

Abstract
Cancer cells often have unstable genomes and increased centrosome and chromosome numbers, which play an important part of malignant transformation in the most recent models tumorigenesis. However, very little is known about divisional failures in cancer cells that may lead to chromosomal and centrosomal amplifications. We show here that cancer cells often failed at cytokinesis due to decreased phosphorylation of the myosin regulatory light chain (MLC), a key regulatory component of cortical contraction during division. Reduced MLC phosphorylation was associated with high expression of myosin phosphatase and/or reduced myosin light chain kinase levels. Furthermore, expression of phosphomimetic MLC largely prevented cytokinesis failure in the tested cancer cells. When myosin light chain phosphorylation was restored to normal levels by phosphatase knockdown multinucleation, and multipolar mitosis were both markedly reduced, resulting in enhanced genome stabilization. Furthermore, both overexpression of myosin phosphatase or inhibition of the myosin light chain kinase (MLCK) in nonmalignant cells can recapitulate some of the mitotic defects of cancer cells, including multinucleation and multipolar spindles, indicating these changes are sufficient to reproduce the cytokinesis failures we see in cancer cells. These results for the first time define the molecular defects leading to divisional failure in cancer cells.
Keywords: cytokinesis, myosin light chain kinase, multinucleation, multipolar spindles, myosin phosphatase, myosin regulatory light chain
Introduction
Chromosomal instability (CIN) is a key mechanism resulting in genomic changes associated with tumorigenesis (Geigl et al., 2008). In many cancer cells, CIN is associated with chromosomal and centrosomal amplification. Tetraploidy, or twice the normal number of chromosomes, has been observed in some cancers, such as myeloid leukemia, malignant gliomas, colonic adenocarcinoma, (Lemez et al., 1998; Park et al., 1995; Takanishi et al., 1996) and tumor-derived cell lines (Olaharski et al., 2006; Shi and King, 2005). Shackney et al. have shown that one of the earliest events in human tumor formation is development of tetraploidy (Shackney et al., 1989). Additionally, Pellman and coworkers found that tetraploidy in primary cells lacking p53, caused by an experimentally-induced failure of cytokinesis, promotes tumorigenesis in mice (Fujiwara et al., 2005). In addition to increased chromosome number, cytokinesis failure also leads to centrosomal amplification producing multipolar mitotic spindles that cannot segregate their chromosomes evenly (King, 2008; Nigg, 2006). Centrosomal amplification is also common in tumor cells, is associated with a more severe prognosis, and centrosomal amplification causes tumor formation in model systems (Sluder and Nordberg, 2004; Wang et al., 2004). These observations suggest that failure of cytokinesis, and the associated chromosomal and centrosomal amplification, could play an important role in human cancer.
While much is known about the basic mechanisms of cytokinesis, how cytokinesis failure contributes to tetraploidy in cancer cells is still unknown. Cytokinesis begins in late anaphase with the assembly of a transient structure called the contractile ring at the equator between the spindle poles. Contraction occurs during telophase, driven by an actin-myosin molecular motor system, and results in the formation of a cleavage furrow. Nonmuscle myosin II is composed of a heavy chain and essential and regulatory light chains (MLC). In higher eukaryotes, cellular myosin is activated by phosphorylation of MLC at Thr18/Ser19 (Komatsu et al., 2000). The phosphorylation at MLC Ser19 is critical for filament assembly, however, diphosphorylation at Thr18 and Ser19 promotes the interaction of myosin with the actin at the cleavage furrow (Ikebe et al., 1988; Scholey et al., 1980). Komatsu and colleagues showed that the expression of unphosphorylated MLC in mammalian cells caused failure of cytokinesis (Komatsu et al., 2000). Thus, MLC phosphorylation is one of the key processes regulating contractile ring formation and completion of cytokinesis.
MLCK and myosin phosphatase are known as critical enzymes that regulate myosin phosphorylation. MLCK phosphorylates MLC on both Ser19 and Thr18 (Ikebe and Hartshorne, 1985). MLCK localizes at cleavage furrows (Poperechnaya et al., 2000) and inhibition of cleavage furrow contractility is observed with either the MLCK inhibitory peptide or ML-7, a specific MLCK inhibitor in sea urchin eggs and crane-fly primary spermatocytes (Lucero et al., 2006; Silverman-Gavrila and Forer, 2001). Additionally, ROCK, a Rho-effector kinase, and citron kinase also regulate MLC phosphorylation in cytokinesis (Amano et al., 1996; Eda et al., 2001; Kosako et al., 1999). Myosin phosphatase is the only known phosphatase that regulates MLC and MYPT1 is the targeting subunit (Ito et al., 2004). Misregulation of any of these kinases or MYPT1 could influence MLC phosphorylation in cancer cells and potentially contribute to cytokinesis failure and chromosome instability.
In this research report, we have examined the failure of cytokinesis in cancer cells and the fate of cells with multipolar spindles by real time microscopic imaging. We observe that the cell divisions fail in cancer cells, resulting in multinucleated cells (a single cell with two or more than two nuclei) which divide with multipolar spindles in the next cell cycle. Furthermore, we investigate the causes of defective cytokinesis in cancer cells. We show here that failure of cytokinesis in cancer cell lines is correlated with low levels of MLC phosphorylation. The phosphorylation reduction is caused by increased MYPT1 and decreased MLCK in oral cancer cells. When MLC phosphorylation was restored to high levels, cytokinesis failure and multipolar division were reduced in oral and liver cancer cells. Additionally, both overexpression of MYPT1 and inhibition of MLCK in primary cells elevated multinucleation and multipolar spindles defects. In conclusion, our results show for the first time that cytokinesis failure in cancer cells are caused by deficiencies in myosin light chain phosphorylation, which is due to the reduction of MLCK, as well as elevation of myosin phosphatase.
Materials and methods
Cell culture
American Type Culture Collection (ATCC) cell lines, HCT116, SK-HEP1, U2OS, HeLa, MES-SA, A549, Human fibroblast (CCL-110), HEK-293, RPE1 were cultured in medium recommended by the supplier. Oral cancer cell lines, UPCI:SCC103 and UPCI:SCC078 were maintained in minimal essential medium (Sigma), supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals), 2mM L-Glutamine, 0.05 mg/ml Gentamycin and 1% non-essential amino acids (Invitrogen) (White et al., 2007). All cells were incubated at 37°C in 5% CO2.
