

Abstract
It is well known that Nb-benzyl tryptophan alkyl esters undergo the Pictet-Spengler reaction with aldehydes to furnish both cis and trans 1,2,3,4-tetrahydro-β-carbolines, with the trans isomer predominating. Epimerization at C-1 took place under acidic conditions to produce, exclusively, the thermodynamically more stable trans diastereomer via internal asymmetric induction. Recent kinetic experiments provided insight into the cis to trans epimerization mechanism involved in the Pictet-Spengler reaction of 1,2,3-trisubsituted tetrahydro-β-carbolines. Since the epimerization reaction had been shown to be sensitive to electronic effects at C-1, the rate data for a series of 1-phenyl-substituted-1,2,3,4-tetrahydro-β-carbolines was investigated via a Hammett study. Analysis of the data supported the presence of a positively charged intermediate with a ρ value of −1.4, although the existence of an iminium ion intermediate or a carbocationic intermediate could not be determined from this data alone. Analysis of the rate of epimerization demonstrated first-order kinetics with respect to TFA following the initial protonation of the substrate. This observation was consistent with the formation of a doubly protonated intermediate as the rate determining step in the carbocation-mediated cis to trans epimerization process. In addition, the observed first-order rate dependence was inconsistent with the retro Pictet-Spengler mechanism since protonation at the indole-2 position was not rate determining as demonstrated by kinetic isotope effects. Based on this kinetic data, the retro Pictet-Spengler pathway was ruled out for the cis to trans epimerization of 1,2,3-trisubstituted-1,2,3,4-tetrahydro-β-carbolines, while the olefinic mechanism had been ruled out by experiments carried out in TFA-d.
Keywords: Pictet-Spengler, reaction mechanism, tetrahydro-β-carbolines, kinetics, carbocations
Introduction
The Pictet-Spengler reaction,1 which involves the cyclization of electron-rich aryl or heteroaryl groups onto imine or iminium ion electrophiles, has long been a standard method for the construction of both tetrahydroisoquinolines1 and tetrahydro-β-carboles.2 These privileged scaffolds appear in a diverse array of biologically active compounds; both of these ring systems are popular choices for combinatorial libraries targeted at drug discovery.3 Recent advances in medicinal chemistry include potential implications for β-carbolines in the treatment of alcoholism.4,5 The enantiospecific Pictet-Spengler reaction, in particular, has stimulated widespread interest in both organic and medicinal chemistry6–19 and has resulted in the total synthesis of a number of natural products including vincamajinine, alstonisine, macralstonidine, (−)-alstophylline, (−)-macralstonine, ajmaline and (−)-raumacline.20 Alternatively, several groups have extended the scope to examine the ratio of cis/trans isomers of the Pictet-Spengler reaction employing tryptophan derivatives with aldehydes and this has been reviewed.21,22 The development of chiral auxiliaries by Reddy8 and Nakagawa23,24 provided the basis for asymmetric Pictet-Spengler reactions. In addition, Nakagawa10 had also explored Brønsted acid-assisted Lewis acids in stoichiometric amounts to induce stereoselectivity in the Pictet-Spengler reaction. Recently, important examples of external asymmetric induction via organocatalysis have been applied to the Pictet-Spengler cyclization. Jacobsen, et al.12,16,17 have developed chiral thiourea catalysts for the asymmetric Pictet-Spengler reaction of tryptamines. Alternatively, List13 and van Maarseveen14,15,18 have investigated chiral phosphoric acid catalysts with much success.
Although progress has been made in regard to synthesis, interestingly, the mechanism of the cis to trans isomerization in the presence of Brønsted acids has not been fully defined. Three potential mechanistic pathways have been proposed and their intermediates are depicted in Figure 1. The olefinic pathway had been previously ruled out with aliphatic substituents at the C-1 position22 and was therefore not considered in the kinetic analysis. A structural basis for the development of the proposed carbocationic intermediate had been described by Han.25 The conformation of the C-ring was found to be important during the epimerization process; ultimately the p-orbital of the developing carbocation was thought to overlap well with the indole π-electrons. In efforts aimed to investigate the mechanism of the isomerization at C-1, a set of readily accessible electron-donating and electron-withdrawing substituted aromatic aldehydes have been reacted with L-tryptophan ethyl ester, followed by Nb-benzylation, to provide optically active cis and trans 1,2,3-trisubsituted-tetrahydro-β-carbolines.26 In a general sense, electron-rich substituents on aromatic rings at the C-1 position enhanced the rate of the cis to trans epimerization.26 This result was in agreement with the proposed carbocationic mechanism of epimerization, although the retro Pictet-Spengler process could not be completely ruled out at that time.26 In the following report, the kinetics of the cis to trans epimerization of 1,2,3-trisubsituted-tetrahydro-β-carbolines was studied to elucidate the mechanism of this epimerization.
Figure 1.

Proposed intermediates in the cis to trans epimerization.
Results and Discussion
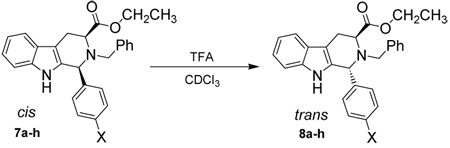
Since the general trend of rates of epimerization was in place, a detailed kinetic study was designed. A Hammett linear free energy relationship is a common method in the application of kinetic data to mechanistic problems.27–28 In order to use this method, the parent 1-phenyl-tetrahydro-β-carboline (7d) was employed as one of the molecules to be studied. This parent substrate was obtained from benzaldehyde and L-tryptophan ethyl ester when stirred under the conditions of the Pictet-Spengler cyclization previously described.26 In addition, substrates which carried a series of substituents with a range of electron densities at the C-1 position were easily obtained by a Pictet-Spengler cyclization, followed by separation of the cis and trans diastereomers and subsequent Nb-benzylation, as previously described (Scheme 1 and Table 1).26 Several of the analogs were very sensitive to acid-induced epimerization; therefore large quantities of base were used during the alkylation reaction.
Scheme 1.

Synthesis of trisubstituted-1,2,3,4-tetrahydro-β-carbolines.
Table 1.
Products from the Pictet-Spengler reaction and Nb-benzylation.
| X | Pictet-Spengler Products | Alkylation Products | |||||
|---|---|---|---|---|---|---|---|
| cis | trans | Yield a,b% | cis | Yielda,c % | trans | Yielda,c % | |
| Et | 5a | 6a | 90 | 7a | 69 | 8a | 68 |
| iPr | 5b | 6b | 91 | 7b | 55 | 8b | 59 |
| tBu | 5c | 6c | 87 | 7c | 61 | 8c | 60 |
| H | 5d | 6d | 95 | 7d | 64 | 8d | 68 |
| F | 5e | 6e | 91 | 7e | 75 | 8e | 72 |
| Cl | 5f | 6f | 76 | 7f d | 85 | 8f d | 82 |
| Br | 5g | 6g | 88 | 7g | 64 | 8g | 68 |
| NO2 | 5h | 6h | 88 | 7h d | 82 | 8h d | 88 |
| OMe | 5i | 6i | 82 | 7i | 71 | 8i | 72 |
Yields are not optimized;
cis + trans;
cis and trans compounds were alkylated individually;
1.5 equiv DIPEA at reflux.
