

SUMMARY
The genome of Streptococcus pneumoniae strains, as typified by the TIGR4 strain, contains several genes encoding proteins putatively involved in α-glucan degradation, modification and synthesis. The extracellular components comprise an ABC-transporter with its solute-binding protein, MalX, and the hydrolytic enzyme SpuA. We show that of the commonly occurring exogenous α-glucans, S. pneumoniae TIGR4 is only able to grow on glycogen in a MalX and SpuA-dependent manner. SpuA is able to degrade glycogen into a ladder of α-1,4-glucooligosaccharides while the high affinity interaction (Ka ~ 106 M−1) of MalX with maltooligosaccharides plays a key role in promoting the selective uptake of the glycogen degradation products that are produced by SpuA. The X-ray crystallographic analyses of apo- and complexed MalX illuminate the protein’s specificity for the degradation products of glycogen and its striking ability to recognize the helical structure of the ligand. Overall, the results of this work provide new structural and functional insight into streptococcal α-glucan metabolism while supplying biochemical support for the hypothesis that the substrate of the S. pneumoniae α-glucan metabolizing machinery is glycogen, which in a human host is abundant lung epithelial cells, a common target for invasive S. pneumoniae.
Keywords: Streptococcus pneumoniae, crystal structure, solute-binding protein, virulence, α-glucan, α-glucanase
INTRODUCTION
Streptococcus pneumoniae is a Gram-positive bacterium that frequently asymptomatically colonizes the nasopharynx of humans. Given the appropriate circumstances, however, this bacterium can become an aggressive pathogen causing a wide variety of diseases including serious invasive infections such as pneumonia and meningitis (Kadioglu & Andrew, 2004). The frequency and severity of S. pneumoniae infections have made it the subject of intense studies aimed at identifying bacterial and host factors that contribute to the prowess of this organism as a pathogen. Several studies, including large-scale signature tagged mutagenesis experiments (Hava & Camilli, 2002, Chen et al., 2007, Polissi et al., 1998, Lau et al., 2001), have revealed a large number of virulence factors, which, surprisingly, have known or predicted roles in carbohydrate metabolism. Among the known ones are the neuraminidases and hyaluronate lyase, which have been well-studied (Jedrzejas, 2007); however, the vast majority of these streptococcal carbohydrate-active virulence factors remain unexamined.
Among the emerging streptococcal virulence factors involved in carbohydrate metabolism are a group of proteins that are putatively involved in α-glucan degradation and synthesis. α-glucans are a group of polysaccharides, for example starch (a composite of amylose and amylopectin) and glycogen, that are defined by their predominant composition of α-linked glucose. Examination of the S. pneumoniae genome, which contains up to 11 genes encoding proteins involved in the breakdown and/or biosynthesis of α-glucans and an additional 5 genes encoding regulatory and transport proteins related to these metabolic functions, indicates the capacity for this bacterium to sense, respond to and metabolize α-glucans (Table 1). Six of these genes have been identified by signature-tagged mutagenesis to be necessary for the full virulence of S. pneumoniae (Hava & Camilli, 2002) (Table 1). The importance of α-glucan utilization pathways to bacterial virulence is somewhat surprising considering that the evolutionary retention of α-glucan metabolizing machinery has been linked to a “free-living” bacterial lifestyle, rather than one of commensalism and/or pathogenesis (Henrissat et al., 2002). Recent studies of α-glucan utilization in S. pyogenes (group A streptococcus or GAS) is beginning to provide insight into this apparent inconsistency. Microarray analysis of the S. pyogenes transcriptome has indicated that α-glucan utilization occurs in deep infections of soft tissue (Graham et al., 2006). Subsequent studies have specifically shown that the maltooligosaccharide specific solute-binding protein, MalE, and maltose/maltotriose specific phosphotransferase system, MalT, enable colonization of the oropharynx through their niche-specific role in the utilization of dietary starch (Shelburne et al., 2007, Shelburne et al., 2008, Shelburne et al., 2006). More strikingly, a recent study has shown that a GAS cyclodextrin glucanotransferase produces extracellular cyclodextrins from exogenous starch, which facilitates host epithelial cell invasion by a process that does not appear to be linked to energy liberation(Shelburne et al., 2009). Despite these advances, beyond an appreciation of the genes in the pneumococcal genome that encode proteins putatively involved in α-glucan metabolism, few details have been confirmed regarding the metabolism of this class of polysaccharide by S. pneumoniae and even less is understood about the apparent involvement of this metabolic process in pneumococcal virulence.
Table 1.
Proteins encoded by the Streptococcus pneumoniae TIGR4 genome that are putatively involved in α-glucan metabolism.
| Protein Name | Locus Tag | CAZy Familya | Cellular location (PSORTf) | Putative virulence factord | Putative activity |
|---|---|---|---|---|---|
| MalA | SP2111 | N/A | Cytoplasm | Unknown | |
| MalR | SP2112 | N/A | Cytoplasm | Negative regulator | |
| MalX | SP2108 | N/A | Membrane/cell wall | ✓ | Solute binding |
| MalC | SP2109 | N/A | Membrane | ABC transporter - permease | |
| MalD | SP2110 | N/A | Membrane | ABC transporter - permease | |
| MalP | SP2106 | GT35b | Cytoplasm | Glycogen phophorylase | |
| MalM | SP2107 | GH77c | Cytoplasm | 4-α-glucanotransferase | |
| GlgA | SP1124 | GT5 | Cytoplasm | Glycogen synthase | |
| SpuA | SP0268 | GH13(12)e | Membrane/cell wall | ✓ | pullulanase |
| SpAMY | SP1382 | GH13(5) | Cytoplasm | ✓ | α-amylase |
| AG13 | SP1883 | GH13(29) | Cytoplasm | ✓ | α-phosphotrehalase |
| Dex | SP0342 | GH13(31) | Cytoplasm | α-1,6-glucosidase | |
| Pul | SP1118 | GH13(14) | Cytoplasm | ✓ | pullulanase |
| SP | SP1894 | GH13(18) | Cytoplasm | Sucrose phosphorylase | |
| GT | SP1121 | GH13(9) | Cytoplasm | 1,4-α-glucan branching enzyme | |
| AG31 | SP0312 | GH31 | Cytoplasm | ✓ | α-glucosidase |
see URL: http://www.CAZY.org (Cantarel et al., 2008).
the prefix GT refers to the glycosyltransferase families
the prefix GH to the glycoside hydrolase families.
as identified by signature tagged mutagenesis (Hava & Camilli, 2002) and/or microarray analysis (Orihuela et al., 2004).
numbers in brackets refers to the subfamily of GH13, which was used to aid in the functional prediction (Stam et al., 2006).
Among the six putative pneumococcal virulence factors suggested to be involved in α-glucan degradation are SpuA, a cell-wall attached protein classified as a pullulanase, and MalX, the lipid-anchored solute-binding protein (SBP) of a maltooligosaccharide specific ATP-binding cassette (ABC) transporter. MalX and SpuA are particularly notable as they are the only extracellular components of the predicted pneumococcal α-glucan metabolism machinery, suggesting their importance in exogenous α-glucan metabolism (Table 1). The genetic structure of S. pneumoniae’s maltodextrin utilization operon was described quite some time ago when it was found through genetic means that MalX was required for the bacterium’s growth on maltotetraose (Puyet & Espinosa, 1993) (Figure 1); however, this was not considered in the context of polysaccharide metabolism. SpuA (S. pneumoniae pullulanase A) was more recently identified through screening a genome-based library against convalescent-phase human serum and confirmed as being an immunogenic cell-surface attached protein with activity on the bacterial α-glucan pullulan (Bongaerts et al., 2000). Though the α-glucanase activity of SpuA was conclusively demonstrated, pullulan is a biologically rare polysaccharide thus leaving the biological substrate of SpuA unknown. Towards identifying a biologically relevant substrate for SpuA, we recently dissected the modular architecture of SpuA’s 1280 amino acids to reveal a complex multimodular structure, which includes two family 41 carbohydrate-binding modules (CBMs) at its N-terminus, and a family 13 glycoside hydrolase catalytic module at its C-terminus (van Bueren et al., 2007, Boraston et al., 2004, Stam et al., 2006)(Figure 1). Significantly, the CBMs were demonstrated bind to glycogen stores in type II alveolar lung cells leading to our prediction that exogenous glycogen, possibly that in lung cells, is a relevant and viable target for S. pneumoniae’s α-glucan metabolising machinery (van Bueren et al., 2007). Should this be the case, the extracellular localization of SpuA and MalX leads to the expansion of this hypothesis to suggest that these two proteins will work in concert and be critical to the depolymerization of exogenous glycogen and the transport its breakdown products. In this work we provide compelling support for these hypotheses through functional analyses of purified recombinant SpuA and MalX, demonstration of their role in S. pneumoniae’s growth on glycogen, and the X-ray crystallographic analysis of the structure of MalX.
Figure 1.