DNA transfection and RNA interference
Cells were transfected with 1-2μg of DNA plasmids as follows by using 6μl FuGENE6 transfection reagent (Roche Diagnostics) per coverslip according to the manufacture's protocol. Cells were lysed or fixed after 22-hour transfection. Site-directed mutagenesis to create the siRNA-resistant silent mutant of GFP-MYPT1was performed using primers 5′-CAG AGA CAA GAG CGG TTT GCT GAC AG-3′ and 5′-CTG TCA GCA AAC CGC TCT TGT CTC TG-3′. siMYPT1 (5′-GAG ACA AGA AAG ATT TGC T-3′,Dharmacon) (Xia et al., 2005) or the control rhodamine siRNA (Qiagen) was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's recommendations. For the 20-day MYPT1 knockdown, cells were seeded on Day0 and siRNA treatment started on Day1. On Day4, half of the cells were analyzed, while the other half were seeded again and the second knockdown cycle started. The whole knockdown process lasted for 20 days. After 20 days, cells were cultured in fresh culture medium without any siRNA for 4 days and analyzed as released cells.
Phosphorylated MLC localization detection
Cells on coverslips were fixed in 3.7% formaldehyde at room temperature and cold acetone at -20°C. Primary antibodies mAb-phosphorylated- Ser19-MLC (Cell Signaling) and rAb-MHC (Sigma) were diluted in PBST and incubated overnight at 4°C. Cells were incubated with secondary antibodies (Invitrogen) and DAPI (Sigma) at room temperature for 2 hours.
Immunoprecipitation and MLCK activity assay
Whole cell lysates were incubated with monoclonal mouse MLCK antibody (Sigma) at 4°C for 2 hours. After 2 hours, Protein A sepharose beads (Amersham Biosciences) were added and incubated at 4°C for 2 hours. Immunoprecipitates were split into two halves. One half of the sample was electrophoresed for immunoblotting. The other half was resuspended in a kinase reaction mixture containing 10mM MOPS, pH 7.0, 1mM DTT, 4mM MgCl2, 0.1mM CaCl2, 1μM calmodulin,0.1 mM [γ32P] ATP, and 15 μM myosin light chain kinase substrate (Sigma) (Poperechnaya et al., 2000). 10 μl reaction mixture was removed at different time points and spotted onto 1 cm2 squares of phosphocellulose (Upstate). The squares were washed 10 times with 2 ml of 75mM phosphoric acid and measured for incorporation of 32P by scintillation counter.
MLC phosphorylation analysis
MLC phosphorylation was measured by urea/glycerol-PAGE and immunoblotting as previously described (Word et al., 1991). Cells were seeded on 6-well plates and harvested by 0.5 ml ice-cold trichloroacetic acid containing 10 mM dithiothreitol (DTT). The pellets were washed three times with diethyl ether and resuspended with 8 M urea, 20 mM Tris–HCl, 23 mM glycine, 10% glycerol, 10 mM EGTA, 1 mM EDTA, and 0.2% bromophenol blue (pH 8.6).
FISH
Four ‘blinded’ flasks of cultured cells labeled, UPCI:SCC103A through D were treated with Colcemid (0.1 mg/mL) for 5 h before harvesting. After mitotic arrest, the cells were processed in accordance with standard cytogenetic laboratory procedures and slides were prepared for cytogenetic analysis. Fluorescence in situ hybridization (FISH) assays were 8 carried out to determine chromosome copy number changes. Two chromosome enumeration probes (CEP), CEP6 labeled with Spectrum Green TM and CEP20 labeled with Spectrum Orange TM (Abbott Molecular, Inc., Des Plaines, IL) were randomly selected. Slides were pretreated with RNase, dehydrated in an ethanol series, denatured in 70% formamide and hybridized overnight at 37°C in a humidified chamber. Posthybridization washes were carried out according to the Abbott Molecular protocol. The slides were stained with DAPI and mounted with antifade (Parikh et al. 2007). At least 500 metaphase cells and interphase nuclei were analyzed for each of the four conditions. All FISH analyses were carried out using an Olympus BX61 epifluorescence microscope (Olympus Microscopes, Melville, KY). The Genus software platform on the Cytovision System was used for image capture and analysis (Applied Imaging, San Jose, CA).
Statistical analysis
All statistical analyses were performed using R statistical package (R version 2.4.1). The group comparisons were conducted by non-parametric Wilcox test. All p-values are one-sided. Error bars, mean ± standard deviation of three different experiments.
Results
Multipolar spindle formation associated with failure of cytokinesis
In order to investigate the origin of multipolar spindle (MPS) formation in aneuploid cells, the cell divisions of HEK-293 and human OSCC tumor cells (UPCI:SCC103) were examined by DIC or fluorescent live cell microscopy. Both cell lines were transiently transected with plasmids expressing GFP-histone H2B and farnesylated-GFP to visualize cell nuclei and membrane respectively. We observed more than 90% of MPS arose in multinucleated cells in both tested cell lines (defined as cells with two or more nuclei, Supplementary Figure 1A), commonly resulting from cytokinesis failure. Additionally, the great majority of multinucleated cells observed to form in these cells were due to a failure of cytokinesis (Figure1A and B; Supplementary Movie1A and B). The frequency of cytokinesis failure was estimated at approximately 10% of mononucleated cells that undergo a bipolar division in both HEK-293 and UPCI:SCC103 cells (Figure 1C). Moreover, when cytokinesis failed, cells formed MPS in the following mitosis (9 out of 9 in HEK-239 cells, Supplementary Figure 1B and Movie 2) and usually failed in cytokinesis again (Supplementary Figure 2). These data confirm that a failure of cytokinesis is observed in these aneuploid cell lines and followed by a multipolar cell division.
Figure 1. Cytokinesis failure at an early stage leads to multinucleation in HEK-293 and oral cancer cells.