Since the synthesis of a series of electronically-altered molecules had been completed (see Supporting Information for details), the Hammett analysis was undertaken. Instead of removing aliquots of the reaction mixture during the epimerization process with TFA as previously executed,26 more detailed observations were obtained by observing the cis/trans ratio in situ. In the present case, 1H NMR spectroscopy had proven useful as a means to observe both of these species. In the following experiments, the cis to trans ratio was determined by integration of 1H NMR spectra while the epimerization of the cis isomer into its trans counterpart was followed by 1H NMR spectroscopy. Beside the less laborious nature of observations of the reaction in the spectrometer, the real strength of this method was the ability to obtain more data over the course of the reaction progress for the rate studies.
Experiments designed to determine useful reaction conditions to study the epimerization process in situ were not trivial. Sampling rate of the detection method (1H NMR), detection interference of either reagents or intermediates, as well as detection of both the cis and trans epimers proved to be of importance in the experimental design. First, the reaction conditions were set up in order to observe the reaction on a reasonable time scale with the ability to monitor the cis to trans conversion. Also, reactants or intermediates must not interfere with the detection of either isomer. In order to obtain useful data, the acid (and its concentration), the cis isomer concentration and the temperature were parameters that were altered.
Initially, the effect of the acid strength on rate was studied. Several acids were employed in the epimerization reaction at different concentrations. Acetic acid proved to be insufficient since epimerization of 7d (12.5 mM) was very slow under the concentrations used (8.7 M). In addition, the significant signal of the acetic acid hydrogen atoms in the 1H NMR spectrum significantly reduced the signals of the tetrahydro-β-carbolines in the spectrum, which indicated acetic acid may not be the optimal catalyst to study the cis to trans epimerization by this method. In an attempt to increase the rate of epimerization, formic acid was employed in higher concentration (13.3 M) than that employed for acetic acid. In this case, the ratio of cis:trans isomers was unable to be determined since the formic acid signal was too strong to permit observation of the signals from the tetrahydro-β-carbolines of interest. To reduce the formic acid signal, the concentration was reduced to 6.6 M. The clarity of the spectrum remained poor and the cis:trans ratio could not be determined; therefore, the use of formic acid was abandoned. Since this issue complicated data analysis, an acid which does not contain unnecessary hydrogen atoms associated with the molecule would be beneficial. The use of deuterated acids would be useful in this case; however the inherent cost of these acids was not the optimal solution. Trifluoroacetic acid (TFA) was chosen based upon this criteria as well as its increased acidity.
Initial attempts with TFA suggested that this acid would be useful with regard to the clarity of the spectrum; however the protons at C-4 were broadened in the 1H NMR spectrum. The integration of these signals could not be used as previously described26 to determine the epimeric ratio; however the triplet from the ethyl ester function of the cis isomer 7d (δ 1.26 ppm) was noticeably shifted upfield as compared to the trans isomer 8d (δ1.16 ppm) (see Figure S1 and Figure S2, Supporting Information). This chemical shift difference permitted the concentration of both isomers to be monitored over the course of the epimerization process.
Due to the increased acidity of TFA as compared to either acetic or formic acid, TFA concentrations of 1.7 M of TFA promoted the rate of the epimerization of the parent 7d ([7d] = 12.5 mM) in less than 1 minute. Since the standard 1H spectrum required approximately 1 minute (16 scans), molar concentrations would not permit observation of the rate of the process. In this regard, the concentration of TFA was reduced significantly by means of dilution in CDCl3 to 2.7 mM while the concentration of 7d was kept constant. After 1 hour, the proton spectrum was taken of the reaction mixture. Approximately 20% of the trans isomer was present, based on the integration of the NMR spectrum. These conditions could be used to monitor the reaction; however, in an effort to minimize reaction times and collect more data in a timely fashion, the TFA concentration was increased. It was found that TFA concentrations up to 67 mM could be used to obtain measurable rate constants when the concentration of the substrate (7d) was equal to 12.5 mM (Table 2).
Table 2.
Effect of TFA concentration on the rate of epimerization.
 | |||
|---|---|---|---|
| [TFA] (mM) | kobs (s−1) a | std dev | std error |
| 13.5 | 0.0016 | 0.0001 | <0.0001 |
| 18.0 | 0.0025 | 0.0001 | 0.0001 |
| 22.5 | 0.0031 | 0.0003 | 0.0002 |
| 27.0 | 0.0049 | 0.0004 | 0.0003 |
| 67.5 | 0.0122 | 0.0007 | 0.0005 |
Average of 2 runs; [7d] = 5.0 mM for all experiments; T = 303.0 K.
Since it was known that electron donating substituents on the phenyl ring at C-1 increased the rate of the epimerization, the 4-methoxy-substituted substrate 7i was subjected to the epimerization process under the same conditions as 7d; [7i] = 12.5 mM, [TFA] = 13.5 mM. Since the rates are compared to one another in a free-energy relationship, all reaction conditions were kept exactly the same. Upon addition of TFA to the cis isomer of 7i, unfortunately, the cis to trans epimerization took place in less than 3 minutes. Analysis of the first proton spectrum in the kinetic experiment showed that all the cis isomer had been converted completely into the trans diastereomer. On the other hand, when catalytic amounts of TFA were employed in order to measure the rate of this reaction 7i→8i, this provided excellent results (see Figure S3, Supporting Information); however, the same reaction conditions were not ideal for phenyl rings with electron-withdrawing substituents at the C-1 position due to the extremely long reaction times. For example, data could not be collected in a reasonable time frame for the p-nitrophenyl analog 7h since epimerization was only 70% complete after 15 hours (Figure S4, Supporting Information). Since the methoxy group was strongly electron donating (σ + = −0.78), it was felt rate data obtained by 1H NMR spectroscopy for this substrate (7i) would not be possible under the same reaction conditions required for the remaining compounds in the series and it was therefore excluded from the kinetic analysis.
Since the p-methoxyphenyl analog 7i reacted too rapidly to obtain accurate rate data, the upper limit of reactivity (electron donating) in order to observe the rate of epimerization by NMR was determined. The σ+ value of an ethyl function located in the para position of a phenyl group was −0.30, which was less able to stabilize a positively charged intermediate when compared to that of a methoxy substituent located in the same position (−0.78); therefore the para-ethyl substituted analog (7a) should not react as fast as the para-methoxy substituted analog (7i). When the 1–4'-ethyl substituted substrate 7a was subjected to the standard epimerization conditions, fortunately the rate could be followed by 1H NMR spectroscopy. Due to this observation, a series of 1-(para-substituted)-phenyl-tetrahydro-β-carbolines could now be employed in the epimerization experiments. Since rate data was obtained for p-ethyl analog 7a, any substituents with a σ+ value greater than −0.30 could be used in the Hammett analysis.