Organization of the Mal operon (A) and the modular structure of SpuA (B). Large arrows represent genes and their direction of transcription in the Mal operon (B). Small arrows indicate the promoters and their directionality. The modules of SpuA are labelled indicating unknown modules (UNK), the family 41 carbohydrate-binding modules (CBM41-1 and CBM41-2), a linker module, and the N-, C-, and (α/β)8-barrel of the catalytic domain. The construct used for the work in this study is shown immediately below the full-length schematic.
RESULTS AND DISCUSSION
SpuA partly depolymerises glycogen
The qualitative analysis of SpuA’s activity on pullulan, a linear α-glucan comprising maltotriose (three α-1,4-glucose residues) units joined by α-1,6-glycosidic linkages, led to the postulation that this protein is active on the α-1,6-linkages in this polysaccharide (Bongaerts et al., 2000). More recently, the related pullulanase from S. agalactiae, SAP, was demonstrated to degrade pullulan into maltotriose by hydrolysis of the α-1,6-linkages (Santi et al., 2008). SAP was also able to depolymerize glycogen into an uncharacterized population of oligosaccharides. To better examine α-glucan degradation by SpuA we cloned and overexpressed a gene fragment encoding the two N-terminal CBMs, the linker module, and the catalytic module of SpuA (see Figure 1), and purified the protein product. Like SAP, SpuA hydrolyzed pullulan to generate a sugar product with a fluorophore-assisted carbohydrate electrophoresis (FACE) mobility (Figure 2A) and thin layer chromatography mobility (not shown) most consistent with maltotriose. No activity on dextran, a pure polymer of α-1,6-linked glucose, could be detected (not shown). Together, these results are consistent with selectivity for α-1,6-linkages but only in the context of stretches of α-1,4-linked glucoses. Pullulan, however, is a relatively rare fungal polysaccharide making it an unlikely biological substrate for streptococcal pullulanases. Thus, we examined the product profile of SpuA treated glycogen, a more relevant substrate as it is a common storage polysaccharide in many organisms including humans, by FACE and high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD), both of which showed the generation of an array of gluco-oligosaccharides having degrees of polymerization (DP) ranging from one (glucose) to over thirty (Figure 2B and C). Furthermore, the electrophoretic mobilities (FACE) and elution positions (HPAEC) of the low molecular weight oligosaccharides produced by the SpuA-catalyzed degradation of glycogen matched those of pure α-1,4-glucooligosaccharide standards with degrees of polymerization (DP) from 2 (maltose) to 8 (maltooctaose)(Figure 2B and C). No evidence of products containing α-1,6-linkages, which did show distinctive properties by HPAEC, could be detected. Given that glycogen comprises α-1,4-linked glucose chains with α-1,6-linked branches of these chains occurring every 8–12 glucose residues, this hydrolysis pattern is again most consistent with the SpuA-catalyzed hydrolysis of α-1,6-glycosidic linkages at the glycogen branch points.
Figure 2.

Product profiles of pullulan and glycogen hydrolyzed by SpuA. A) FACE analysis of product produced by SpuA treatment of pullulan. The lanes are as follows: 1, maltooligosaccharide standards (M2–M6 indicate maltose to maltohexaose, inclusively); 2 and 3, SpuA treated pullulan; 4, untreated pullulan. B) FACE analysis of product produced by SpuA treatment of glycogen. The lanes are as follows: 1, maltooligosaccharide standards; 2 untreated glycogen; and 3, SpuA treated glycogen. C) HPAEC-PAD analysis of glycogen treated by SpuA. The upper trace shows a mixture of sugar standards - the unique elution positions of the sugars were first determined by analysis of the individual sugars. The peak labels are as follows: G, glucose; I2, isomaltose; I3, isomaltotriose; M2-M8, maltose to maltooctaose; P, panose; and GM3M3; 63-α-D-glucosyl-maltotriosyl-maltotriose. The lower trace shows the product profile of SpuA treated glycogen. Untreated glycogen (not shown) showed only a peak corresponding to the small peak indicated in the lower trace of panel C by an asterisk (*). The peaks are labelled as in the upper trace according to the deduced identity of the sugars.
MalX interacts tightly with maltooligosaccharides
Though the genetic analysis of the pneumococcal maltodextrin utilization operon revealed the importance of MalX in maltodextrin utilization the direct interaction of this protein with sugars has not been demonstrated. To facilitate analysis of MalX the gene fragment encoding a soluble form of the protein was expressed in E. coli and the overproduced protein purified. The interaction of MalX with maltooligosaccharides was quantified by both UV difference binding analysis and isothermal titration calorimetry (Figure 3, Table 2). Initial UV difference scans revealed a complex UV difference profile comprising at least four peaks, consistent with a composite spectrum resulting from the involvement of both tryptophan residues and tyrosine residues in carbohydrate recognition by this protein (Boraston et al., 2001)(Figure 3A). Both methods gave experimental stoichiometries consistent with 1:1 binding and revealed relatively high-affinity interactions in the 1 × 105–106 M−1 range for linear oligosaccharides increasing in size from maltotriose to maltooctaose, all of which is in agreement with other maltodextrin binding proteins (Shelburne et al., 2007, Thomson et al., 1998)(Table 2). The protein’s affinity for maltose, which could only be approximated by UV difference due to very weak binding, was approximately 3 orders of magnitude weaker than for maltotriose. The very weak affinity of MalX for maltose suggests that this protein would not able to facilitate the transport this disaccharide. For linear maltooligosacchardes larger than maltose, the association constants were very similar with only a roughly 2-fold difference observed between the highest and lowest affinities. MalX also had a very low affinity for maltotetraitol, which contains a linearized and oxidized reducing end sugar, indicating that MalX is selective for maltooligosaccharides with an intact reducing end.
Figure 3.

Representative binding experiments showing the interaction MalX with maltotriose. A) The UV difference spectrum induced by the binding of maltotriose to MalX. Peaks and troughs in the spectrum are labelled. B) A representative isotherm of maltotriose to MalX produced by a UV difference titration. Each curve corresponds to a different peak-to-trough wavelength pair used to measure the change in UV absorbance upon ligand binding: circles, 287.1–282.7 nm pair; and squares, 295.3–290.0 nm pair. Solid lines show the fits to a one-site binding model accounting for ligand depletion. C) Representative isothermal titration calorimetry titration for maltotriose. The upper panel shows the unintegrated data corrected for baseline. The lower panel shows the integrated heats fit to a one-site binding model.
Table 2.
Quantitative analysis of MalX binding toα-glucooligosaccharides.
| Ligand | UV Difference (at 20°C) |
Isothermal titration calorimetry (at 25°C) |
||||||
|---|---|---|---|---|---|---|---|---|
| Ka (μM−1) | ΔG (kcal mole−1) | n | Ka (μM−1) | ΔG(kcal mole−1) | n | ΔH(kcal mole−1) | ΔS (cal mole−1 K−1) | |
| Maltose | ~0.0005 | ~−3.6 | 1* | ND | ND | ND | ND | ND |
| Maltotriose | 1.27 (±0.20) | −8.18 (±0.09) | 0.96 (±0.02) | 0.51 (±0.06) | −7.65 (±0.07) | 0.90 (±0.01) | −6.32 (±0.23) | 4.94 (± 0.93) |
| Maltotetraose | 0.47 (±0.09) | −7.62 (±0.10) | 1.00 (±0.06) | 0.28 (±0.03) | −7.31 (±0.06) | 1.06 (±0.10) | −4.86 (±0.47) | 8.65 (±1.40) |
| Maltopentaose | 0.47 (±0.09) | −7.60 (±0.11) | 1.02 (±0.00) | 0.29 (±0.01) | −7.33 (±0.02) | 1.02 (±0.01) | −4.27 (±0.09) | 10.70 (±0.30) |
| Maltohexaose | 0.83 (±0.08) | −7.94 (±0.05) | 0.90 (±0.04) | 0.38 (±0.00) | −7.48 (±0.01) | 1.01 (±0.01) | −5.14 (±0.15) | 8.26 (±0.51) |
| Maltoheptaose | 0.83 (±0.09) | −7.94 (±0.06) | 0.91 (±0.02) | 0.38 (±0.03) | −7.47 (±0.06) | 0.97 (±0.03) | −4.48 (±0.30) | 10.49 (±1.07) |
| Maltooctaose | 0.40 (±0.06) | −7.63 (±0.08) | 0.95 (±0.04) | ND | ND | ND | ND | ND |
| Maltotetraitol | 0.005 (±0.000) | −5.12 (0.02) | 1* | ND | ND | ND | ND | ND |
the stoichiometry (n) was set constant at one during the non-linear regression analysis of the data.
ND, not determined.