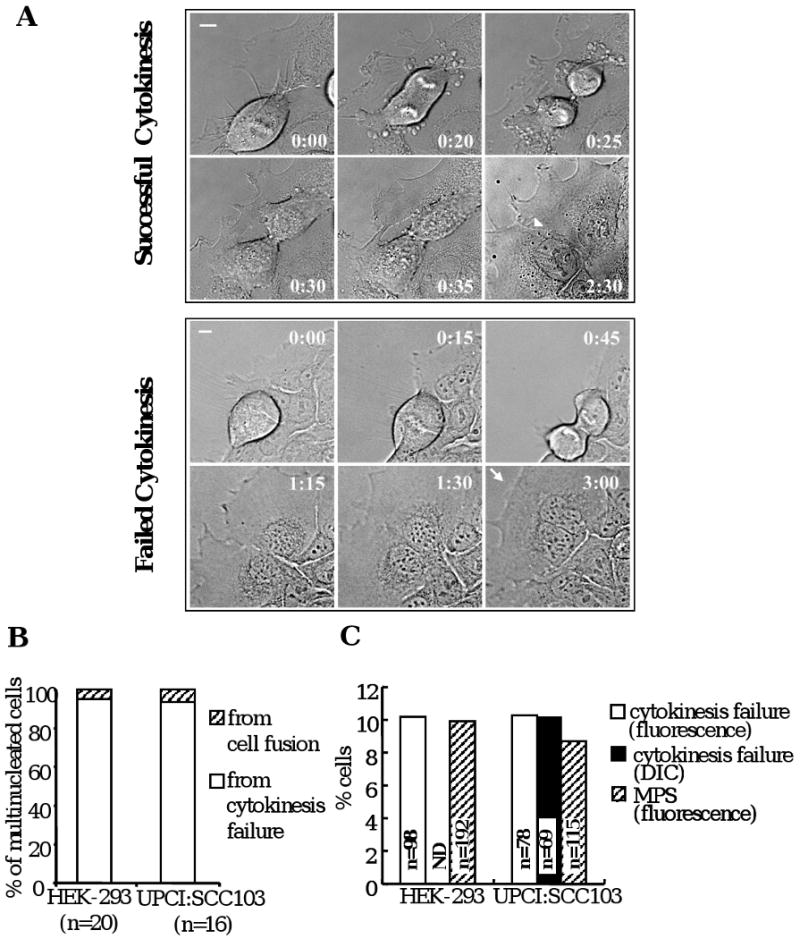
(A) Examples of successful and failed cytokinesis in UPCI:SCC103 cells as viewed by DIC optics. Arrow head: plasma membrane border between two daughter cells; Arrow: a continuous plasma membrane. Time: hours: minutes; Bar: 10μm (following figures using the same formats). (B) Majority of multinucleated cells are from cytokinesis failure. (C) The frequency of cells with cytokinesis failure by real-time DIC or fluorescence microscopy compared to the frequency of multipolarity.
MLC phosphorylation is deficient in cancer cells
In order to dissect where the cells failed in cytokinesis, UPCI:SCC103 cells were transfected with a plasmid expressing GFP-actin and examined by live-cell microscopy to view cleavage furrow and contractile ring formation. The recombinant GFP-actin colocalized with myosin, suggesting that the protein is active (Supplementary Figure 3A). We observed that 8.1% of UPCI:SCC103 cells failed in cytokinesis (n=62) and most exhibited cleavage furrow and contractile ring formation defects (Supplementary Figure 3B, Supplementary Movies 3A and B), suggesting that these cancer cells primarily failed at an early stage of cytokinesis with abnormal contractility.
MLC phosphorylation at Thr18/Ser19 is one of the key regulatory steps in contractile ring formation and contractility (Komatsu et al., 2000; Moussavi et al., 1993). We observed that phosphorylation of MLC in cancer cells was different from normal cells. The cancer cells universally showed low levels of MLC phosphorylation compared to normal cells (p< 0.001) (Figure 2A). All tested non-cancer cell lines, RPE-hTERT (retinal pigment epithelial cells immortalized with human telomerase, hereafter RPE1), fibroblast and OKF-hTERT (normal human oral keratinocyte immortalized with human telomerase) showed high MLC phosphorylation but low multinucleation frequency, while the tested different cancer cell lines clustered in the area of low MLC phosphorylation and high multinucleation (Figure 2B). These observations suggest that the deficiency in MLC phosphorylation could be a cause of cytokinesis failure in cancer cells.
Figure 2. The phosphorylation of MLC is defective and results in cytokinesis failure in cancer cells.
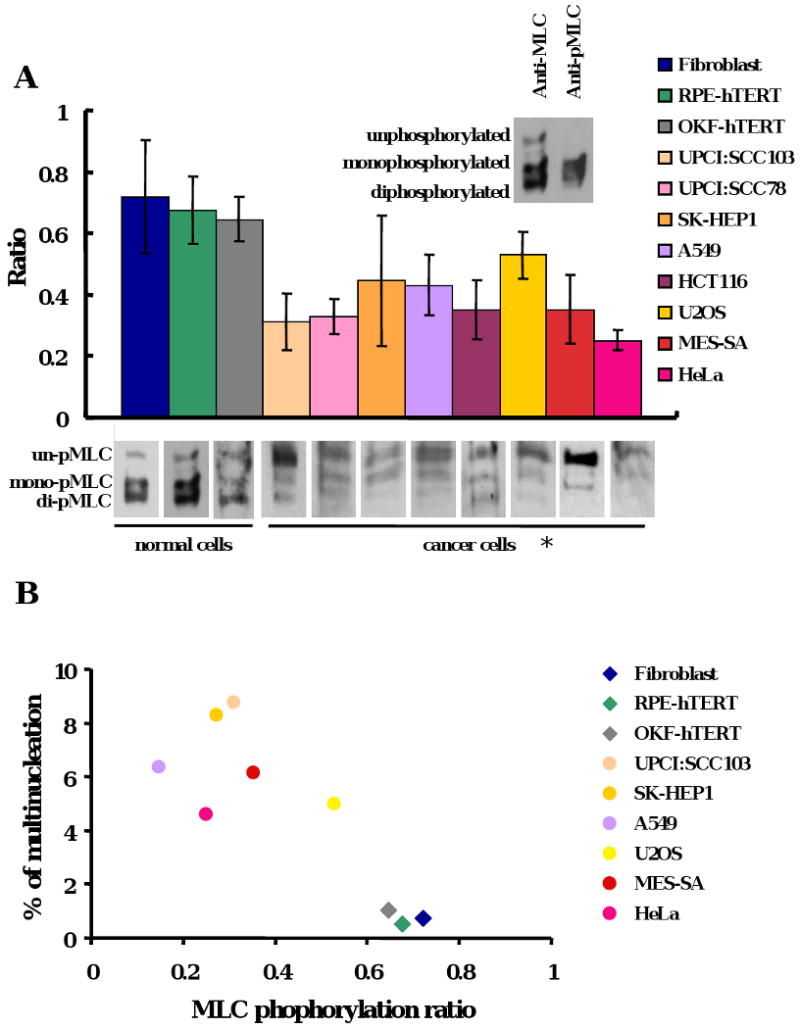
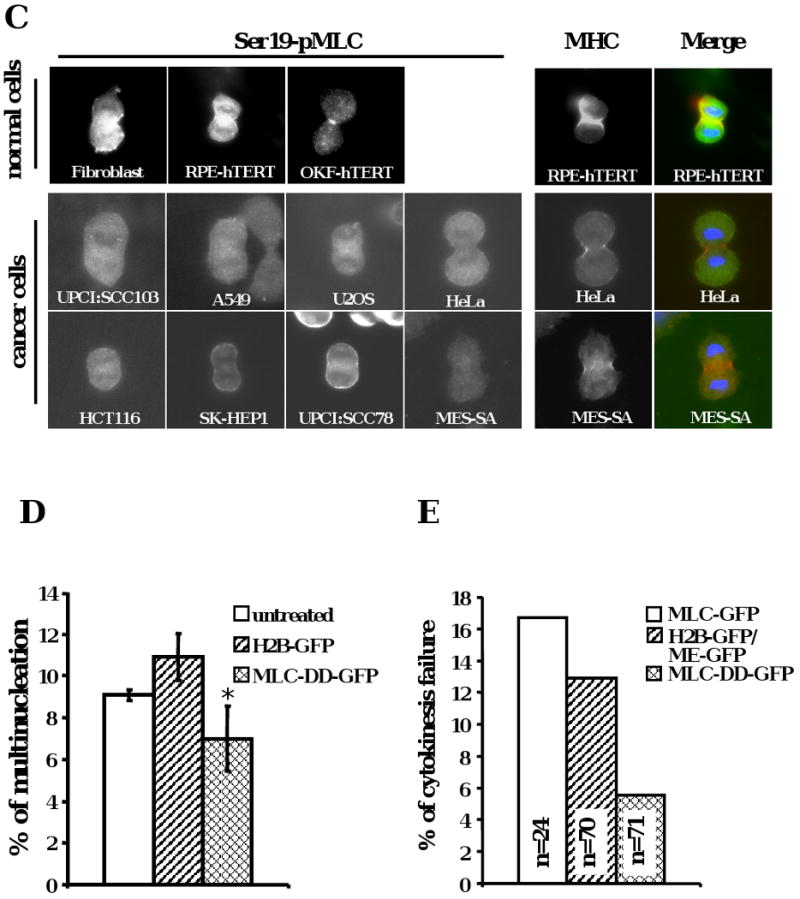
(A) The phosphorylated MLC levels in cancer cells were low compared to normal cells (*p <0.001). MLC phosphorylation was analyzed by urea glycerol gel electrophoresis from asynchronous cells. The data were means +/- standard deviation from more than three experiments. Ratio = (mono-pMLC + di-pMLC) / (un-pMLC + mono-pMLC + di-pMLC). (B) The ratio of MLC phosphorylation was correlated with the frequency of multinucleation in various cell lines. Diamonds: normal cells; dots: cancer cells. The data were means from more than three experiments. (C) Localization of phosphorylated MLC in anaphase cells from different cell lines. Left panel, Ser19-phosporylated MLC. Right panel, blue, DNA; red, MHC; green, Ser19-phosphorylated MLC. (D) Multinucleation was reduced after phosphomimetic MLC expression in UPCI:SCC103 cells (* p < 0.01). Multinucleation of cells was examined 48 hours after transient transfection with indicated plasmids. The data includes +/- standard deviation from more than three experiments. (E) Cytokinesis was rescued by phosphomimetic MLC overexpression. UPCI:SCC103 were transfected and bipolar cell divisions were followed 24 hours after transfection by live cell imaging.
To test this hypothesis, the phosphorylation of MLC was examined in UPCI:SCC103 cells and compared to primary RPE1 cells. We first determined whether MLC phosphorylation was induced during mitosis in the cancer cells. UPCI:SCC103 and RPE1 cells were synchronized at metaphase by Colcemid and released for different time points. The phosphorylation of MLC was elevated in mitosis of both cell types. However, the level of phosphorylation declined more rapidly in the cancer cells (Supplemental Figure 3C). The lower levels of MLC phosphorylation in the mitotic cancer cells is partially obscured by the higher frequency of mitotics in tumor lines. Therefore, to further compare the MLC phosphorylation levels in the different cell types, we compared by immunofluorescence the relative intensity of the phosphoepitope of MLC (Figure 2C, left panel). These results are consistent with diminished MLC phosphorylation in the cancer cells during division. Furthermore, the phosphorylated MLC in cancer cells did not co-localize completely with the myosin heavy chain (MHC) (Figure 2C, right panel). In conclusion, we observed that failure of cytokinesis in cancer cell lines was associated with decreased phosphorylation of MLC during mitosis.