In this regard, the series of molecules illustrated in Scheme 1 and Table 1 (7a-7h) was employed to determine if a linear free energy relationship existed for the epimerization reaction, which, in turn, would provide insight into the mechanism of the epimerization. Each kinetic experiment was set up with the following conditions: [cis] = 12.5 mM, [TFA] = 13.5 mM, T = 303 K. Data for compounds 7a–g were obtained over a reasonable time frame, i.e. complete epimerization occurred in 15–30 minutes for this series of substrates. Because the rates could be observed for this series of molecules, the data was manipulated further; each spectrum of a kinetic experiment was integrated manually to obtain the concentration of the cis isomer and was then plotted for each compound over time. Essentially, a first-order decay of the cis isomer was observed for each compound; however, the initial region of each plot appeared to be more linear in nature than an actual exponential decay (see Figure S5, Supporting Information). It was thought this could be due to an insufficient amount of TFA present during the initial stages of the reaction. If this were the case, a lower concentration of TFA would increase this effect; the linear portion of the plot would extend further throughout the process. To test this hypothesis, the acid concentration was reduced to catalytic amounts (see Figure S6, Supporting Information). Indeed, as the amount of acid catalyst was reduced, the reaction plot became more linear over the epimerization process. This observation can be understood because the catalytic amount of TFA had cycled via equilibrium, presumably, through the epimerization process to the point wherein equimolar concentrations were present between the cis isomer and TFA. Just past the equimolar point, TFA would become an excess reagent, which would then make the reaction essentially pseudo first-order, which resulted in a first-order exponential decay.
Since it was now known that a slight excess of TFA (13.5 mM) was insufficient to produce a pseudo first-order decay from time = 0, the concentration of TFA was increased to 27.0 mM. Typically, 10 equivalents of reagent were required to satisfy pseudo first-order conditions; however, since this epimerization reaction was catalytic, an excess of TFA completely protonated the cis isomer. A study of the amount of TFA required to completely protonate the cis isomer indicated that 1.5 equivalents was the minimum amount of TFA that would produce a first-order decay as the cis isomer was converted into the trans diastereomer over time (see Figure S7, Supporting Information).
Once the final reaction conditions were set, the series of para-substituted substrates (7a–h) were subjected to the epimerization process and followed by 1H NMR spectroscopy. The plots fit a first-order model very well (Figure S8), which permitted extraction of the pseudo first-order rate constants (see Table 3). Since all of the kinetic experiments were carried out under the same reaction conditions, a Hammett plot could be constructed. Here, the observed rate constants were compared directly to that of the unsubstituted parent compound, 7d. As mentioned earlier, electron donating substituents (either by resonance or by induction) increased the rate of epimerization, and therefore, are expected to produce a negative slope in the Hammett plot. In fact, this was exactly what was observed (see Figure 2). Since the data was scattered around the linear regression line, it was clear the data did not fit the Hammett linear free energy relationship well when the standard σpara values were employed. When σ+ para values were used, however, the data correlated much better (Figure 3). The key substrate (7e) which was used to confirm that σ+ para values should be used in this mechanistic study had been prepared (see Supporting Information). The σpara value for a fluorine atom in the para position of a benzene ring was 0.06 while the σ+ para value was −0.07. If the parent 7d epimerized at C-1 faster than the p-fluoro analog 7e, σ+ para values should be employed in the Hammett plots. Indeed, this was the exact observation. Hence, analysis of this observation indicated that a positively charged intermediate was in direct resonance with the substituent at the para (4') position under study and the Hammett ρ value was found to be −1.4. The negative slope was in agreement with the expected value since positive charge builds up throughout the course of the reaction process. However, the observed ρ value of −1.4 was considerably less than typical ρ values for benzylic cations. The decreased sensitivity to changes in the substitution pattern could be attributed to electron delocalization from the neighboring indole ring, which also stabilized the proposed carbocationic intermediate.
Table 3.
Observed rate constants of the cis to trans isomerization.
| Compound | Substituent | kobsa (s−1) | std dev | std error |
|---|---|---|---|---|
| 7a | Et | 0.0073 | 0.0006 | 0.0003 |
| 7b | iPr | 0.0062 | 0.0003 | 0.0002 |
| 7c | tBu | 0.0062 | 0.0004 | 0.0002 |
| 7d | H | 0.0026 | <0.0001 | 0.00005 |
| 7e | F | 0.0018 | <0.0001 | 0.00002 |
| 7f | Cl | 0.0017 | <0.0001 | 0.00001 |
| 7g | Br | 0.0015 | 0.0001 | 0.00006 |
| 7h | NO2 | 0.0001 | <0.0001 | 0.00001 |
average of 3 experiments.
Figure 2.

Hammett plot with σpara values.
Figure 3.

Hammett plot with σ + para values.
The Hammett plot produced here could not be used to unequivocally distinguish between the carbocationic mechanism or the retro Pictet-Spengler pathway for the epimerization process. Both the carbocationic intermediate as well as the iminium ion intermediate are in direct resonance with a substituent at the para position of the phenyl ring at C-1. However, by careful examination of both proposed mechanisms, differences could be noticed when the concentration of TFA was altered.
Observation of the data shown in Figure 4 clearly indicated the rate of epimerization of 7d increased as the concentration of TFA increased and indicated first-order kinetics after protonation of the starting material. Since this reaction was carried out with excess quantities of acid catalyst, the rate should not change to a significant degree as the catalyst (TFA) concentration was altered if one catalyst molecule was involved in the epimerization process. However, this was not the case which suggested that the acid catalyst was more involved in the epimerization mechanism than a single protonation. In this case, it was believed that TFA was involved in a double protonation. In the proposed carbocationic mechanism, the initial protonation took place at the Nb-nitrogen atom. As this mechanistic route progressed and the carbocationic intermediate was formed, a second protonation took place on the Nb-nitrogen atom (see Scheme 2). First-order kinetics was observed with respect to the concentration of TFA as would be expected when k −1 ≫ k 2[TFA].
Figure 4.

Effect of the TFA concentration on the rate.
Scheme 2.

Carbocation-mediated mechanism including a second protonation.
In the carbocationic mechanism, a doubly protonated intermediate such as 9 would form following C–N bond cleavage (Scheme 2). The rate at which 9 would form would increase as the concentration of TFA was increased. In order for cyclization to take place, 9 would have to be deprotonated, which would be slowed in the presence of a higher concentration of TFA; however, the rate of the reaction would not be affected to a large extent as the concentration of such an intermediate would remain small over time due to its instability. Therefore, this step was not considered as the rate determining step. In this case, a rate enhancement would be observed for each mechanistic step when the TFA concentration was increased. This was exactly what was observed (Figure 4). In fact, after initial protonation of the starting material (as detected by 1H NMR spectroscopy), first-order kinetics with respect to the concentration of TFA was observed, therefore the rate was dependent upon k 2 and the concentration of TFA.
As reported by Shudo, et al,30 doubly charged intermediates have been proposed in the tetrahydroisoquinoline series. In these cases, strong acids, such as TFA and trifluoromethanesulfonic acid (TfOH) were used to catalyze the Pictet-Spengler reaction. The acid dependence of these reactions suggested that a dicationic electrophile (di-protonated imine) was the reactive intermediate.30 The same concept could be applied to the cis to trans epimerization mechanism under study here, as shown in Scheme 2. The second protonation would take place after the carbocation had formed. During the time of the second protonation, the C-3/C-4 bond would have time to rotate to the more stable trans configuration, followed by Nb-deprotonation and re-cyclization to form the trans product. This type of mechanism would be consistent with the rate data shown in Figure 4 since the reaction rate was first-order in the concentration of TFA even after initial protonation of the starting material.
