10.1038/ni1044. [DOI] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- Blazar B, Taylor P, Panoskaltsis-Mortari A, Vallera D. Rapamycin inhibits the generation of graft-versus-host disease- and graft-versus-leukemia-causing T cells by interfering with the production of Th1 or Th1 cytotoxic cytokines. J Immunol. 1998;160:5355–5365. [PubMed] [Google Scholar]
- Cannons JL, Yu L, Hill B, Mijares L, Dombroski D, Nichols KE, Antonellis A, Koretzky G, Gardner K, Schwartzberg PL. SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity. 2004;21:693–706. doi: 10.1016/j.immuni.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Carriere A, Cargnello M, Julien L, Gao H, Bonneil E, Thibault P, Roux P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol. 2008;18:1269–1277. doi: 10.1016/j.cub.2008.07.078. [DOI] [PubMed] [Google Scholar]
- Chan TO, Tsichlis P. PDK2: a complex tail in one Akt. Sci STKE. 2001;2001:PE1. doi: 10.1126/stke.2001.66.pe1. [DOI] [PubMed] [Google Scholar]
- Constant S, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- Corn R, Aronica M, Zhang F, Tong Y, Stanley S, Kim SR, Stephenson L, Enerson B, McCarthy S, Mora A, Boothby M. T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J Immunol. 2003;171:1816–1824. doi: 10.4049/jimmunol.171.4.1816. [DOI] [PubMed] [Google Scholar]
- Corn R, Hunter C, Liou HC, Siebenlist U, Boothby M. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci USA. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe G, Kole TP, Zheng Y, Zarek P, Matthews K, Xiao B, Worley P, Kozma S, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Liu F. PDK2: the missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol. Endocrinol. Metab. 2005;289:E187–E196. doi: 10.1152/ajpendo.00011.2005. [DOI] [PubMed] [Google Scholar]
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton A, Mao Y, Miao R, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- Guertin D, Stevens D, Thoreen C, Burds A, Kalaany N, Moffat J, Brown M, Fitzgerald K, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Gwinn D, Shackelford D, Egan D, Mihaylova M, Mery A, Vasquez D, Turk B, Shaw R. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SH, Kim DH, Kim IS, Kim JH, Lee MN, Lee HJ, Jang SK, Suh PG, Ryu SH. PLD2 forms a functional complex with mTOR/raptor to transduce mitogenic signals. Cell Signal. 2006;18:2283–2291. doi: 10.1016/j.cellsig.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton H, Alessi D, Cantrell DA. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat Immunol. 2004;5:539–545. doi: 10.1038/ni1062. [DOI] [PubMed] [Google Scholar]
- Huang J, Lo P, Zal T, Gascoigne N, Smith B, Levin S, Grey HM. CD28 plays a critical role in the segregation of PKC theta within the immunologic synapse. Proc Natl Acad Sci USA. 2002;99:9369–9373. doi: 10.1073/pnas.142298399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg M, Hall A, Hall M. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jiang H, Al Rasebi Z, Mensah-Brown E, Shahin A, Xu D, Goodyear C, Fukada S, Liu F, Liew F, Lukic M. Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J Immunol. 2009;182:1167–1173. doi: 10.4049/jimmunol.182.2.1167. [DOI] [PubMed] [Google Scholar]
- Juntilla M, Wofford J, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proc Natl Acad Sci USA. 2007;104:12105–12110. doi: 10.1073/pnas.0705285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane L, Andres P, Howland K, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2:37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- Kane L, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- Konstandin M, Wabnitz G, Aksoy H, Kirchgessner H, Dengler T, Samstag Y. A sensitive assay for the quantification of integrin-mediated adhesiveness of human stem cells and leukocyte subpopulations in whole blood. J Immunol Methods. 2007;327:30–39. doi: 10.1016/j.jim.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Kopf H, de la Rosa G, Howard O, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819–1824. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchroo V, Das M, Brown J, Ranger A, Zamvil S, Sobel R, Weiner H, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Lane P. Role of OX40 signals in coordinating CD4 T cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J Exp Med. 2000;191:201–206. doi: 10.1084/jem.191.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letschka T, Kollmann V, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Obermair G, Fresser F, Leitges M, Hermann-Kleiter N, Kaminski S, Baier G. PKC-θ selectively controls the adhesion-stimulating molecule Rap1. Blood. 2008;112:4617–4627. doi: 10.1182/blood-2007-11-121111. [DOI] [PubMed] [Google Scholar]