MLC phosphorylation deficiency causes cytokinesis failure in oral cancer cells
To test whether the deficiency of phosphorylation of MLC is the cause of the defective contractile ring and failed cytokinesis in the tumor cells, a phosphomimetic MLC plasmid (Thr18 and Ser19 were replaced with Asp) was transfected into UPCI:SCC103 cells. This recombinant MLC protein expressed abundantly and localized properly with the MHC, the same as wild-type MLC (Supplementary Figure 3D). The multinucleation frequency decreased marginally, but statistically significantly, after phosphomimetic MLC expression in the oral cancer cells (Figure 2D, p < 0.01). This modest reduction may be due to residual multinucleated cells in the population. Therefore, to test more directly the frequency of cytokinesis failure, UPCI:SCC103 cells were visualized by live microscopy. Cell division failure decreased markedly after introduction of the phosphomimetic MLC compared to the control cells with wild-type MLC-GFP or H2B-GFP and farnesylated-GFP expression (Figure 2E, Supplementary Figure 3F and G, Supplementary Movie 5A, B and Movie 6A, B). In addition to oral cancer cells, multinucleation all significantly reduced after posphomimetic MLC was expressed in A549 (lung cancer cell line), SK- HEP1(liver cancer cell line), U2OS (osteosarcoma cell line) and HeLa (cervical cancer cell line) cells (Supplementary Figure 3E). These observations indicate that deficient phosphorylation of MLC caused cytokinesis failure in this cancer cell line.
Myosin phosphatase is an important regulator of MLC phosphorylation and cytokinesis completion in cells
To understand why MLC phosphorylation is decreased in malignant cells, MLC kinases and phosphatase expression was examined by immunoblotting. ROCK1 expressions showed no consistent differences between normal and cancer cells (Figure 3) and citron kinase expression overall was low (data not shown). Additionally, defective ROCK1 or overexpression of citron kinase has not previously been shown to cause contractile ring defects (Kosako et al., 1999; Madaule et al., 1998). On the other hand, MYPT1, the myosin targeting subunit, showed upregulated expression in many of the tested cell lines (Figure 3). Hence, we designed an siRNA targeting MYPT1, to knock down the targeting subunit in tumor cells (Supplementary Figure 4A). After siMYPT1 treatment, the phosphorylation ratio of MLC in UPCI:SCC103 cells elevated to approximately 80%, which was comparable to normal cells, showing that phosphorylation was limited due to the activity of this protein (Figure 4A, p < 0.01). This increase was not caused by upregulation of MLC expression after siRNA treatment (Supplementary Figure 4B). When the phosphorylation levels increased, the multinucleation frequency decreased (Figure 4B, p<0.01). In order to confirm that this reduction is due to down-regulation of myosin phosphatase subunit and not other off-target effects, a recombinant siRNA-resistant chicken MYPT1 protein was expressed in the siMYPT1-treated UPCI:SCC103 cells. This recombinant MYPT1-GFP protein colocalized with endogenous MHC and the expression restored multinucleation to untreated levels (Figure 4B and Supplementary Figure 4A). MYPT1 knockdown did not cause significant apoptosis or changes in the mitotic index (data not shown). These results indicated that the multinucleation in the OSCC cell line was caused by decreased MLC phosphorylation, resulting from MYPT1 overexpression. To confirm that the reduction in multinucleation was caused by a correction of cytokinesis defects, we examined cell divisions by live cell imaging. The frequency of cytokinesis failure in bipolar cell divisions in UPCI:SCC103 cells dropped from 8.4% to 3.2% after MYPT1 down-regulation by siRNA treatment (Figure 4C). We hypothesized that the extended siMYPT1 treatment in UPCI:SCC103 cells would sharply reduce the multinucleation and spindle multipolarity defects in these cells. We observed that both multinucleation and multipolarity were markedly decreased after 20-day siMYPT1 treatment compared to scrambled siRNA treatment controls (Figure 4D, p < 0.005, Supplementary Figure 4C). In addition, the chromosomal instability was measured by the frequency of the deviation from the modal number of chromosomes after MYPT1 was knocked down in oral cancer cells (Thompson and Compton, 2008). Consistent with the reduction in spindle defects, chromosomal instability was also reduced (Figure 4E, Supplementary Table 1), indicating that defective MLC phosphorylation and failed cytokinesis is a major cause of multinucleation, multipolarity, and chromosomal instability in these oral cancer cells.