The poor binding to maltose but maximal affinity for maltotriose suggests the presence of three major subsites within the binding site of MalX that accommodate the three sugar units of the reducing end of the oligosaccharide. A comparison of the energetic components reveals there is a slight general trend of decreasing enthalpies with small favourable entropic compensations as the oligosaccharide length increases resulting in small overall changes in the free energy. This thermodynamic signature closely resembles that reported for S. pyogenes MalE (Shelburne et al., 2007) and shows that in the longer ligands sugar subunits positioned towards the non-reducing end do not contribute significantly to the binding energy. The binding of linear maltooligosaccharides to E. coli MalE had more variability for the same range with but maltotriose still displaying the strongest interaction (Quiocho et al., 1997).
SpuA and MalX are critical to growth on glycogen
The biochemical properties of SpuA and MalX in isolation are therefore consistent with their respective hypothesized functions in the depolymerization of glycogen and transport of the degradation products. At present, however, it is unknown if S. pneumoniae can catabolize glycogen or whether SpuA and MalX participate in this. Towards addressing these questions we initially assessed the ability of S. pneumoniae to grow in liquid culture on various highly polymerized α-glucans. As expected, S. pneumoniae grew well on control substrates, glucose and maltotriose, but virtually not at all in the absence of a carbohydrate source (Figure 4A). No growth was observed on components of starch: amylopectin (Figure 4A) and amylose (not shown, results identical to amylopectin). Significant growth was found on glycogen, though reduced in comparison to glucose, but surprisingly little growth was observed with pullulan as a substrate (Figure 4A). To determine the importance of SpuA and MalX to growth on glycogen, Δspua and Δmalx strains were created by replacing the respective genes in S. pneumoniae TIGR4 with a chlorampenicol resistance cassette. These strains grew readily on glucose and maltotriose but neither were able to grow on glycogen (Figure 4C and D). The addition of purified recombinant SpuA to the culture improved the ability of wild-type to grow on glycogen and restored the ability of the Δspua strain to grow on glycogen to a similar level as wild-type with added SpuA (Figure 4B and D). Thus, the α-glucan degrading machinery of S. pneumoniae is clearly selective for glycogen with SpuA and MalX having critical importance in the metabolism of this polysaccharide when it is supplied as an exogenous substrate.
Figure 4.

Growth of S. pneumoniae (TIGR4) on α-glucans. A) Growth of wild-type on various α-glucans. B) Growth of wild-type on glycogen. C) Growth of the Δmalx strain. D) Growth of the Δspua strain. In all panels, the symbol type represents different growth substrates: closed squares - glucose; open squares - glycogen; closed circles - no substrate; open circles - maltotriose; closed triangles - glycogen with added recombinant SpuA; open triangles - pullulan; open diamonds - amylopectin. In panels C and D the glycogen and no substrate curves overlap. Error bars represent the standard deviations measurements made from growth cultures performed in triplicate. Where error bars are not visible the error was within the size of the symbol.
MalX mediates the size-dependent selective uptake of maltooligosaccharides
The activity of SpuA, likely on the α-1,6-branch points of glycogen, results in a population of maltooligosaccharides with a wide array of sizes raising the question of whether all of these oligosaccharides or only a sub-population could be utilized by the bacterium. We addressed this question by the HPAEC-PAD analyses of the residual α-glucans remaining in the culture supernatants of the pneumococcus when grown on glycogen. Wild-type S. pneumoniae TIGR4 cultures grown with glycogen contained an array of α-glucooligosaccharides whose degrees of polymerization (DP), but not relative abundance, were consistent with the in vitro depolymerization of glycogen by purified SpuA (Figure 5A). The majority of the α-glucooligosaccharides were larger than eight sugar units in length with only small amounts of shorter oligosaccharides identified by their elution positions as maltohexaose, maltoheptaose, and maltooctaose (Figure 5A). As expected, the Δspua strain showed no soluble α-glucooligosaccharides in the culture supernatant (Figure 5A). Though it did not grow well on glycogen the Δmalx strain was still clearly able to depolymerize glycogen to some extent resulting in α-glucooligosaccharides increasing in size from maltotetraose to maltooctaose (identified by their elution position) and larger oligosaccharides (Figure 5A). In contrast to the wild-type cultures, the most abundant α-glucooligosaccharides produced by the Δmalx strain were maltopentaose, maltohexaose, and maltoheptaose. The differences in the relative abundances of residual α-glucooligosaccharides observed for the Δmalx and wild-type strains, as judged by peak areas, indicated that the wild-type strain is effective at almost completely depleting α-glucooligosaccharides up to eight glucose units in length (Figure 5A). Oligosaccharides of 9-11 glucose units in length were only partially depleted while the bacterium appeared to have little to no ability to deplete oligosaccharides longer than 11 glucose units in length.
Figure 5.

Analysis of maltooligosaccharides in the culture supernatants of S. pneumoniae grown in the presence of glycogen. A) Upper trace, oligosaccharides present in the culture supernatant of wild-type S. pneumoniae TIGR4 grown in ACGHY media with glycogen; middle trace, oligosaccharides present in the culture supernatant of the Δspua strain grown in the same medium; lower trace, oligosaccharides present in the culture supernatant of the Δmalx strain. B) Upper trace, ACGHY media containing glycogen, supplemented with recombinant SpuA (indicated as rSpuA) and incubated as for other cultures but not inoculated with bacteria. Second trace, oligosaccharides present in the culture supernatant of wild-type S. pneumoniae TIGR4 grown in ACGHY media with glycogen and supplemented with recombinant SpuA. Third trace, oligosaccharides present in the culture supernatant of Δspua grown in ACGHY media with glycogen and supplemented with recombinant SpuA. Lower trace, oligosaccharides present in the culture supernatant of the Δmalx strain grown in ACGHY media with glycogen and supplemented with recombinant SpuA. In all panels, oligosaccharide identities for maltooligosaccharides with degrees of polymerization from 2 to 8 were determined from comparison to known standards; these are labelled M2-M8. Oligosaccharides M9-M28 are inferred to be α-1,4-glucooligosaccharides with degrees of polymerization from 9 to 28.
To enhance our ability to observe the depletion of glucooligosaccharides from the culture we supplemented cultures with purified recombinant SpuA to enhance glycogen degradation. In all cases, glycogen was effectively depolymerized and yielded roughly 10-times more oligosaccharide, as approximated by HPAEC-PAD signal, than in the absence of exogenous SpuA (Figure 5B). Bacteria-free media samples containing glycogen with added recombinant SpuA yielded a pattern of oligosaccharide production identical to the in vitro activity of purified recombinant SpuA. Relative to the bacteria-free sample and the Δmalx culture, the wild-type strain and the Δspua strain, whose glycogen degrading capacity was restored by the exogenous addition of SpuA, completely depleted the culture of oligosaccharides smaller than nine glucose units in length and partially depleted those from ~9-11 glucose residues. This is consistent with the results observed without the exogenous addition of SpuA.
Curiously, the Δmalx strain was still able to grow effectively on maltotriose and its growth on glycogen was partially restored by the addition of exogenous purified SpuA (Figure 4C). Given that the mutant Δmalx strain is clearly compromised in its ability to transport α-glucooligosaccharides, the improved growth of this strain in the presence of added SpuA and its ability to grow on maltotriose was somewhat surprising. An explanation is suggested by the comparison of the pattern of glucooligosaccharides generated by purified recombinant SpuA in the absence of bacteria and in the presence of the Δmalx strain; the Δmalx strain appears to retain the capacity to deplete the culture supernatant of maltose and maltotriose, sugars that are produced in some abundance in media containing purified recombinant SpuA (Figure 5B). This is consistent with our observation that the Δmalx strain grew comparably to wild-type on maltotriose and suggests the presence of a second system that transports α-glucooligosaccharides smaller than maltotetraose. Indeed, a phosphoenolpyruvate-dependent phosphotransferase transporter system (PTS) performing exactly this function, MalT, has recently been identified in S. pyogenes (Shelburne et al., 2008). The genome of S. pneumoniae TIGR4 encodes a putative protein (locus tag SP_0758) with 70% amino acid sequence identity to MalT. Based on this similarity, we postulate SP_0758 to participate in maltose and maltotriose transport in S. pneumoniae and thus compensate for the low affinity of MalX for maltose. This hypothesis is given conceptual support by the S. pyogenes system (Shelburne et al., 2008) and also the presence of a similar maltose/maltodextrin transport system in Bacillus subtilis that incorporates a homologous PTS transporter specific for maltose and maltotriose while an ABC-transporter with a maltodextrin binding protein is specific for maltotriose and longer maltodextrins (Schonert et al., 2006).
S. pneumoniae is apparently unable to grow on amylopectin or amylose, which collectively comprise starch, indicating the bacterium’s inability to utilize exogenous α-glucans that are primarily composed of α-1,4-linked glucose. This is consistent with its lack of a predicted extracellular α-1,4-glucanase. S. pneumoniae is, however, clearly able to utilize exogenous glycogen as an energy source in a manner that is absolutely dependent upon the SpuA-catalyzed hydrolysis of α-1,6-branch points in this polysaccharide. By hydrolysis of this substrate, which has branch points on average every 8–12 glucose units, SpuA generates a relatively large amount of smaller maltodextrins. A subset of these maltodextins, DP 3–8, are apparently optimal ligands for MalX while those with DP 9-11 appear to be less optimal. Though amylopectin also has α-1,6-branches, these occur only roughly every 24–30 glucose residues, which would give SpuA hydrolysis products of a high DP that would not be ligands for MalX. Thus, SpuA’s specificity and MalX’s selectivity appear to create a system tailored for glycogen metabolism.