On the other hand, if the retro Pictet-Spengler pathway was involved, the initial protonation step (step A, Scheme 3) to form 10 would increase in rate as the concentration of TFA increases. However, the rate of the subsequent deprotonation to form 11 (step B, Scheme 3) would decrease in rate as the concentration of TFA was increased. When a high concentration of TFA was present, the concentration of 10 would increase over the course of the reaction. In all of these steps, the Nb-nitrogen would remain protonated since its pKa was much greater than the carbon at the indole-2 position. In fact, the retro Pictet-Spengler mechanism cannot proceed with this Nb-nitrogen atom protonated. Although the protonated (10) and unprotonated (11) forms are in equilibrium, the likelihood of the free amine being present becomes increasingly smaller as the concentration of TFA increases. Therefore, deprotonation (step B, Scheme 3) was presumably the rate determining step. This was contrary to the data depicted in Figure 4 and was inconsistent with first-order kinetics with respect to the concentration of TFA.
Scheme 3.

Retro Pictet-Spengler mechanism.
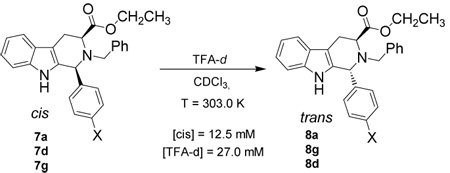
To determine if protonation at the indole-2 position was or was not the rate determining step in the retro Pictet-Spengler process, kinetic isotope experiments were designed and executed. Since primary KIEs are observable when an acid is involved in the rate determining step, such experiments, in support of the other kinetic experiments, would provide useful mechanistic information about the epimerization process. If, in fact, this protonation was the rate determining step, strong primary kinetic isotope effects (KIE) would be observed. The rate would be noticeably decreased when the epimerization was run in TFA-d. Since protonation/deprotonation of the nitrogen atom is fast (due to basicity), the isotope effect would not be observed for these mechanistic steps. In fact, similar experiments have been carried out by O’Connor, et al. in regard to the biosynthesis of indole alkaloids.31 A deuterium atom was synthetically installed at the indole-2 position of tryptamine, followed by a Pictet-Spengler reaction with propanal under acidic conditions. KIEs between 2.0 and 2.6 were observed by O’Connor, et al., which suggested that deprotonation at the indole-2 position was the rate determining step in the Pictet-Spengler reaction.31 Similar results were expected in these experiments if, in fact, protonation (or deprotonation) at the indole-2 position was the rate determining step in the retro Pictet-Spengler process. Each experiment was set up to keep the same reaction conditions as the reactions run earlier in TFA: that is to say, the concentration of starting cis isomer, concentration of TFA-d and reaction temperature (303 K) in the NMR spectrometer (Scheme 4) were maintained as before. A small primary kinetic isotope effect (1.2–1.3) was observed (Table 4). The KIEs observed here were much smaller than those observed by O’Connor, et al.31 This data was inconsistent with the retro Pictet-Spengler mechanism for the cis to trans epimerization process since a strong KIE was not observed. Therefore, it was concluded that protonation at the indole-2 position was not the rate determining step in the cis to trans epimerization of 1-(4'-substituted)-phenyl-tetrahydro-β-carbolines.
Scheme 4.

cis to trans Epimerization process in TFA-d on 1-(substituted)-phenyl-tetrahydro-β-carbolines 7a, 7d and 7g.
Table 4.
KIE Experiments on 1-(substituted)-phenyl-tetrahydro-β-carbolines.
 | ||||
|---|---|---|---|---|
| Compound | Substituent X |
kobs ave (s−1) |
k-dobs ave (s−1) |
KIE |
| 7a | Et | 0.0073 | 0.0060 | 1.21 |
| 7d | H | 0.0018 | 0.0015 | 1.20 |
| 7g | Br | 0.0016 | 0.0013 | 1.30 |
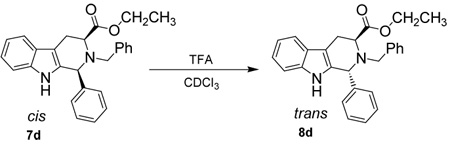
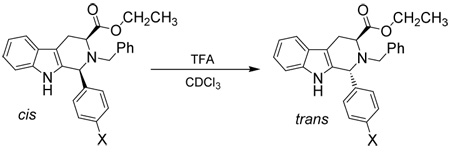
In addition to a study of the rates of epimerization for 1-(4’-substituted)-phenyl-tetrahydro-β-carbolines, chemical experiments were also conducted in order to determine the mechanism of the cis to trans epimerization. Czerwinski32 had shown that deuterium was not incorporated into the trans product in the presence of deuterated TFA which confirmed that the olefinic mechanism had not occurred in the cis to trans isomerization. This same experiment was carried out in the 1-(4'-substituted)-phenyl-tetrahydro-β-carboline series (7a, 7d, 7g) to determine if different substituent patterns altered the outcome of that process. The cis substrate (7a, 7d, 7g) was dissolved in CDCl3 and TFA-d was added (Scheme 4). The cis to trans epimerization process was allowed to take place and the 1H NMR spectrum was taken following aqueous work-up. Full integration of the C-1 proton was observed in the spectrum. This was identical to the result observed by Czerwinski32 which unequivocally indicated that the olefinic mechanism did not occur in the 1-(4’-substituted)-phenyl-tetrahydro-β-carboline series which was in agreement with the earlier work of Joule, et al.33 Therefore, this mechanism did not occur with 1-alkyl or 1-aryl substituents.
If the cis to trans epimerization proceeded through the retro Pictet-Spengler mechanism, Nb-benzyl tryptophan ethyl ester 15 would be an expected product if the reaction was carried out in the presence of water. Here, the iminium ion intermediate would be hydrolyzed to the tryptophan ethyl ester derivative 15 and the corresponding aldehyde. In this vein, the parent 7d (12.5 mM) was dissolved in CDCl3. Prior to the addition of TFA, 1.0 volume equivalent of water (13.9 M) was added to the reaction medium. The TFA (27.0 mM) was then added and the epimerization reaction was allowed to take place (Scheme 5). The reaction was monitored by TLC and the 1H NMR spectrum was acquired when the cis to trans epimerization was complete. At no time was Nb-benzyl trytophan ethyl ester 15 or the corresponding aryl aldehyde observed by TLC nor by 1H NMR spectroscopy. Hydrolysis of the ethyl ester was also not observed. With a large excess of water present in the reaction medium, Nb-benzyl tryptophan ethyl ester would be expected if the iminium ion was formed in a retro Pictet-Spengler process since this would be favored entropically. Since the epimerization was complete for the parent 7d, the same reaction was carried out for both the p-ethylphenyl, 7a, and p-bromophenyl, 7g, substrates. The same result was realized in both of these cases, which suggested that the mechanism of epimerization did not change as the electronic nature at the C-1 position was varied. This data was consistent with the carbocationic mechanism since hydrolysis of a proposed iminium ion which would arise in the retro Pictet-Spengler process was not observed.