Figure 3. Expression of kinases and phosphatase involved in regulation of MLC phosphorylation in different cell lines.
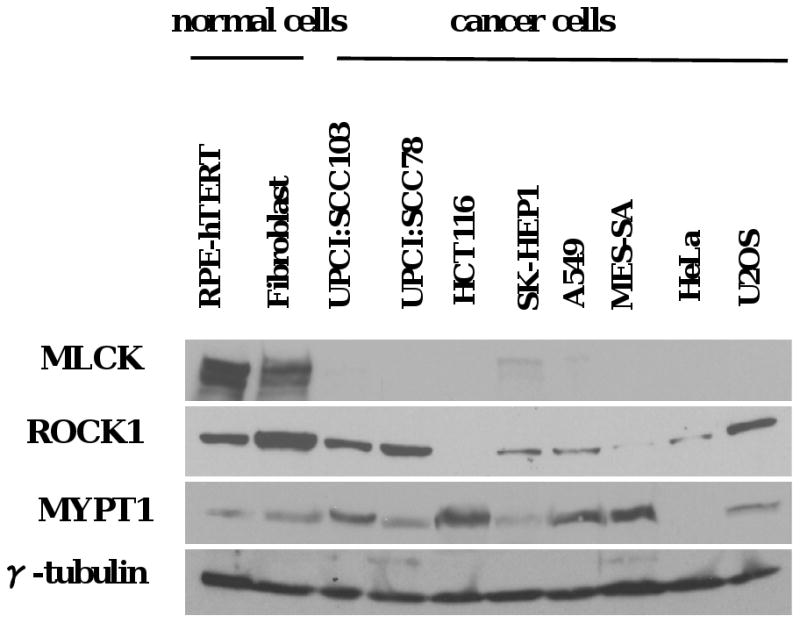
Cells were lysed and MLCK, ROCK1, MYPT1 and γ-tubulin expression was examined by immunoblotting. γ-tubulin was used as a loading control.
Figure 4. siRNA-mediated myosin phosphatase knockdown reduces cytokinesis failure, multinucleation, multipolar mitosis and chromosomal instability in cancer cell lines.
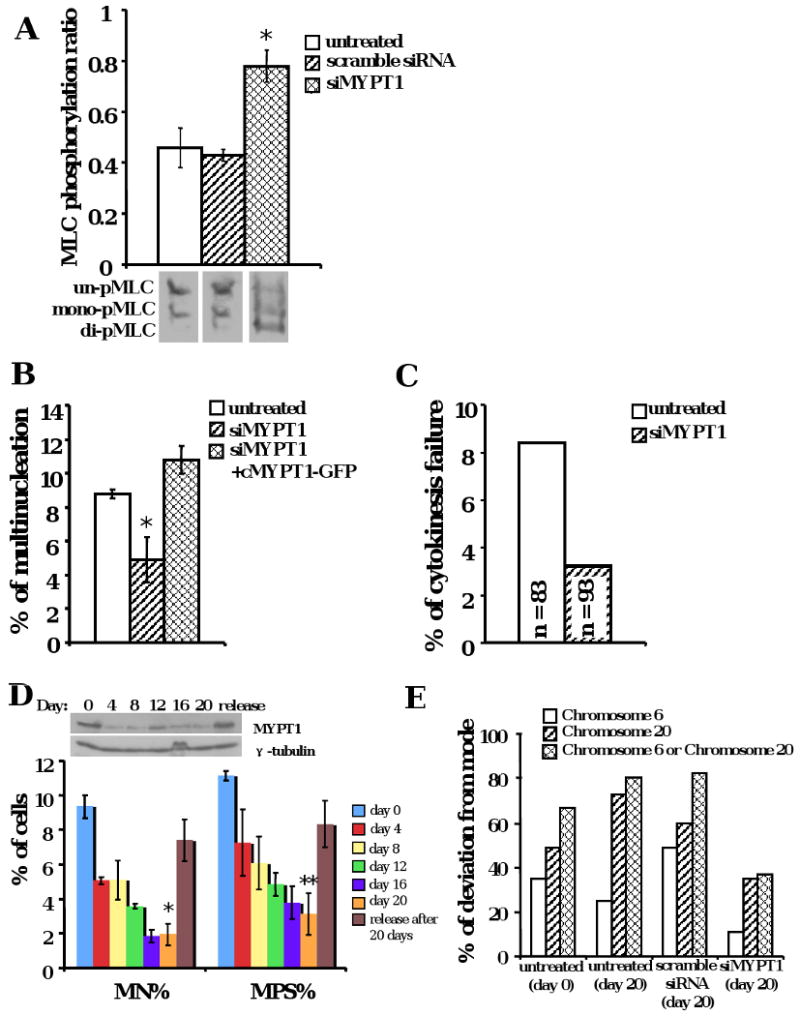
(A) MYPT1 knockdown elevated MLC phosphorylation in oral cancer cells (*p < 0.01). UPCI:SCC103 cells were treated by siMYPT1 and phosphorylation of MLC was examined by urea glycerol gel electrophoresis. The data were means +/- standard deviation from more than three experiments. (B) Multinucleation frequency was reduced by siMYPT1 knockdown and was restored by resistant MYPT1-GFP protein expression in oral cancer cells (*p < 0.01). UPCI:SCC103 cells were treated by siMYPT1 and transfected with resistant MYPT-GFP plasmids. Cells with both siRNA and plasmid were quantitated for multinucleation. The data were means +/- standard deviation from more than three experiments. (C) MYPT1 knockdown markedly rescued cytokinesis failure in oral cancer cells. UPCI:SCC103 cells were transfected with siMYPT1. Bipolar cell divisions were examined by DIC time-lapse microscopy. (D) Multinucleation and multipolarity were reduced after 20-day siMYPT1 treatment in oral cancer cells (*p < 0.005, **p < 0.005). The data were means +/- standard deviation from more than three experiments. UPCI:SCC103 cells were treated with siMYPT1 for 20 days. Multinucleation and multipolarity were quantified every four days. (E) Chromosomal instability was reduced after 20-day siMYPT1 treatment in oral cancer cells. The copy number of chromosomes 6 and 20 were enumerated by FISH. Modal chromosome numbers were chosen as the value that occurred the most frequently in each cell type. The frequency of deviation from the mode was calculated by the percentage of non-modal number in each cell type, which was used to measure the chromosomal instability of the cell.