ABC-transporters, particularly the E. coli maltodextrin transporter as a model for carbohydrate specific transporters, are well understood (Oldham et al., 2007, Procko et al., 2009). On the strength of this knowledge, it should be noted that MalX possess a biological function that is dependently linked to other components of an archetypal ABC-transporter. Because MalX resides in a three gene operon where it is upstream of the two genes encoding the permease components of the ABC-transporter (Figure 1), we cannot formally rule out the possibility of a polar effect from replacement of the malx gene, which disrupts expression of the permease genes and thus contributes to the phenotype of the Δmalx strain. However, in light of a full understanding that MalX must function in vivo as part of a multicomponent complex whose activity is dependent upon its complete assembly and cooperativity of the parts, the influence of possible polar effects becomes unimportant. That is, from the perspective of maltooligosaccharide uptake, polar and non-polar mutations in the ABC-transporter components would appear phenotypically identical, thus any gene replacement in the transporter operon would probe overall transporter function rather than individual component function. Along similar lines it might be argued that the selectivity of the transporter may actually be determined by the permease components. We find this unlikely on two bases. First, permease-dependent selectivity would result in the default possibility that MalX could form complexes with tight-binding but non-transportable substrates (e.g. very long maltooligosaccharides), which would be extremely inefficient as this would effectively clog the system. Second, and likely related to the first, because, where substrate-binding proteins are employed, they initiate transport by substrate recognition and delivery of the substrate to the permease and thus are generally recognized as both critical to function and as specificity-determining components of ABC-transporters (Davidson et al., 2008). Nevertheless, to provide additional evidence of MalX’s maltooligosaccharide binding specificity to further support our contention that MalX defines the observed selective uptake of glycogen-derived maltooligosaccharides, we pursued structural studies of this protein.
MalX recognizes the helical conformation of maltooligosaccharides
MalX was crystallized in complex with maltoheptaose and its crystal structure determined by X-ray diffraction to 2.0 Å resolution. The protein displays a conventional α/β Type II solute binding protein fold with the two globular domains separated by a hinge region that criss-crosses between them at three distinct positions (147–150, 288–301, and 371–377) (Figure 6A). The ligand in the maltoheptaose complexed protein was identified by unambiguous electron density for seven glucose subunits in the MalX binding site, which allowed this sugar to be confidently modeled (Figure 6B). The maltoheptaose ligand adopts a signature conformation corresponding to a full turn of a left-handed α-helix. Each monosaccharide constituent adopts its favourable 4C1 pyranosyl conformation with α-1,4 linkages displaying an average glycosidic bond angle of 111.7° ± 0.8. The torsional constraints of the helix, which have an average ϕ angle of −124.3° and ψ angle of 104.6°, within the binding site are similar to that observed for numerous other non-catalytic maltooligosaccharide binding proteins that have open binding sites (Koropatkin et al., 2008, Quiocho et al., 1997, Tonozuka et al., 2007). This suggests that the engulfing of the ligand by MalX does not impose significant constraints on the conformation of the α-glucan chain.
Figure 6.
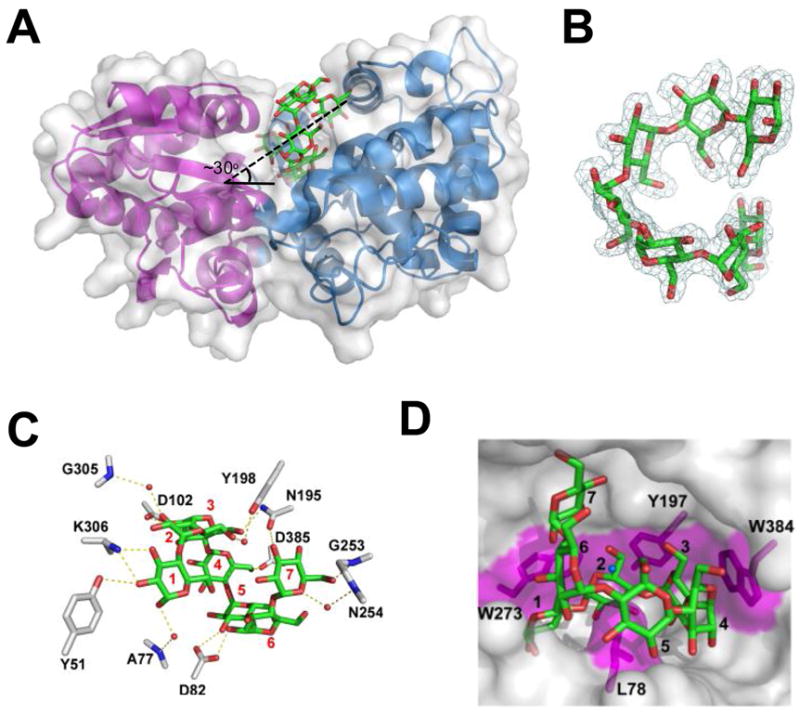
The three-dimensional structure of MalX in complex with maltohepatose at 2.0 Å resolution. A) MalX is shown from a side projection in a ribbon format with the small N-terminal domain colored purple and the large C-terminal domain colored blue. The solvent accessible surface is shown in transparent grey and maltohepatose is shown as green sticks. The approximate angle of the entry for the sugar is indicated. B) Electron density of maltohepatose shown as a maximum-likelihood (Murshudov et al., 1997)/σa-weighted (Read, 1986) 2Fobs-Fcalc electron density map contoured at 1.0 σ (0.28e−/Å3). C) Direct and water-mediated hydrogen bonds within the binding site of MalX. Side chains are represented as grey sticks, maltoheptaose as green sticks, and waters as red spheres. The binding subsites are labelled. D) Solvent accessible surface representation of the binding site. Putative hydrophobic interactions, including the three “stacking” interactions and a leucine “pin,” are shaded in magenta. The ligand subsites are numbered as in (C).
When the protein is viewed from the side projection maltoheptaose enters the binding site on a ~30° angle to the horizontal plane of the protein (Figure 6A). The oligosaccharide is pinned in the binding site by a leucine residue that protrudes from the base of the MalX binding site and pierces the ring of the ligand in a manner much like that observed for the carbohydrate-binding module from rat AMP-activated protein kinase (Polekhina et al., 2005). This trapped form of the protein reveals the presence of at least seven subsites in which extensive direct and water-mediated hydrogen bonds are made between the protein and sugar (Figure 6C), as well as classic the protein-carbohydrate interaction whereby aromatic amino acid side-chains pack in a parallel fashion against the pyranose rings of the sugar ligand (Figure 6D). The core binding site is defined by the first three subsites characterized by the presence of three aromatic amino acid side chains, which are structurally conserved in all described maltodextrin-binding proteins (Quiocho et al., 1997, Tonozuka et al., 2007). In MalX these are Tyr197 (subsite 2), which is flanked by two tryptophans, Trp273 (subsite 1) and Trp384 (subsite 3) (Figure 6D). These residues, whose observed interaction with ligand are consistent with the complex composite UV difference spectra, are all present on the large C-terminal domain and form the “aromatic cradle” that has been reported for several other unrelated α-glucan binding protein families (Boraston et al., 2006, Koropatkin et al., 2008, Lammerts van Bueren et al., 2004, Lammerts van Bueren et al., 2007). This concave surface beautifully complements the unique dimensions of curved surface of the twisting maltooligosaccharide helix, providing selectivity in ligand recognition.
Of the core binding site, subsite 1 appears to be particularly important with Lys306 Nζ and Tyr51 Oη making several direct hydrogen bonds with the O2 and O3 of the glucose residue bound in this subsite (Figure 6C). Because the architecture of this subsite legislates against recognition of the non-reducing end due to steric clashes with the C6-hydroxymethyl group of a non-reducing terminal sugar, this provides the protein with specificity for the reducing end of maltooligosaccharides. However, MalX’s low affinity for maltose indicate that subsites 1 and 2 provide less than one-half of the change in binding free-energy whereas subsite 3, as suggested by the protein’s maximal affinity for maltotriose, provides the majority of the binding energy. Thus, together, subsites 1-3 contribute the selectivity for the reducing end of the sugar, specificity for the curved conformation of the ligand, and the energy that drives the interaction. In contrast, though subsites 4-7 in MalX provide some hydrogen bonds to the ligand, they do not significantly contribute to the binding energy of the interaction, as indicated by the similar affinities of MalX for maltooligosaccharides ranging in size from maltotetroase to maltoheptaose.