Scheme 5.

cis to trans Epimerization in the presence of water.
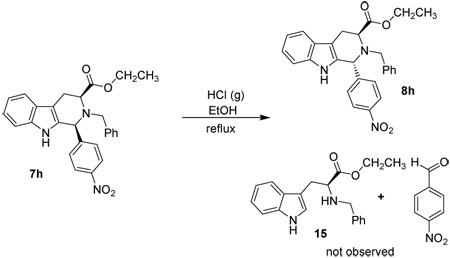
In a separate earlier experiment, Hamaker34 had shown that a retro Pictet-Spengler mechanism was forced to occur if a strong electron withdrawing group was installed on the indole Na-nitrogen atom. This result was observed when a Na-sulfonamide derivative 16 was reacted in ethanolic HCl. The Nb-benzyl tryptophan derivative 21 was the exclusive product of this process in the Na-sulfonamido case of Hamaker;34 the trans isomer 18 was not observed (see Scheme 6). It was felt this result was observed because the electron withdrawal of the sulfonamide retarded stabilization of the carbocationic intermediate 17 which prevented cleavage of the C–N bond. However, continued heating in ethanolic HCl resulted in the production of the tryptophan alkyl ester 21, presumably via a retro Pictet-Spengler reaction through iminium ion intermediate 20. Since the retro Pictet-Spengler mechanism had been postulated to take place in the Na-sulfonamide case, the cis to trans epimerization of the Na-H case, 7h, was carried out under similar reaction conditions (Table 4). In this case, hydrolysis of the tetrahydro-β-carboline ring system to give Nb-benzyl tryptophan ethyl ester 15 or the corresponding aryl aldehyde did not occur (see Table 5). After 3 hours at reflux in concentrated ethanolic HCl, a mixture of cis and trans isomers was detected by TLC. This was confirmed by 1H NMR spectroscopy following aqueous work-up. The products of this reaction were then re-subjected to the same reaction conditions overnight. The following day, decomposition was observed by TLC, but no Nb-benzyl tryptophan ethyl ester 15 or para-nitrobenzaldehyde was observed by TLC nor by NMR spectroscopy at any time during these reactions.
Scheme 6.


Proposed mechanism of the retro Pictet-Spengler process observed by Hamaker.34
Table 5.
cis to trans Epimerization of 1-(4’-nitro)-tetrahydro-β-carboline 7h under the conditions of Hamaker.34
 | ||||
|---|---|---|---|---|
| Ethanolic HCl | Time | 7h % |
8h % |
15 % |
| saturated solution | 3 h | 90 | 10 | < 1a |
| 10 h c | ND | ND | ND | |
| 1:2 dilution with EtOH |
2 d | 60 | 40 | < 1 b |
| 4 d | 25 | 75 | < 1 b | |
Analyzed by 1H NMR;
by TLC;
decomposition occurred.
To minimize the possibility of decomposition, a 1:2 dilution of concentrated ethanolic HCl with EtOH was used as the solvent/reagent system. After 2 days at reflux, a mixture of cis and trans isomers was again observed by TLC in the 1-(4’-nitro)-tetrahydro-β-carboline case, 7h. Additional ethanolic HCl was added to the reaction mixture in order to increase the rate of epimerization. The appearance of more of the the trans 1-(4'-nitro)-tetrahydro-β-carboline diastereomer 8h was observed after 4 hours. The reaction was continued for an additional 2 days at which point a mixture of cis and trans isomers remained, accompanied by slight decomposition as observed by TLC. The reaction was stopped at this point since it was clear that hydrolysis of an iminium ion intermediate from a retro Pictet-Spengler mechanism did not occur vs. an authentic sample of 15.
Electron withdrawal at the C-1 position via the para nitro group did not promote a retro Pictet-Spengler process as was the case with the Na-sulfonamide species of Hamaker.34 In the cases with an indole, or an electron rich indole, electron density flowed away from the indole ring during the formation of the proposed carbocationic intermediate. This was contrary to a retro Pictet-Spengler type mechanism wherein electrons flow into the indole ring when the iminium ion was formed.34 Therefore, it was reasonable to believe that the retro Pictet-Spengler mechanism did not occur in the cis to trans epimerization process of 1,2,3,4-tetrahydro-β-carbolines when indole, or an electron rich indole was employed.
Enthalpies and entropies of activation were determined from kinetic data. Such data was acquired for the parent 7d at temperatures ranging from 298 to 313 K (Table 6). The rate data collected as shown in Table 6 was then plotted according to the Arrhenius equation (Figure 5) to obtain energy of activation (Ea) data for the parent substrate, 7d, and was determined to be 153.7 kJ/mol. Since the Arrhenius equation correlated rate data directly to the energy of activation, the differences in rates, namely the ρ value from Figure 3, was employed to determine the Ea for the series of compounds under study. The activation energies ranged from 152.3 to 153.8 kJ/mol for the series of 1-(4'-substituted)-phenyl-1,2,3,4-tetrahydro-β-carbolines (see Table 7).
Table 6.
Temperature effect on rate.
 | |
|---|---|
| Temperature K |
kobsa s−1 |
| 313.0 | 0.0132 |
| 308.0 | 0.0059 |
| 303.0 | 0.0018 |
| 300.0 | 0.0012 |
| 298.0 | 0.0007 |
[cis] = 12.5 mM; [TFA] = 27.0 mM;
average of 3 experiments.
Figure 5.


Arrhenius plot.
Table 7.
Energies of activation.
 | |||
|---|---|---|---|
| Compound | Substituent X |
σρ | Ea kJ/mol |
| 7a | Et | 0.4187 | 152.3 |
| 7b | iPr | 0.3908 | 152.3 |
| 7c | tBu | 0.3629 | 152.3 |
| 7d | H | 0.0000 | 152.7 |
| 7e | F | 0.0977 | 152.6 |
| 7f | Cl | −0.1535 | 152.9 |
| 7g | Br | −0.2093 | 152.9 |
| 7h | NO2 | −1.1025 | 153.8 |
[cis] = 12.5 mM; [TFA] = 27.0 mM. Data shown in grey were calculated based on the Hammett plot and assumed negligible change in ΔS‡
In order to obtain more specific thermodynamic data, the rate data from Table 6 for 1-phenyl-tetrahydro-β-carboline 7d was employed to construct an Eyring plot (Figure 6). The enthalpy of activation (ΔH‡) and the entropy of activation (ΔS‡) were extracted from the linear plot and were determined to be 150.2 kJ/mol and 199 J/K·mol, respectively and the data is depicted in Table 8. Since both ΔH‡ and ΔS‡ were determined, the Gibb’s free energy of activation (ΔG‡) was calculated to be 89.9 kJ/mol. It was important to realize that the Hammett free energy relationship correlated rate data to the free energy of the reaction. Since the Hammett plot was constructed and ΔG‡ had been determined for 1-phenyl-tetrahydro-β-carboline 7d, ΔG‡ for the remaining compounds studied in the Hammett analysis were determined and the data is presented in Table 8. The difference in ΔG‡ across the series of compounds was found to be 8.8 kJ/mol. Presumably, ΔS‡ remained relatively constant throughout the series of compounds studied in the Hammett analysis. Due to the relatively small effect of ΔS‡ on ΔG ‡, small changes in entropy would not affect ΔG‡ to a significant degree. If this were true, ΔH‡ was calculated for the remaining compounds and the data is shown in Table 8.