Next, we asked whether MYPT1 expression was also the cause for multinucleation in other cancer types. Another cancer cell line (SK-HEP1), with relatively low levels of MYPT1 expression was tested with siRNA knockdown. Similar to oral cancer cells, liver cancer cells showed decreased multinucleation and multipolarity percentages after 20-day siMYPT1 treatment (Supplementary Figure 4D), suggesting that elevated myosin phosphatase activity is a cause of cytokinesis failure in different tumor cell types.
We next asked whether overexpression of MYPT1 was sufficient to cause cytokinesis failure and result in multinucleation in non-malignant cells. Full-length chicken MYPT1 fused with GFP was transiently expressed in RPE1 cells. After MYPT1-GFP overexpression, multinucleation frequency increased approximately four-fold (Figure 5A, p < 0.005). Similarly, overexpression of MYPT1 in UPCI:SCC103 cells gave rise to additional multinucleated cells (Figure 5B, p < 0.005). HEK-293 cells stably expressing MYPT1-GFP protein also showed elevated multinucleation and multipolarity frequencies (data not shown). Taken together, these results indicate that the overexpression of MYPT1 is an essential and sufficient cause of multinucleation and multipolarity in cancer cell lines.
Figure 5. Overexpression of MYPT1 in cells increases the multinucleation and multipolarity.
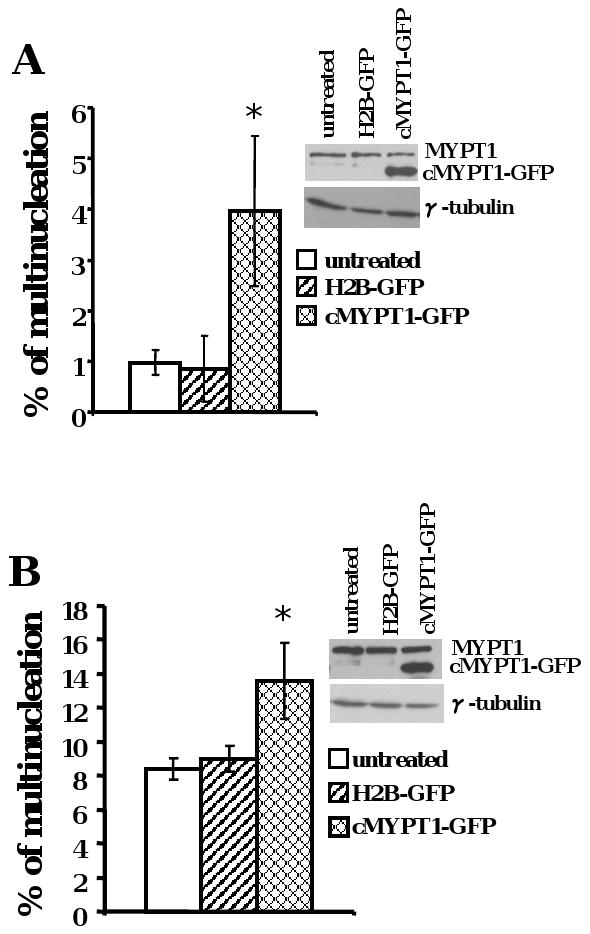
(A) RPE1 cells (*p <0.005) (B) UPCI:SCC103 cells (*p <0.005). The data were means +/- standard deviation from more than three experiments.
MLCK is downregulated and inhibited in oral cancer cells
We observed that MLC phosphorylation was defective in oral cancer cells and resulted in cytokinesis failure. Therefore, we hypothesized that cytokinesis would be rescued if we restored the expression and activity of MLCK in oral cancer cells. To test this hypothesis, MLCK-EGFP protein was expressed in UPCI:SCC103 cells (Supplementary Figure 6A). To confirm activity of the MLCK-EGFP fusion protein, MLCK was immunoprecipitated from whole cell lysates and MLCK activity assays were performed in vitro (Poperechnaya et al., 2000). ML-7, an MLCK specific inhibitor, was used to show specificity of the assay (Supplementary Figure 5A). Recombinant MLCK-EGFP was as active as endogenous MLCK by the MLC phosphorylation assay in vitro (Supplementary Figure 5B and C). However, the MLC phosphorylation ratio did not increase in these cells (Supplementary Figure 6B). Furthermore, we observed no reduction, but an increase of multinucleation (Figure 6A), and no change in cytokinesis failure or contractile ring defects (Supplementary Figure 6C; Supplementary Movie 4A and B). This suggested that MLCK might be inhibited in UPCI:SCC103 cells. To confirm this hypothesis, equal amounts of endogenous MLCK were immunoprecipitated from UPCI:SCC103 and RPE1 cells and MLCK activity was compared in vitro. The MLCK activity in oral cancer cells was lower than noncancer, but also epithelial origin, RPE cells (Figure 6B), indicating that MLCK was inhibited in the UPCI:SCC103 cells. Similarly, MLCK immunoprecipitated from primary fibroblasts showed more activity than U2OS cells, both cell lines of mesenchymal origin (Supplementary Figure 5D). Taken together, these results show that both MLCK expression and activity are down-regulated in oral cancer cells.
Figure 6. MLCK deficiency induces cytokinesis failure cells.