This ligand-stabilized “closed” conformation of MalX displays closest structural similarity to the B chain of Thermotoga maritima maltotriose-binding protein in complex with maltotriose (2GHA chain B, rms = 1.8 Å for 360 aligned Cα) (unpublished), the Thermoactinomyces vulgaris MBP in complex with β-cyclodextrin (2ZYK, Chain D, Z-value = 44.9, rmsd = 1.8 Å for 361 Cα) (Tonozuka et al., 2007) and the E. coli MBP in complex with maltotriose (3MBP, Z-value = 42.3, rmsd = 2.2 Å for 353 aligned Cα(Quiocho et al., 1997), though there is still significant structural similarity with other maltodextrin-binding proteins (Cuneo et al., 2006, Diez et al., 2001). In keeping with this structural identity, the spatial positioning of subsites 1-4 in MalX are generally conserved with the four core subsites described for other maltodextrin-binding proteins, except the E. coli maltodextrin-binding protein, which lacks subsite 1 (Cuneo et al., 2009). This difference in the presence of subsite 1 may be origin of MalX’s inability to bind maltotetraitol whereas the E. coli maltodextrin binding protein appears able to accommodate this reduced sugar (Duan et al., 2001). Though the structure of MalX was determined in complex with a longer linear maltooligosaccharide than any other maltodextrin-binding protein determined to date, its binding site is only approximately 4–6 Å wider than other maltodextrin-binding proteins determined in complex with maltotriose or maltotetraose. This appears consistent, however, with the approximation that each glucose residue in an maltooligosaccharide extends the linear length of the sugar by ~5.5 Å but only increases the depth of its helical structure by ~1.5 Å. Thus, a maltooligosaccharide that is increased in relative length by 3–4 sugars might be expected to prop open the binding site by an additional 4.5–6 Å, as we observed. This hints at the structural changes that would be required by this class of proteins to accommodate ligands of varying size.
Recognition of longer maltooligosaccharides requires structural plasticity
The ligand bound structure of MalX shows the maltoheptaose molecule to be neatly sequestered in its binding site with both ends of the saccharide engaging the end-walls of the binding site (figure 6A and D). While this is consistent with the protein’s high affinity for maltotriose to maltoheptaose and with the ability of S. pneumoniae to deplete these sugars from culture when grown on glycogen, it suggests that maltoheptaose represents the upper limit on the length of maltodextrin recognized by MalX. Maltooctaose, however, is clearly bound tightly by the protein and is depleted from culture, while sugars of DP 9-11 are partly depleted from the culture suggesting they must be recognized by MalX. Therefore, despite the suggestion from the MalX-maltoheptaose structure that due to steric clashes maltooctaose could not be accommodated in this conformation of the protein, MalX can clearly bind maltooligosaccharides longer than maltoheptaose. To provide insight into the conformational changes in MalX that govern ligand recognition we determined the X-ray crystal structure of MalX in its apo open form to 2.9 Å resolution. The bending at the hinge of MalX results in the binding site of the open form being roughly 6.5 Å wider than in the maltoheptaose complexed form (Figure 7A). Strikingly, the binding site of the protein maintains a shape that complements the contours of a helical maltooligosaccharide and, in particular, due to the presence of the aromatic cradle in one lobe of the protein the conformation of this key binding determinant is maintained (Figure 7A). Furthermore, a maltooligosaccharide with a DP of 12 can be fit into this binding site while maintaining the key interactions with the aromatic cradle (Figure 7B). The upper limit on the length of the oligosaccharide is imposed by the physical limits of this open binding site as the ends of longer sugar would collide with the end-walls of the binding site. Though only an approximation, this apparent structural limit approaching a sugar of 12 glucose units is quite consistent with the observed pattern of maltooligosaccharide depletion from S. pneumoniae cultures grown on glycogen. From this we propose that MalX can accommodate the size of the sugar ligand through the opening and closing of its binding site. Conceptual support for this comes from the E. coli maltodextrin-binding protein, which is known to maintain a degree of sugar binding activity even in its open conformation (Duan et al., 2001). This postulated mechanism of ligand accommodation, however, does require some shifting of the sugar in the binding site likely resulting in the loss of some specific intermolecular interactions and thus reduced binding affinity. We propose that the partial depletion of the DP 9-11 maltodextrins from the culture medium reflects a lower binding affinity for these sugars imposed by the more open conformation of MalX necessary to accommodate them while the lack of depletion of longer oligosaccharides reflects the protein’s complete inability to accomodate these sugars. The good agreement between our structural and functional data and the observed depletion pattern of glycogen-derived maltooligosaccharides from S. pneumoniae cultures argues strongly for MalX defining the selective uptake of maltooligosaccharides with a DP greater than 2 but less than 12.
Figure 7.
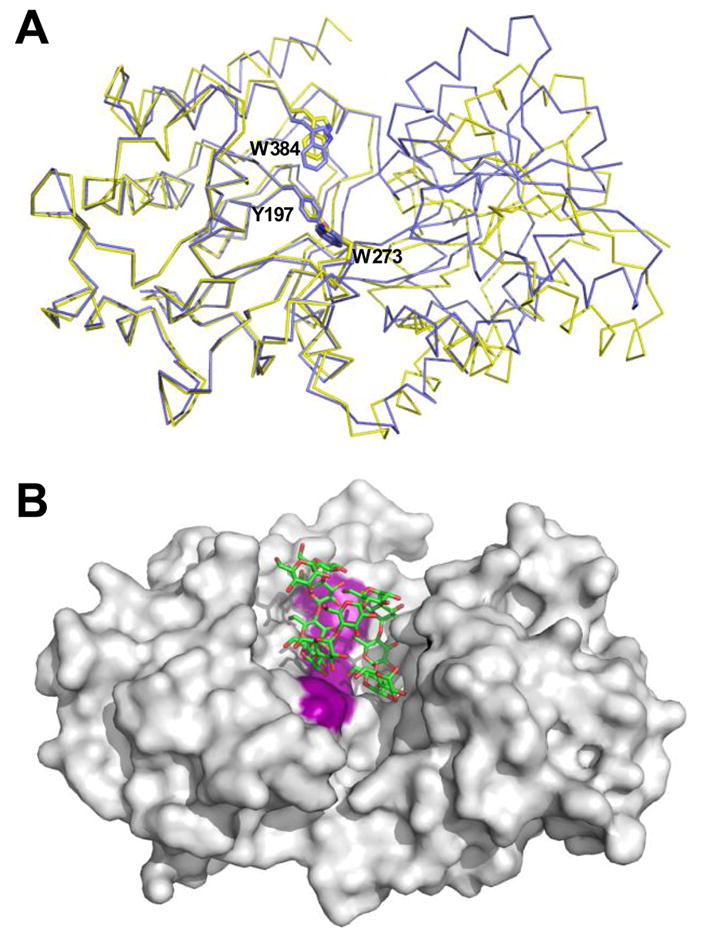
Structural changes and ligand accommodation in MalX. A) Overlay of MalX in the maltoheptaose complexed form (shown as a blue Cα trace) and the apo form (yellow) made by overlapping only the large C-terminal domain. The amino acids making up the aromatic binding platform are shown in stick representation and labelled. B) solvent accessible surface of the apo form of MalX with an idealized maltododecaose molecule (green sticks) modelled into the active site. The aromatic binding platform is colored purple for reference.
The Biological Role of SpuA and MalX
Although S. pneumoniae does appear to have a multicomponent system for the metabolism of α-glucans, the only relevant exogenous form of α-glucan this organism is able to effectively use as an energy source is glycogen through a pathway that is schematically outlined in Figure 8. SpuA facilitates the depolymerization of glycogen through hydrolysis of its branch points, while MalX selectively transports a subset of the glycogen degradation products. SpuA and MalX are critical to the utilization of exogenous glycogen, which is consistent with their positions as the only extracellular components of S. pneumoniae’s α-glucan metabolizing machinery, though a second PTS transporter may play a less important role in the uptake of the maltose and maltotriose fractions produced from glycogen degradation. Given that S. pneumoniae appears to be an obligate commensal, one can only conclude that this pathway exists to utilize glycogen present within the host. Energy liberation to maintain bacterial growth is likely to be the primary role of glycogen utilization. While glycogen released as a result of tissue destruction is one possible source of the polysaccharide, S. pneumoniae is also a skilled invader of lung cells, which it does without necessarily destroying the cell (Kadioglu et al., 2001), making the intracellular glycogen of host cells a possible target. This suggests a second and potentially more insidious outcome of S. pneumoniae’s attack on host glycogen. It has been postulated that type II alveolar cells are a preferred target of S. pneumoniae (Cundell & Tuomanen, 1994). In these cells glycogen is a precursor for surfactant production, which is a key component of the innate immune system within the lung mucosa (Ridsdale & Post, 2004). By depleting the stores of glycogen in surfactant producing cells invading S. pneumoniae may also cripple the production of surfactant. In this light, the specific degradation of lung cell glycogen may promote infection by both supporting bacterial growth and by lowering the lungs’ defences to S. pneumoniae invasion. Further investigation into the function of virulence factors within this pathway, therefore, is paramount towards understanding the mechanisms by which glycogen is depolymerised and the implications that this has for S. pneumoniae pathogenesis and host immunity.