Figure 6.


Eyring plot.
Table 8.
Thermodynamic data.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | Substituent X |
σ+ | σρ | Δ G‡ kJ/mol |
Δ H‡a kJ/mol |
Δ S‡a J/K mol |
| 7a | Et | −0.30 | 0.4187 | 87.5 | 146.7 | 200 |
| 7b | iPr | −0.28 | 0.3908 | 87.7 | 147.0 | 200 |
| 7c | tBu | −0.26 | 0.3629 | 87.8 | 147.2 | 200 |
| 7d | H | 0.00 | 0.0000 | 89.9 c | 150.2 b | 199 b |
| 7e | F | −0.07 | 0.0977 | 89.4 | 149.4 | 200 |
| 7f | Cl | 0.11 | −0.1535 | 90.8 | 150.4 | 200 |
| 7g | Br | 0.15 | −0.2093 | 91.2 | 150.4 | 200 |
| 7h | NO2 | 0.79 | −1.1025 | 96.3 | 156.9 | 200 |
[cis] = 12.5 mM; [TFA] = 27.0 mM ; at 303.0 K;
if ΔS‡ remains reasonably constant;
determined graphically from the Eyring plot;
calculated from ΔH‡ and ΔS‡. Data shown in grey were calculated based on the Hammett plot and assumed negligible change in ΔS‡.
Conclusions
Three potential mechanistic pathways have been considered for the cis to trans epimerization of 1,2,3-trisubstituted-1,2,3,4-tetrahydro-β-carbolines. One of these, the olefinic pathway, had been ruled out by Czerwinski32 and in the present study by deuterium labeling experiments. Results from epimerizations carried out in TFA-d indicated that no deuterium was incorporated at the C-1 position, which would be necessary if the olefinic pathway was to take place. The remaining two mechanisms of epimerization, namely the carbocationic pathway and the retro Pictet-Spengler pathway, were compared on a kinetic basis.
A series of 1-(4'-substituted)-phenyl-tetrahydro-β-carbolines were synthesized by the Pictet-Spengler cyclization in order to study the cis to trans epimerization mechanism. In order to study this trend in more detail, the epimerization reaction was followed by 1H NMR spectroscopy. The observed pseudo first-order rate constants were determined graphically which permitted a quantitative comparison of the rates of epimerization of each of the 1-(4'-substituted)-phenyl-tetrahydro-β-carbolines studied (7a-7h). The observed rate constants were employed to construct a Hammett plot, which correlated to a line very well when σ+ values were utilized. A ρ value of −1.4 was observed for the series of compounds studied and the negative slope was consistent with a positively charged intermediate. The magnitude of the ρ value was found to be considerably smaller than those expected for a benzylic cation, however this effect was attributed to electron delocalization from the neighboring indole ring, thus stabilizing the proposed carbocationic intermediate.
It was believed that a second protonation took place at the Nb-nitrogen atom after formation of the carbocationic intermediate, which resulted in a dicationic intermediate. The observed first-order kinetics with respect to TFA (after initial protonation of the substrate) was consistent with the formation of the dicationic intermediate in the carbocationic mechanism as this was determined to be the rate determining step. On the other hand, pseudo first-order kinetics was not expected in the retro Pictet-Spengler process since the formation of the iminium ion intermediate required the Nb-nitrogen atom to be unprotonated.
Kinetic isotope experiments were devised and carried out where small primary kinetic isotope effects of 1.2–1.3 were observed for three separate tetrahydro-β-carbolines. The determined KIEs were considerably smaller than those reported by O’Connor31 for deprotonation at the indole-2 position in the Pictet-Spengler mechanism. This implied that protonation at the indole-2 position was not involved in the rate determining step of the cis to trans epimerization process. Therefore, the retro Pictet-Spengler mechanism was ruled out since the rate of epimerization was dependent upon the concentration of TFA (first-order kinetics after protonation of the substrate) and protonation at the indole-2 position was not involved in the rate determining step.
In separate, chemical experiments, Nb-benzyl tryptophan ethyl ester was not observed when 1-(4'-substituted)-phenyl-tetrahydro-β-carbolines were epimerized with TFA in the presence of water or under the ethanolic HCl conditions of Hamaker.34 Presumably the iminium ion intermediate did not form in either case, and thus the retro Pictet-Spengler process did not occur since neither of these two products was observed by TLC or 1H NMR spectroscopy.
In conclusion, a Hammett study alone was insufficient to distinguish between a carbocation-mediated pathway and a retro Pictet-Spengler pathway in the 1-(4’-substituted)-phenyl-1,2,3,4-tetrahydro-β-carboline series. However, analysis of both rate data and chemical experiments proved to be inconsistent with the retro Pictet-Spengler mechanism and it was thus ruled out. On the other hand, the data was found to be consistent with a carbocationic mechanism for the C-1/N-2 bond scission process in the isomerization of cis Nb-benzyl tetrahydro-β-carbolines into their more stable trans diastereomers. This process has important implications in regard to the total synthesis of indole alkaloids by internal asymmetric induction.
Experimental Section
General procedure for the preparation of both cis and trans diastereomers via the Pictet-Spengler reaction of aromatic aldehydes under acidic conditions.26
L-Tryptophan ethyl ester (13.5 g, 0.058 mol) was added to a 500-mL three-neck round-bottom flask containing dry benzene (300 mL). The flask was attached to a Dean-Stark trap topped by a reflux condenser for azeotropic removal of water during the course of the reaction. Trifluoroacetic acid (0.12 mol, 2.0 equiv) was added and this was followed by addition of the aldehyde in dry benzene (0.070 mol, 1.2 equiv). The mixture was allowed to heat to reflux for 4–8 h under argon until all of the tryptophan ethyl ester was consumed as indicated by TLC (silica gel, hexanes:EtOAc, 3:1). The reaction mixture was then cooled to r.t. and the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (300 mL) and washed with a solution of cold, saturated NaHCO3 (3 × 300 mL) to remove TFA. The organic layer was separated and dried (Na2SO4). Analysis by TLC (silica gel, hexanes:EtOAc, 3:1) of the crude oil indicated the presence of two major components along with some unreacted aldehyde. The residue was chromatographed on silica gel (gradient elution, EtOAc: hexane = 1:10, 2:10, 3:10, 4:10) to provide the pure cis and trans diastereoisomers, respectively.