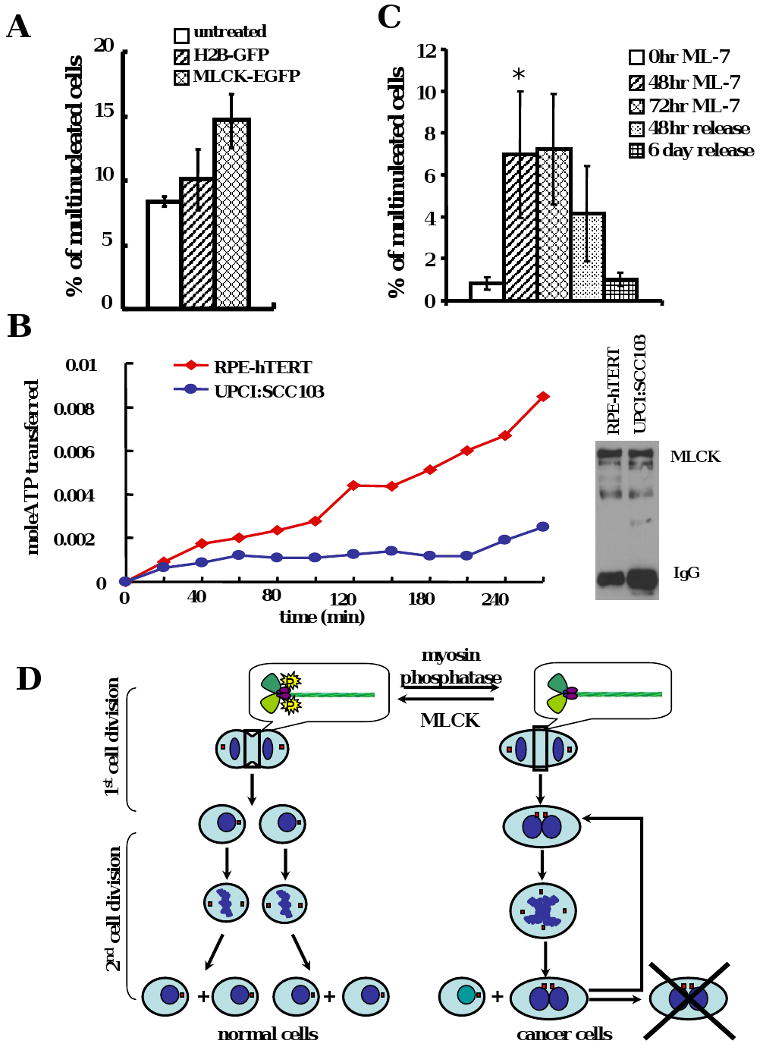
(A) The multinucleation frequency increased slightly after MLCK overexpression in UPCI:SCC103 cells. The data were means +/- standard deviation from more than three experiments. (B) The endogenous MLCK activity was inhibited in oral cancer cells. The same amount of MLCK was immunoprecipitated from UPCI:SCC103 cells and RPE1 cells and the MLCK activity was examined in vitro. (C) Multinucleation frequency increased after ML-7 treatment and recovered after release from ML-7 treatment in RPE1 cells (*p < 0.005). The data were means +/- standard deviation from more than three experiments. (D) The model of how cytokinesis failure contributes to chromosomal instability in cancer cells.
To examine if MLCK inhibition is sufficient to block completion of cytokinesis in non-malignant cells, RPE1 cells were treated with 35μM ML-7 for 48 hours. Stress fibers disappeared and MLC phosphorylation decreased sharply (Supplementary Figure 7A and B), which was not due to reduced expression of endogenous MLC (Supplementary Figure 7C). Furthermore, the multinucleation increased to approximately 7% after 48- and 72-hour ML-7 treatment (Figure 6C, p<0.005). This multinucleation phenotype was diminished when the RPE1 cells were released after 72-hour treatment (Figure 6C). These results confirmed that a reduction in MLCK activity in non-transformed cells could reproduce the MLC phosphorylation defects and multinucleation seen in malignant cells.
Discussion
Polyploidy is an important characteristic of major solid tumor tissues and tumor-derived cells (Lemez et al., 1998; Lothschutz et al., 2002; Olaharski et al., 2006; Park et al., 1995; Shi and King, 2005; Takanishi et al., 1996). The data presented here demonstrate that cytokinesis failure is a primary mechanism for the increased ploidy of oral carcinoma cells and the molecular defects leading to cytokinesis failure in these and other cancer cell types. We present four lines of evidence to support this conclusion: 1) the ratios of MLC phosphorylation are universally reduced, and this reduction is correlated with increased multinucleation in the tested cancer cell lines. 2) phosphomimetic MLC overexpression overcomes the endogenous defective MLC phosphorylation and rescues cytokinesis failure. 3) when MLC phosphorylation is restored to normal levels by phosphatase knockdown, cytokinesis failure and multinucleation decrease. Finally 4) the reduction of MLC phosphorylation by phosphatase overexpression or MLCK inhibition is sufficient to cause multinucleation and multipolar mitoses in nonmalignant cells. These results demonstrate that the decreased MLC phosphorylation is an important cause of cytokinesis failure in cancer cells, resulting in multinucleation and multipolar division.
Pellman and colleagues have shown that the tetraploid p53-null cells generate malignant myoepitheliomas in nude mice. They have linked cytokinesis failure to carcinogenesis for the first time in vivo (Fujiwara et al., 2005). However, how cytokinesis fails in cancer cells remains unclear. With the results of this study, we propose a model of the mechanism for the accumulation of multinucleated cells: in normal cells, MLC phosphorylation is regulated by MLCK and myosin phosphatase at the appropriate time during mitosis to ensure the completion of cytokinesis. If the balance of MLC phosphorylation is changed by MLCK inhibition or myosin phosphatase overexpression, cytokinesis fails early in division, leading to multinucleated cells. These cells inherit extra centrosomes causing them to divide with multipolar spindles in the next mitosis. By going into multipolar cell division, mother multinucleated cells usually generate a mixture of multinucleated and mononucleated daughter cells. These multinucleated cells could enter a new cell cycle or apoptosis. The mononucleated cells are likely to inherent one centrosome and an abnormal chromosome set, which may lead to a “normal” bipolar cell division in the next cell cycle and contribute to a stable genome change (Figure 6D).
Recent microarray analyses have shown that myosin light chain kinase (MLCK) gene expression is low in various tumors, such as colon, lung, prostate, bladder, brain, breast, ovarian, liver, melanoma, and multiple myeloma, compared to normal controls, but high in head-neck and lymphoma (www.oncomine.org) (Rhodes et al., 2007). Down-regulated MLCK transcription and expression is also observed in mesenchymal tumor cells and transformed chicken embryo fibroblasts, respectively (Schenker and Trueb, 1998; Van Eldik et al., 1984). These results confirm that the inhibited MLCK we observe in cancer cell lines is also relevant for tumor tissue. When we increased the level of phosphorylated MLC in the MLCK-downregulation background by knockdown of MYPT1, we rescued the cytokinesis failure in oral cancer cells. Thus, we conclude that both overexpression of MYPT1 and inhibition of MLKC is a cause of low MLC phosphorylation and cytokinesis failure in cancer cells.
In the last decade, additional myosin light chain kinases including Rho-kinase and citron kinase have been shown to localize at the cleavage furrow (Amano et al., 1996; Eda et al., 2001; Kosako et al., 1999). However, we did not detect significant consistent differences of ROCK1 and citron kinase expressions in the tested cell lines (Figure 3, data not shown). Moreover, we have also examined the RhoA expression, which is the activator of ROCK and citron kinase, and observed the same levels in cancer cells compared to normal cells (data not shown). But we can not rule out that the defective cytokinesis in cancer cells is also influenced by abnormal regulations of these kinases.