Figure 8.

Schematic summary of exogenous glycogen utilization in S. pneumoniae showing the degradation of glycogen (1), specific recognition and transport of the products (2–4), and processing of the internalized glycan products (5). The proteins putatively involved in processing the internalized glycans were proposed as the most likely candidates based on their predicted functions. The role of SP_0758, the MalT homolog, is putative. The putative transported products of SP_0758 are indicated as phosoph-sugars consistent with the normal function of a carbohydrate specific PTS. The yellow circles represent the nucleotide binding/ATPase component of the ABC transporter whose gene is apparently not present in the maltodextrin metabolon. Green linked hexagons represent the peptidoglycan layer to which SpuA is attached. The capsule is omitted for clarity.
EXPERIMENTAL PROCEDURES
Cloning, recombinant production, and purification of SpuA and MalX
The DNA fragment encoding nearly full length SpuA without the N-terminal signal peptide and C-terminal LPXTG motif was amplified from S. pneumoniae genomic DNA (TIGR strain BAA-334D) using the oligonucleotide primers SpuAF and SpuAR (Supplementary Table 1). The amplified DNA product encoding amino acids 135-1245 was cloned into pET28a plasmid vector via engineered 5′ NheI and 3′ XhoI restriction sites to generate pSpuA.
To produce MalX in a soluble form, the malx gene was engineered to lack the secretion signal peptide and lipoprotein attachment motif. The malx gene fragment encoding amino acids 31-423 of MalX was amplified by PCR from S. pneumoniae TIGR4 genomic DNA (American Type Culture Collection BAA-334D) using the forward primer MALXF and reverse primer MALXR (Supplementary Table 1). The DNA fragments were inserted into pET 28a via engineered 5′ NheI and 3′ XhoI restriction sites to generate pMalX. Both pSpuA and pMalX encodes the protein of interest fused to an N-terminal His6-tag via a thrombin protease cleavage site.
Recombinant polypeptides were produced in E. coli BL21 Star (DE3) cells (Invitrogen) using LB media supplemented with 50 μg ml−1 of kanamycin. Cultures were grown at 37 °C until they reached an optical density at 600 nm of 0.5–0.7 whereupon protein production was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation after 4 hours of additional growth at 37 °C then disrupted using a French pressure cell in 20 mM Tris-HCl, pH 8.0, 0.5 M NaCl supplemented with Protease Inhibitor Cocktail (Roche). Polypeptides were purified from the cell lysate, previously cleared by centrifugation, by immobilized metal affinity chromatography using 2 ml of Ni2+ affinity resin (GE Healthcare). Purified protein was concentrated and buffer exchanged into 20 mM Tris-HCl, pH 8.0, in a stirred cell ultrafiltration device with a 5000 molecular weight cut-off membrane (Millipore).
Determination of Protein Concentration
The concentration of purified SpuA and MalX were determined by UV absorbance at 280 nm using the calculated extinction coefficients (Gasteiger et al., 2003) of 172930 M−1 cm−1 and 61310 M−1cm−1, respectively.
Bacterial strains and growth conditions. S. pneumoniae
TIGR4 strain (ATCC BAA-334) was used as the parental strain. S. pneumoniae was routinely cultured on Tryptic Soy Agar (TSA), 6% (v/v) defibrinated sheep blood from a freezer stock stored at −80°C in Todd Hewitt with 0.5% (w/v) yeast extract (THY) supplemented with 16% (v/v) glycerol. S. pneumoniae was also grown on THY broth for the transformations and a semi-defined media containing casein hydrolase, salts, vitamins, amino acids, catalase, and albumin (AGCH) with 0.2% (w/v) yeast extract (AGCHY) for the growth curves (Lacks, 1966, Chan et al., 2003). When necessary, chloramphenicol was added to 5 μg ml−1. Unless otherwise stated, S. pneumoniae was grown in a candle jar at 37 °C with no shaking.
Generation of Δmalx and Δspua strains
A PCR ligation technique was used to replace malX and spua with a chloramphenicol resistant cassette (Schell et al., 2002). Briefly, regions flanking the gene to be replaced and the chloramphenicol resistant cassette were amplified by PCR with primers containing specific restriction sites (see Supplementary Table 1): flanking region 1 has a 3′ NheI site, flanking region 2 has a 5′ XhoI site, and the chloramphenicol resistant cassette has both a 5′ NheI site and a 3′ XhoI site (CAM-F and CAM-R). Each amplicon was digested with the particular restriction enzyme and then all three digested amplicons used in one ligation reaction. This ligation reaction was subsequently used to transform S. pneumoniae TIGR4 creating gene replacement strains. The S. pneumoniae transformation protocol was adapted from Bricker and Camilli (Bricker & Camilli, 1999). Bacteria were grown overnight in THY broth to an approximate OD600nm of 0.7. A 1/50 dilution was then made into the pre-induction growth medium of THY, 0.5% (w/v) glycine, 11 mM HCl and grown at 37 °C in a candle jar until an OD600nm of 0.04 was reached. The media was then supplemented with 10 mM NaOH, 0.2% (w/v) BSA, 1 mM CaCl2, and 100 ng ml−1 Competence Stimulating Peptide 2 (CSP-2) in their respective order and incubated in a candle jar at 37°C for 14 minutes. At this time, the entire ligation reaction was added to the media and subsequently incubated for 1 hour at 37 °C in a candle jar. The transformation reaction was then diluted 1/4 with THY broth and incubated further for 2 hours. The transformations were finally plated on sheep blood agar supplemented with chloramphenicol and incubated overnight in a candle jar at 37 °C. Colonies were re-streaked onto new blood agar plates and the location of the chloramphenicol cassette was confirmed by several PCR analyses.
Growth curves
The protocol for the growth curves was adapted from Battig et al. (Battig et al., 2006). Wild-type, Δmalx, and Δspua S. pneumoniae TIGR4 strains were streaked onto blood agar plates and incubated at 37 °C in a candle jar for approximately 12 hours. Overnight cultures were prepared by inoculating 10 colonies into AGCHY broth supplemented with 1% (w/v) glucose and incubated for 9–10 hours at 37°C in a candle jar. The overnight cultures were diluted 1/50 into fresh AGCHY with 1% (w/v) glucose and incubated at 37 °C in a candle jar until reaching an OD600nm of 0.5–0.6. Cells were then washed with AGCHY broth and subsequently diluted 1/50 into AGCHY with an additional 1% (w/v) of glucose, glycogen, amylose, maltotriose, pullulan, or starch, each in triplicate. The cells were incubated at 37 °C in a candle jar; optical densities at 600 nm were read at regular intervals. Where appropriate, cultures were supplemented with 1 μM purified recombinant SpuA.
Carbohydrate analysis
The procedure for FACE was adapted from Jackson et. al. (Jackson, 1993). Enzyme reactions were carried out by incubating 1% (w/v) solutions of glycogen or pullulan in PBS with 30 μg of SpuA. Immediately following the reactions, 10 μl (100 μg) of polysaccharide was removed from each tube and dried in a Speedvac for 45 minutes at 50 °C. Labeling of the sugar products was carried out by adding 5μl of a solution of 0.15% ANTS in 15% acetic acid and 5μl of 1 M NaCNBH3 in DMSO to the dried samples. The reaction was incubated overnight in the dark at 37 °C then dried. The ANTS-labeled products were then resuspended in 50 μl deionized water plus 50 μl of 0.01% Thorin I loading dye (Sigma) in 20% glycerol. Approximately 1–3 μg of ANTS-labeled product were loaded onto a 28% polyacrylamide (19:1) gel with a 10% stacking gel and electrophoresed at a constant 15 mA for 105 minutes at 4°C in native running buffer (25 mM Tris-HCl, 0.2M glycine) in a XCell SureLock Mini-Cell system (Invitrogen). Gels were immediately visualized and imaged under UV light.
HPAEC-PAD analysis was performed using a Dionex ICS3000 instrument equipped with a CarboPac PA-200 column and pulsed amperometric detection. Samples were loaded onto the column as 20 μl volumes followed by elution with a linear gradient of sodium acetate (0–300 mM) in 100 mM sodium hydroxide over 30 minutes. Glycogen (1% w/v) was treated with SpuA as above then diluted 1000-times in sterile distilled water followed by centrifugation at 10000 × g prior to HPAEC analysis. The residual carbohydrates present in the supernatant of S. pneumoniae cultures, grown as above, were analyzed by clearing the culture of cells by centrifugation and preparing samples of the supernatant by diluting 1000-times in sterile distilled water followed by centrifugation at 10000 × g. Individual carbohydrate standards of glucose, α-1,4-glucooligosaccharides from maltose to maltooctaose, panose, isomaltose, isomaltotriose, and 63-α-D-glucosyl-maltotriosyl-maltotriose were run in 1/1000 dilutions of distilled water, PBS or AGCHY medium as appropriate and were performed on the day of each experiment. Peaks resulting from the standard sugars did not display any overlap in elution position. All resulting chromatograms were corrected for background by subtraction of the chromatogram of a 1/1000 dilution of water, PBS or AGCHY medium, with or without glycogen, run over the column. Carbohydrates were identified by comparison of the distinctive peak elution positions with those of the standards. Experiments were performed at least two times and were highly reproducible.