- Lin X, O'Mahony A, Mu Y, Geleziunas R, Greene WC. Protein kinase C-θ participates in NF-κB activation induced by CD3–CD28 costimulation through selective activation of IκB kinase beta. Mol Cell Biol. 2000;20:2933–2940. doi: 10.1128/mcb.20.8.2933-2940.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Graham C, Parravicini V, Brown M, Rivera J, Shaw S. Protein kinase C θ is expressed in mast cells and is functionally involved in Fcε receptor I signaling. J Leukoc Biol. 2001;69:831–840. [PubMed] [Google Scholar]
- Locksley RM. Nine lives: plasticity among T helper cell subsets. J Exp Med. 2009;206:1643–1646. doi: 10.1084/jem.20091442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;127:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsland B, Soos T, Spath G, Littman DR, Kopf M. Protein kinase C θ is critical for the development of in vivo T helper (Th)2 cell but not Th1 cell responses. J Exp Med. 2004;200:181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald P, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell E, Bally M, Foster L, Dedhar S. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008;68:1618–1624. doi: 10.1158/0008-5472.CAN-07-5869. [DOI] [PubMed] [Google Scholar]
- Mora A, Stephenson L, Enerson B, Youn J, Keegan AD, Boothby M. New programming of IL-4 receptor signal transduction in activated T cells: Stat6 induction and Th2 differentiation mediated by IL-4Rα lacking cytoplasmic tyrosines. J Immunol. 2003;171:1891–1900. doi: 10.4049/jimmunol.171.4.1891. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali S, Kim D, Guertin D, Latek R, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali S, Sengupta S, Sheen J, Hsu P, Bagley A, Markhard A, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin D, Ali S, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Scheid M, Marignani P, Woodgett J. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota C, Woo J, Lindner J, Shelton K, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Sinclair L, Finlay D, Feijoo C, Cornish G, Gray A, Ager A, Okkenhaug K, Hagenbeek T, Spits H, Cantrell DA. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Arendt C, Ellmeier W, Schaeffer E, Sunshine M, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR. PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, So T, Lebedeva S, Croft M, Altman A. Impaired IL-4 and c-Maf expression and enhanced Th1-cell development in Vav1-deficient mice. Blood. 2005;106:1286–1295. doi: 10.1182/blood-2004-10-4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valmori D, Tosello V, Souleimanian N, Godefroy E, Scotto L, Wang Y, Ayyoub M. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J Immunol. 2006;177:944–949. doi: 10.4049/jimmunol.177.2.944. [DOI] [PubMed] [Google Scholar]
- Wang D, Matsumoto R, You Y, Che T, Lin X, Gaffen S, Lin X. CD3/CD28 costimulation-induced NF-κB activation is mediated by recruitment of protein kinase C-θ, Bcl10, and IκB kinase β to the immunological synapse through CARMA1. Mol Cell Biol. 2004;24:164–171. doi: 10.1128/MCB.24.1.164-171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zamorano J, Keegan AD, Boothby M. HMG-I(Y) phosphorylation status as a nuclear target regulated through insulin receptor substrate-1 and the I4R motif of the interleukin-4 receptor. J Biol Chem. 1997;272:25083–25090. doi: 10.1074/jbc.272.40.25083. [DOI] [PubMed] [Google Scholar]
- Wood J, Schneider H, Rudd C. TcR and TcR-CD28 engagement of protein kinase B (PKB/AKT) and glycogen synthase kinase-3 (GSK-3) operates independently of guanine nucleotide exchange factor VAV-1. J Biol Chem. 2006;281:32385–32394. doi: 10.1074/jbc.M604878200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ochando J, Yopp A, Bromberg J, Ding Y. IL-6 plays a unique role in initiating c-Maf expression during early stage of CD4 T cell activation. J Immunol. 2005;174:2720–2729. doi: 10.4049/jimmunol.174.5.2720. [DOI] [PubMed] [Google Scholar]
- Zhang D, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart A, Mao X, Killeen N. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.