cis-1-Phenyl-1,2,3,4-tetrahydro-9H-β-carboline-3-carboxylic acid ethyl ester (5d)
The general procedure was followed and the cis/trans diastereomers were separated by column chromatography on silica gel. L-tryptophan ethyl ester·HCl (20.1 g, 0.075 mol), benzaldehyde (8.3 mL, 0.081 mol) and TFA (13.2 mL, 0.18 mol) were used to obtain the cis isomer (95 % overall yield, cis + trans). 5d: mp 161–163 °C; −8.89 (c 0.48, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 7.7 Hz, 1H), 7.54 (s, 1H), 7.47-7.41 (m, 5H), 7.27-7.24 (m, 1H), 7.23-7.17 (m, 2H), 5.31 (s, 1H), 4.38-4.29 (m, 2H), 4.02 (dd, J = 11.2, 4.2 Hz, 1H), 3.30 (ddd, J = 15.1, 4.2, 1.8 Hz, 1H), 3.11-3.06 (m, 1H), 2.68 (br s, 1H), 1.41 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 173.2, 141.1, 136.6, 135.1, 129.4, 129.13, 129.10, 127.6, 122.4, 120.1, 118.7, 111.4, 109.4, 61.7, 59.2, 57.4, 26.1, 14.7; MS (EI) m/e (rel intensity): 320 (M+, 69), 247 (65), 218 (100), 169 (42), 144 (36), 77 (45); Anal. Calcd for C20H20N2O2: C 74.98, H 6.29, N 8.74; Found: C 75.21, H 6.40, N 8.66. This material was employed in a later step.
cis-1-(4-Isopropyl-phenyl)-1,2,3,4-tetrahydro-9H-β-carboline-3-carboxylic acid ethyl ester (5b)
The general procedure was followed and the cis/trans diastereomers were separated by column chromatography on silica gel. L-tryptophan ethyl ester·HCl (20.1 g, 0.074 mol), 4-isopropylbenzaldehyde (13.6 mL, 0.090 mol) and TFA (13.2 mL, 0.18 mol) were used to obtain the cis isomer (91 % overall yield, cis + trans). 5b: mp 195–197 °C; −15.56 (c 1.02, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.63-7.61 (m, 1H), 7.58 (s. 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 7.25-7.23 (m, 1H), 7.22-7.17 (m, 2H), 5.26 (s, 1H), 4.37-4.30 (m, 2H), 4.00 (dd, J = 11.2, 4.2 Hz, 1H), 3.29 (dd, J = 14.2, 4.2 Hz, 1H), 3.10-3.05 (m, 1H), 3.00 (p, J = 6.9 Hz, 1H), 2.60 (br s, 1H), 1.40, (t, J = 7.2 Hz, 3H), 1.34 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 173.2, 149.8, 138.5, 136.6, 135.4, 129.1, 128.9, 127.6, 127.5, 127.3, 122.3, 120.0, 118.7, 111.4, 109.3, 61.6, 58.8, 57.5, 34.4, 26.2, 24.5, 24.4, 14.7; MS (EI) m/e (rel intensity): 362 (M+, 94), 289 (87), 261 (54), 218 (100), 169 (32), 144 (33); Anal. Calcd for C23H26N2O2: C 76.21, H 7.23, N 7.73; Found: C 76.35, H 7.35, N 7.61. This material was employed in a later step.
General procedure for the alkylation of the Nb-H tetrahydro-β-carbolines.26
Hunig’s base (N,N-diisopropylethylamine, DIPEA; 87.0 mmol, 15 equiv), and benzyl bromide (7.0 mmol, 1.2 equiv) were added to a solution of tetrahydro-β-carboline (5a–e, 5g, 5i, 8a–e, 8g; 5.8 mmol, 1 equiv) in dry acetonitrile (10 mL). This mixture was cooled to 0°C and allowed to come to r.t. overnight. The solvent was removed under reduced pressure and the residue was dissolved in cold EtOAc (10 mL) to remove the salt of the Hunig’s base which had precipitated. The solution was filtered and the filtrate was dried (Na2SO4) and concentrated in vacuo to yield a brown oil. Analysis of the crude material by TLC indicated the presence of a major component along with some unreacted benzyl bromide and starting material. The residue was purified on a silica gel column via flash chromatography (hexane:EtOAc 7:1) to yield the pure Nb-benzyl tetrahydro-β-carboline isomer, respectively, as an oil which was then triturated with hexanes to form a pale yellow solid or white solid.
cis-2-Benzyl-1-(4-ethyl-phenyl)-1,2,3,4-tetrahydro-9H-β-carboline-3-carboxylic acid ethyl ester(7a)
The modified general procedure was followed. The cis Nb-H analog 5a (0.50 g, 1.1 mmol), DIPEA (3.3 mL, 18.9 mmol) and benzyl bromide (0.2 mL, 1.5 mmol) were used to obtain the cis Nb-benzyl isomer (69 %). 7a: −86.49 (c 0.38, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 7.8 Hz, 1H), 7.39-7.24 (m, 9H), 7.20 (d, J = 8.1, 2H), 7.18-7.16 (m, 2H), 4.99 (s, 1H), 4.13 (d, J = 14.8 Hz, 1H), 3.98 (d, J = 14.8 Hz, 1H), 3.87 (dd, J = 7.9, 3.8 Hz, 1H), 3.86-3.75 (m, 2H), 3.43 (dd, J = 15.6, 6.3 Hz, 1H), 3.11 (dd, J = 15.2, 4.9 Hz, 1H), 2.69 (dd, J = 15.2, 7.6 Hz, 2H), 1.29 (t, J = 7.6 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 173.8, 144.5, 138.8, 137.8, 136.8, 134.1, 129.8, 128.5, 128.4, 127.44, 127.37, 122.2, 119.9, 118.8, 111.2, 107.6, 62.0, 61.5, 61.1, 57.2, 29.0, 24.6, 16.0, 14.4; MS (EI) m/e (rel intensity): 438 (M+, 31), 365 (100), 248 (30), 218 (54); Anal. Calcd for C29H30N2O2: C 79.42, H 6.89, N 6.39; Found: C 79.51, H 6.92, N 6.35.
cis-2-Benzyl-1-(4-tert-butyl-phenyl)-1,2,3,4-tetrahydro-9H-β-carboline-3-carboxylic acid ethyl ester (7c)
The modified general procedure was followed. The cis Nb-H analog 5c (0.52 g, 1.4 mmol), DIPEA (3.6 mL, 20.6 mmol) and benzyl bromide (0.3 mL, 2.1 mmol) were used to obtain the cis Nb- benzyl isomer (61 %). 7c: mp 155–156 °C; +48.57 (c 0.14, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 7.8 Hz, 1H), 7.41-7.32 (m, 7H), 7.29-7.25 (m, 4H), 7.20-7.15 (m, 2H), 5.02 (s, 1H), 4.12 (d, J = 14.7 Hz, 1H), 4.01 (d, J = 14.7 Hz, 1H), 3.88 (dd, J = 6.5, 5.2 Hz, 1H), 3.86-3.81 (m, 1H), 3.72-3.67 (m, 1H), 3.44 (dd, J = 15.6, 6.6 Hz, 1H), 3.11 (dd, J = 14.9, 4.4 Hz, 1H), 1.36 (s, 9H), 1.12 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 173.8, 151.2, 139.0, 137.3, 136.7, 133.9, 129.6, 129.5, 128.5, 127.4, 125.7, 122.1, 119.9, 118.8, 111.2, 107.8, 61.7, 61.1, 61.0, 57.5, 35.0, 31.8, 24.2, 14.4; MS (EI) m/e (rel intensity): 466 (M+, 25), 393 (90), 275 (26), 218 (100); Anal. Calcd for C31H34N2O2: C 79.79, H 7.34, N 6.00; Found: C 79.85, H 7.53, N 5.93.