Binding analysis
Qualitative and quantitative UV difference binding studies were performed and analyzed as described previously using a bimolecular binding model that accounted for ligand depletion and included a term for stoichiometry (n) (Boraston et al., 2001, Lammerts van Bueren et al., 2004). UV difference titrations were performed in 50 mM Tris-HCl, pH 7.5, at 20 °C. The protein concentration used was 12.5 μM. When the protein concentration was 5 or more times in excess of the Kd (i.e giving C-values (Wiseman et al., 1989) greater than 5) the n was determined experimentally from the fits of the data in addition to the association constant (Ka). When this criterion was not met the n value was fixed as a constant of 1 during the data fitting. All titrations were performed in triplicate.
Isothermal titration calorimetry (ITC) was performed as described previously using a VP-ITC (MicroCal, Northampton, MA) (Ficko-Blean & Boraston, 2006, Abbott & Boraston, 2007). Briefly, protein samples were extensively dialyzed against buffer (50 mM Tris-HCl, pH 7.5) then concentrated in a stirred ultrafiltration cell as above. Sugar solutions were prepared by mass in buffer saved from the ultrafiltration step. Both protein and sugar solutions were filtered and degassed immediately prior to use. The protein concentration used was ~100 μM giving C-values in excess of 50. Titrations were performed in triplicate at 25 °C. The data were fit with a single binding site model to determine Ka, n, and ΔH (change in enthalpy). ΔS (change in entropy) was calculated using ΔG=ΔH−TΔS. Errors represent the standard deviations of the triplicate determinations.
Crystallization, data collection and structure solution
Prior to crystallization, MalX was further purified by gel filtration chromatography using Sephacryl-200 resin and 20 mM Tris-HCl, pH 8.0, as a buffer. Crystals of MalX (10 mg ml−1) in complex with maltoheptaose were grown using the hanging-drop vapor diffusion method at 18 °C in 1.6 M ammonium sulfate, 0.1 M MES, pH 6.5, and 10% (v/v) dioxane. MalX was crystallized in its apo form in 1.6 M sodium citrate, pH 6.5, with 3% (v/v) ethanol. Both crystals were cryoprotected by a short soak in the crystallization solution supplemented with 25% ethylene glycol and flash cooled at 113 K directly in a nitrogen stream. X-ray diffraction data sets were collected on a Rigaku R-AXIS 4++ area detector coupled to a MM-002 X-ray generator with Osmic “blue” optics and an Oxford Cryostream 700. Data were processed with CrystalClear/d*trek (Pflugrath, 1999).
The structure of MalX in complex with maltoheptaose was solved by molecular replacement. Using the coordinates of the T. maritima maltodextrin-binding protein in complex maltotriose (PDB code 2gha) as a search model, PHASER (Read, 2001) was able to find a clear solution for the single MalX molecule in the asymmetric unit. Two rounds of successive manual building with COOT (Emsley & Cowtan, 2004), refinement with REFMAC (Murshudov et al., 1997), solvent flattening, and submission to ARP/wARP (Perrakis et al., 1999) ultimately produced phases sufficient for ARP/wARP to build an 80% complete model with docked sidechains. This model was corrected and completed manually using COOT and refined with REFMAC. The apo structure of MalX was solved by molecular replacement using separate N- and C-terminal lobe models derived from MalX-maltoheptaose complex as search models. The two molecules in the asymmetric unit were manually corrected using COOT and refined with REFMAC.
Water molecules were added using the REFMAC implementation of COOT:FINDWATERS and inspected visually prior to deposition. Five percent of the observations were flagged as “free” and used to monitor refinement procedures (Brunger, 1992). Model validation was performed with SFCHECK (Vaguine et al., 1999) and PROCHECK (Laskowski et al., 1993). All data collection and model statistics are given in Table 3. Coordinates and structure factors have been deposited with the PDB code of (to be added in proof/revision).
Table 3.
Data collection and structure statistics.
| MalX | MalX + maltoheptaose | |
|---|---|---|
| Data collection | ||
| Space Group | C2 | P21212 |
| Resolution (Å) | 20.00–2.90 | 20.00–2.00 |
| Cell Dimensions | ||
| (a, b, c) | 181.3, 56.9, 113.7 | 68.4, 127.9, 52.0 |
| (α, β, γ) | 90.0, 128.8, 90.0 | 90.0, 90.0, 90.0 |
| Rmerge | 0.107 (0.417) | 0.060 (0.313) |
| I/σI | 10.9 (3.4) | 17.3 (3.5) |
| Completeness (%) | 96.4 (96.1) | 92.7 (94.1) |
| Redundancy | 4.4 (4.4) | 4.3 (4.1) |
| # Reflections | 85794 | 124651 |
| # unique reflections | 19623 | 29101 |
| Refinement | ||
| Rwork/Rfree | 0.22/0.27 | 0.21/0.25 |
| No. atoms | ||
| Protein | 2888(A); 2852(B) | 2858 |
| Ligand/ion atoms | 0 | 10 (2 SO4) |
| Water molecules | 59 | 309 |
| B-factors | ||
| Protein | 36.5(A); 50.0(B) | 30.2 |
| Ligand/ion | N/A | 75.0 |
| Water | 29.6 | 38.7 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.007 | 0.010 |
| Bond angles (°) | 1.062 | 1.267 |
| Ramachandran statistics (%) | ||
| Preferred | 94.0 | 92.3 |
| allowed | 5.6 | 7.7 |
| outliers | 0.4 | 0.0 |
Highest resolution shell is shown in parenthesis.
Supplementary Material
Acknowledgments
We are grateful to Namrita Sohdi, Jasleen Bains, and Caitlin Wright for their technical assistance. This work was supported by grants from the Canadian Lung Association and the Canadian Institutes of Health Research. MAH and ALvB were supported by doctoral fellowships from the Natural Sciences and Engineering Research Council of Canada and the Michael Smith Foundation for Health Research (MSFHR). ABB is a Canada Research Chair in Molecular Interactions and a MSFHR Career Scholar.
References
- Abbott DW, Boraston AB. Specific recognition of saturated and 4,5-unsaturated hexuronate sugars by a periplasmic binding protein involved in pectin catabolism. J Mol Biol. 2007;369:759–770. doi: 10.1016/j.jmb.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Battig P, Hathaway LJ, Hofer S, Muhlemann K. Serotype-specific invasiveness and colonization prevalence in Streptococcus pneumoniae correlate with the lag phase during in vitro growth. Microbes Infect. 2006;8:2612–2617. doi: 10.1016/j.micinf.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Bongaerts RJ, Heinz HP, Hadding U, Zysk G. Antigenicity, expression, and molecular characterization of surface-located pullulanase of Streptococcus pneumoniae. Infect Immun. 2000;68:7141–7143. doi: 10.1128/iai.68.12.7141-7143.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine tuning polysaccharide recognition. Biochem J. 2004;382:769–782. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraston AB, Healey M, Klassen J, Ficko-Blean E, Lammerts van Bueren A, Law V. A structural and functional analysis of alpha-glucan recognition by family 25 and 26 carbohydrate-binding modules reveals a conserved mode of starch recognition. J Biol Chem. 2006;281:587–598. doi: 10.1074/jbc.M509958200. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Warren RA, Kilburn DG. beta-1,3-Glucan binding by a thermostable carbohydrate-binding module from Thermotoga maritima. Biochemistry. 2001 Dec 4;40(48):14679–14685. doi: 10.1021/bi015760g. [DOI] [PubMed] [Google Scholar]
- Bricker AL, Camilli A. Transformation of a type 4 encapsulated strain of Streptococcus pneumoniae. FEMS Microbiol Lett. 1999;172:131–135. doi: 10.1111/j.1574-6968.1999.tb13460.x. [DOI] [PubMed] [Google Scholar]
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2008;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PF, O’Dwyer KM, Palmer LM, Ambrad JD, Ingraham KA, So C, Lonetto MA, Biswas S, Rosenberg M, Holmes DJ, Zalacain M. Characterization of a novel fucose-regulated promoter (PfcsK) suitable for gene essentiality and antibacterial mode-of-action studies in Streptococcus pneumoniae. J Bacteriol. 2003;185:2051–2058. doi: 10.1128/JB.185.6.2051-2058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ma Y, Yang J, O’Brien CJ, Lee SL, Mazurkiewicz JE, Haataja S, Yan JH, Gao GF, Zhang JR. Genetic requirement for pneumococcal ear infection. PLoS ONE. 2007;3:e2950. doi: 10.1371/journal.pone.0002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundell DR, Tuomanen EI. Receptor specificity of adherence of Streptococcus pneumoniae to human type-II pneumocytes and vascular endothelial cells in vitro. Microb Pathog. 1994;17:361–374. doi: 10.1006/mpat.1994.1082. [DOI] [PubMed] [Google Scholar]
- Cuneo MJ, Changela A, Beese LS, Hellinga HW. Structural adaptations that modulate monosaccharide, disaccharide, and trisaccharide specificities in periplasmic maltose-binding proteins. J Mol Biol. 2009;389:157–166. doi: 10.1016/j.jmb.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Cuneo MJ, Changela A, Warren JJ, Beese LS, Hellinga HW. The crystal structure of a thermophilic glucose binding protein reveals adaptations that interconvert mono and di-saccharide binding sites. J Mol Biol. 2006;362:259–270. doi: 10.1016/j.jmb.2006.06.084. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev. 2008;72:317–364. doi: 10.1128/MMBR.00031-07. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez J, Diederichs K, Greller G, Horlacher R, Boos W, Welte W. The crystal structure of a liganded trehalose/maltose-binding protein from the hyperthermophilic Archaeon Thermococcus litoralis at 1.85 A. J Mol Biol. 2001;305:905–915. doi: 10.1006/jmbi.2000.4203. [DOI] [PubMed] [Google Scholar]
- Duan X, Hall JA, Nikaido H, Quiocho FA. Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: flexibility of tertiary structure and ligand binding. J Mol Biol. 2001;306:1115–1126. doi: 10.1006/jmbi.2001.4456. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ficko-Blean E, Boraston AB. The interaction of carbohydrate-binding module from a Clostridium perfringens N-acetyl-beta-hexosaminidase with its carbohydrate receptor. J Biol Chem. 2006;281:37748–37757. doi: 10.1074/jbc.M606126200. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MR, Virtaneva K, Porcella SF, Gardner DJ, Long RD, Welty DM, Barry WT, Johnson CA, Parkins LD, Wright FA, Musser JM. Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am J Pathol. 2006;169:927–942. doi: 10.2353/ajpath.2006.060112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hava DL, Camilli A. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol. 2002;45:1389–1406. [PMC free article] [PubMed] [Google Scholar]
- Henrissat B, Deleury E, Coutinho PM. Glycogen metabolism loss: a common marker of parasitic behaviour in bacteria? Trends Genet. 2002;18:437–440. doi: 10.1016/s0168-9525(02)02734-8. [DOI] [PubMed] [Google Scholar]
- Jackson P. Fluorophore-assisted carbohydrate electrophoresis: a new technology for the analysis of glycans. Biochem Soc Trans. 1993;21:121–125. doi: 10.1042/bst0210121. [DOI] [PubMed] [Google Scholar]
- Jedrzejas MJ. Unveiling molecular mechanisms of bacterial surface proteins: Streptococcus pneumoniae as a model organism for structural studies. Cell Mol Life Sci. 2007;64:2799–2822. doi: 10.1007/s00018-007-7125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu A, Andrew PW. The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol. 2004;25:143–149. doi: 10.1016/j.it.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Kadioglu A, Sharpe JA, Lazou I, Svanborg C, Ockleford C, Mitchell TJ, Andrew PW. Use of green fluorescent protein in visualisation of pneumococcal invasion of broncho-epithelial cells in vivo. FEMS Microbiol Lett. 2001;194:105–110. doi: 10.1111/j.1574-6968.2001.tb09454.x. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Martens EC, Gordon JI, Smith TJ. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure. 2008;16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacks S. Integration efficiency and genetic recombination in pneumococcal transformation. Genetics. 1966;53:207–235. doi: 10.1093/genetics/53.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerts van Bueren A, Finn R, Ausio J, Boraston AB. Alpha-glucan recognition by a new family of carbohydrate-binding modules found primarily in bacterial pathogens. Biochemistry. 2004;43:15633–15642. doi: 10.1021/bi048215z. [DOI] [PubMed] [Google Scholar]
- Lammerts van Bueren AL, Higgins M, Wang D, Burke RD, Boraston AB. Identification and structural basis of binding to host lung glycogen by streptococcal virulence factors. Nat Struct Mol Biol. 2007;14:76–84. doi: 10.1038/nsmb1187. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Lau GW, Haataja S, Lonetto M, Kensit SE, Marra A, Bryant AP, McDevitt D, Morrison DA, Holden DW. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol Microbiol. 2001;40:555–571. doi: 10.1046/j.1365-2958.2001.02335.x. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, Tuomanen EI. Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun. 2004;72:5582–5596. doi: 10.1128/IAI.72.10.5582-5596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55 (Pt 10):1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Gupta A, van Denderen BJ, Feil SC, Kemp BE, Stapleton D, Parker MW. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure. 2005;13:1453–1462. doi: 10.1016/j.str.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Polissi A, Pontiggia A, Feger G, Altieri M, Mottl H, Ferrari L, Simon D. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect Immun. 1998;66:5620–5629. doi: 10.1128/iai.66.12.5620-5629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procko E, O’Mara ML, Bennett WF, Tieleman DP, Gaudet R. The mechanism of ABC transporters: general lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 2009;23:1287–1302. doi: 10.1096/fj.08-121855. [DOI] [PubMed] [Google Scholar]
- Puyet A, Espinosa M. Structure of the maltodextrin-uptake locus of Streptococcus pneumoniae. Correlation to the Escherichia coli maltose regulon. J Mol Biol. 1993;230:800–811. doi: 10.1006/jmbi.1993.1202. [DOI] [PubMed] [Google Scholar]
- Quiocho FA, Spurlino JC, Rodseth LE. Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor. Structure. 1997;5:997–1015. doi: 10.1016/s0969-2126(97)00253-0. [DOI] [PubMed] [Google Scholar]
- Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst A. 1986;42:140–149. [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Ridsdale R, Post M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L743–751. doi: 10.1152/ajplung.00146.2004. [DOI] [PubMed] [Google Scholar]
- Santi I, Pezzicoli A, Bosello M, Berti F, Mariani M, Telford JL, Grandi G, Soriani M. Functional characterization of a newly identified group B Streptococcus pullulanase eliciting antibodies able to prevent alpha-glucans degradation. PLoS One. 2008;3:e3787. doi: 10.1371/journal.pone.0003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen MC, Desiere F, Bork P, Delley M, Pridmore RD, Arigoni F. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99:14422–14427. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonert S, Seitz S, Krafft H, Feuerbaum EA, Andernach I, Witz G, Dahl MK. Maltose and maltodextrin utilization by Bacillus subtilis. J Bacteriol. 2006;188:3911–3922. doi: 10.1128/JB.00213-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, 3rd, Fang H, Okorafor N, Sumby P, Sitkiewicz I, Keith D, Patel P, Austin C, Graviss EA, Musser JM, Chow DC. MalE of group A Streptococcus participates in the rapid transport of maltotriose and longer maltodextrins. J Bacteriol. 2007;189:2610–2617. doi: 10.1128/JB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, 3rd, Keith DB, Davenport MT, Beres SB, Carroll RK, Musser JM. Contribution of AmyA, an extracellular alpha-glucan degrading enzyme, to group A streptococcal host-pathogen interaction. Mol Microbiol. 2009;74:159–174. doi: 10.1111/j.1365-2958.2009.06858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, 3rd, Keith DB, Davenport MT, Horstmann N, Brennan RG, Musser JM. Molecular characterization of group A Streptococcus maltodextrin catabolism and its role in pharyngitis. Mol Microbiol. 2008;69:436–452. doi: 10.1111/j.1365-2958.2008.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, 3rd, Sumby P, Sitkiewicz I, Okorafor N, Granville C, Patel P, Voyich J, Hull R, DeLeo FR, Musser JM. Maltodextrin utilization plays a key role in the ability of group A Streptococcus to colonize the oropharynx. Infect Immun. 2006;74:4605–4614. doi: 10.1128/IAI.00477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam MR, Danchin EG, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of alpha-amylase-related proteins. Protein Eng Des Sel. 2006;19:555–562. doi: 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Thomson J, Liu Y, Sturtevant JM, Quiocho FA. A thermodynamic study of the binding of linear and cyclic oligosaccharides to the maltodextrin-binding protein of Escherichia coli. Biophys Chem. 1998;70:101–108. doi: 10.1016/s0301-4622(97)00113-0. [DOI] [PubMed] [Google Scholar]
- Tonozuka T, Sogawa A, Yamada M, Matsumoto N, Yoshida H, Kamitori S, Ichikawa K, Mizuno M, Nishikawa A, Sakano Y. Structural basis for cyclodextrin recognition by Thermoactinomyces vulgaris cyclo/maltodextrin-binding protein. FEBS J. 2007;274:2109–2120. doi: 10.1111/j.1742-4658.2007.05753.x. [DOI] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.