Procedure for the 1H NMR kinetics experiments
The 1,2,3-trisubstitued-1,2,3,4-tetrahydro-β-carboline (7a–h; 1.25×10−5 mol, 12.5 mM final concentration) was dissolved into CDCl3 (800 µL) and then transferred to an NMR tube. The proton spectrum was taken and then the tube was ejected. The TFA (200 µL of a 135 mM solution in CDCl3, 27.0 mM final concetration) was quickly added to the tube and the tube injected into the spectrometer with an internal temperature of 303.0 K. The sample was locked and shimmed before starting the kinetic experiment. A series of 50 spectra were taken over the course of 48 minutes for all compounds except for the 4-nitro compound (7h) where 50 spectra were taken over an 11 hour period. The resulting 2D data set was then phased manually and then divided into 50 equal slices, each representing one 1H spectrum. Each spectrum was integrated with the trans isomer calibrated to one proton. The ratio of cis to trans isomers was then determined, which allowed for the calculation of the cis isomer concentration. The concentration data was then plotted against time. A first-order exponential decay was realized and the pseudo first-order rate constant was determined graphically.
Supplementary Material
Acknowledgement
The authors greatly acknowledge Professors Graham Moran, Guilherme Indig and Alan Schwabacher for helpful discussions as well as Dr. F. Holger Försterling for technical support. This work was supported in part by NIMH MH046851 and the Lynde and Harry Bradley Foundation.
Footnotes
Supporting Information Available: Supplemental figures, complete experimental procedures, synthetic procedures and compound characterization data as well as 1H and 13C NMR spectra for all compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Pictet A, Spengler T. Ber. Dtsch. Chem. Ges. 1911;44:2030. [Google Scholar]
- 2.Tatsui G. Yakugaku Zasshi. 1928;48:453. [Google Scholar]
- 3.Bunin BA, Dener JM, Kelly DE, Paras NA, Tario JD, Tushup SP. J. Comb. Chem. 2004;6:487. doi: 10.1021/cc0340776. [DOI] [PubMed] [Google Scholar]
- 4.Harvey SC, Foster KL, McKay PF, Carroll MR, Seyoum R, Woods JE, II, Grey C, Jones CM, McCane S, Cummings R, Mason D, Ma C, Cook JM, June HL. J. Neurosci. 2002;22:3765. doi: 10.1523/JNEUROSCI.22-09-03765.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster KL, McKay PF, Seyoum R, Milbourne D, Yin W, Sarma P, Cook JM, June HL. Neuropsychopharmacology. 2004;29:269. doi: 10.1038/sj.npp.1300306. [DOI] [PubMed] [Google Scholar]
- 6.Ungemach F, DiPierro M, Weber R, Cook JM. J. Org. Chem. 1981;46:164. [Google Scholar]
- 7.Czarnocki Z, Suh D, MacLean DB, Hultin PG, Szarek WA. Can. J. Chem. 1992;70:1555. [Google Scholar]
- 8.Reddy MS, Cook JM. Tetrahedron. Lett. 1994;35:5413. [Google Scholar]
- 9.Waldmann H, Schmidt G, Henke H, Burkard M. Angew. Chem. Int. Ed. 1995;34:2402. [Google Scholar]
- 10.Yamada H, Kawate T, Matsumizu M, Nishida A, Yamaguchi K, Nakagawa M. J. Org. Chem. 1998;63:6348. doi: 10.1021/jo980810h. [DOI] [PubMed] [Google Scholar]
- 11.Gremmen C, Willemse B, Wanner MJ, Koomen GJ. Org. Lett. 2000;2:1955. doi: 10.1021/ol006034t. [DOI] [PubMed] [Google Scholar]
- 12.Taylor MS, Jacobsen EN. J. Am. Chem. Soc. 2004;126:10558. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]
- 13.Seayad J, Seayad AM, List B. J. Am. Chem. Soc. 2006;128:1086. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- 14.Wanner MJ, van der Haas RNS, de Cuba KR, van Maarseveen JH, Hiemstra H. Angew. Chem. Int. Ed. 2007;46:7485. doi: 10.1002/anie.200701808. [DOI] [PubMed] [Google Scholar]
- 15.Wanner MJ, Boots RNA, Eradus B, de Gelder R, van Maarseveen JH, Hiemstra H. Org. Lett. 2009;11:2579. doi: 10.1021/ol900888e. [DOI] [PubMed] [Google Scholar]
- 16.Reisman SE, Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2008;130:7198. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klausen RS, Jacobsen EN. Org. Lett. 2009;11:887–890. doi: 10.1021/ol802887h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sewgobind NV, Wanner MJ, Ingemann S, de Gelder R, van Maarseveen JH, Hiemstra H. J. Org. Chem. 2008;73:6405. doi: 10.1021/jo8010478. [DOI] [PubMed] [Google Scholar]
- 19.Shi X-X, Liu S-L, Xu W, Xu Y-L. Tetrahedron Asymm. 2008;19:435. [Google Scholar]
- 20.Edwankar CR, Edwankar RV, Rallapalli S, Cook JM. Nat. Prod. Commun. 2008;3:1839. [Google Scholar]
- 21.Cox ED, Cook JM. Chem. Rev. 1995;95:1797. [Google Scholar]
- 22.Czerwinski KM, Cook JM. In: Advances in Heterocyclic Natural Products Synthesis. Pearson W, editor. Vol. 3. Greenwich, CT: JAI Press; 1996. p. 217. [Google Scholar]
- 23.Soe T, Kawate T, Fukui N, Hino T, Nakagawa M. Heterocycles. 1996;42:347. [Google Scholar]
- 24.Kawate T, Yamanaka M, Nakagawa M. Heterocycles. 1999;50:1033. [Google Scholar]
- 25.Han DM, Försterling FH, Deschamps JR, Parrish D, Liu XX, Yin WY, Huang SM, Cook JM. J. Nat. Prod. 2007;70:75. doi: 10.1021/np060391g. [DOI] [PubMed] [Google Scholar]
- 26.Kumpaty HJ, Van Linn ML, Kabir MS, Forsterling FH, Deschamps JR, Cook JM. J. Org. Chem. 2009;74:2771. doi: 10.1021/jo8028168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammett LP. Chem. Rev. 1935;17:125. [Google Scholar]
- 28.Hammett LP. J. Am. Chem. Soc. 1937;59:96. [Google Scholar]
- 29.Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165. [Google Scholar]
- 30.Yokoyama A, Ohwada T, Shudo K. J. Org. Chem. 1999;64:611. [Google Scholar]
- 31.Maresh JJ, Giddings L, Friedrich A, Loris EA, Panjikar S, Trout BL, Stockigt J, Peters B, O’Connor SE. J. Am. Chem. Soc. 2008;130:710. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czerwinski KM. Ph.D thesis. University of Wisconsin-Milwaukee; 1995. [Google Scholar]
- 33.Gaskell AJ, Joule JA. Tetrahedron. 1967;23:4053. doi: 10.1016/s0040-4020(01)97916-5. [DOI] [PubMed] [Google Scholar]
- 34.Hamaker LK. Ph.D thesis. University of Wisconsin-Milwaukee; 1995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.