The regulation of phosphorylation is a major target of cancer therapeutics, and the data presented here suggests MLC as a potential candidate. However, myosin activity is relevant to many aspects of cellular growth and survival. It has been shown that the MLCK inhibitor, ML-7, prevents the growth of tumors in mice and rats, which might be caused by less motility and adhesion due to decreased MLC phosphorylation (Gu et al., 2006; Kaneko et al., 2002; Tohtong et al., 2003). However, our data suggests that therapeutic MLCK inhibition might also result in an increase in cytokinesis defects and aneuploidy, which would constitute a serious side-effect and cause severe problems in long-term human therapy. The other potential regulatory candidate is MYPT1. Based on our studies, we have concluded that down regulation of MYPT1 rescues cytokinesis failure and reduces chromosomal instability in cancer cells. Interestingly, Xia et al. have shown that knockdown of MYPT1 in HeLa cells inhibits cell migration and adhesion (Xia et al., 2005). Taken together, these findings suggest that an inhibitor of MYPT1 might be a useful potential target for cancer therapy.
Supplementary Material
Acknowledgments
The authors thank Dr. David J. Hartshorne (University of Arizona) for kindly providing UPCI:SCC103 cell line and HEK293 with MYPT1 expression cell line respectively. We are also grateful to Dr. Jeffrey D. Hildebrand (University of Pittsburgh), Dr. Anne R. Bresnick (Albert Einstein College of Medicine), Dr. Kathleen Kelly (National Cancer Institute), Dr. David J. Hartshorne (University of Arizona) and Dr. James T. Stull (University of Texas Southwestern Medical Center) for plasmids and antibodies. We also thank Dr. Xiaojing Wang (University of Pittsburgh) for helpful discussions concerning statistical analysis and Dr. Jian Huang (University of Texas Southwestern Medical Center) for helpful suggestions about urea glycerol electrophoresis. This study was supported in part by NIH grants R01DE016086 to WSS and SMG and R01DE14729 to SMG and WSS. Cytogenetic analyses were carried out in the UPCI Cytogenetics Facility, supported in part by NIH grant P30CA47904 to Ronald B. Herberman/SMG.
References
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Eda M, Yonemura S, Kato T, Watanabe N, Ishizaki T, Madaule P, Narumiya S. Rho-dependent transfer of Citron-kinase to the cleavage furrow of dividing cells. J Cell Sci. 2001;114:3273–84. doi: 10.1242/jcs.114.18.3273. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet. 2008;24:64–9. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Gu LZ, Hu WY, Antic N, Mehta R, Turner JR, de Lanerolle P. Inhibiting myosin light chain kinase retards the growth of mammary and prostate cancer cells. Eur J Cancer. 2006;42:948–57. doi: 10.1016/j.ejca.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–31. [PubMed] [Google Scholar]
- Ikebe M, Koretz J, Hartshorne DJ. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J Biol Chem. 1988;263:6432–7. [PubMed] [Google Scholar]
- Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- Kaneko K, Satoh K, Masamune A, Satoh A, Shimosegawa T. Myosin light chain kinase inhibitors can block invasion and adhesion of human pancreatic cancer cell lines. Pancreas. 2002;24:34–41. doi: 10.1097/00006676-200201000-00005. [DOI] [PubMed] [Google Scholar]
- King RW. When 2+2=5: the origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu S, Yano T, Shibata M, Tuft RA, Ikebe M. Effects of the regulatory light chain phosphorylation of myosin II on mitosis and cytokinesis of mammalian cells. J Biol Chem. 2000;275:34512–20. doi: 10.1074/jbc.M003019200. [DOI] [PubMed] [Google Scholar]
- Kosako H, Goto H, Yanagida M, Matsuzawa K, Fujita M, Tomono Y, Okigaki T, Odai H, Kaibuchi K, Inagaki M. Specific accumulation of Rho-associated kinase at the cleavage furrow during cytokinesis: cleavage furrow-specific phosphorylation of intermediate filaments. Oncogene. 1999;18:2783–8. doi: 10.1038/sj.onc.1202633. [DOI] [PubMed] [Google Scholar]
- Lemez P, Michalova K, Zemanova Z, Marinov I, Trpakova A, Moravcova J, Jelinek J. Three cases of near-tetraploid acute myeloid leukemias originating in pluripotent myeloid progenitors. Leuk Res. 1998;22:581–8. doi: 10.1016/s0145-2126(97)00177-x. [DOI] [PubMed] [Google Scholar]
- Lothschutz D, Jennewein M, Pahl S, Lausberg HF, Eichler A, Mutschler W, Hanselmann RG, Oberringer M. Polyploidization and centrosome hyperamplification in inflammatory bronchi. Inflamm Res. 2002;51:416–22. doi: 10.1007/pl00000323. [DOI] [PubMed] [Google Scholar]
- Lucero A, Stack C, Bresnick AR, Shuster CB. A global, myosin light chain kinase-dependent increase in myosin II contractility accompanies the metaphase-anaphase transition in sea urchin eggs. Mol Biol Cell. 2006;17:4093–104. doi: 10.1091/mbc.E06-02-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–4. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- Moussavi RS, Kelley CA, Adelstein RS. Phosphorylation of vertebrate nonmuscle and smooth muscle myosin heavy chains and light chains. Mol Cell Biochem. 1993;127-128:219–27. doi: 10.1007/BF01076773. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–23. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, Eastmond DA. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 2006;27:337–43. doi: 10.1093/carcin/bgi218. [DOI] [PubMed] [Google Scholar]
- Park SH, Maeda T, Mohapatra G, Waldman FM, Davis RL, Feuerstein BG. Heterogeneity, polyploidy, aneusomy, and 9p deletion in human glioblastoma multiforme. Cancer Genet Cytogenet. 1995;83:127–35. doi: 10.1016/0165-4608(95)00040-v. [DOI] [PubMed] [Google Scholar]
- Poperechnaya A, Varlamova O, Lin PJ, Stull JT, Bresnick AR. Localization and activity of myosin light chain kinase isoforms during the cell cycle. J Cell Biol. 2000;151:697–708. doi: 10.1083/jcb.151.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenker T, Trueb B. Down-regulated proteins of mesenchymal tumor cells. Exp Cell Res. 1998;239:161–8. doi: 10.1006/excr.1997.3896. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Taylor KA, Kendrick-Jones J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature. 1980;287:233–5. doi: 10.1038/287233a0. [DOI] [PubMed] [Google Scholar]
- Sellers JR. Regulation of cytoplasmic and smooth muscle myosin. Curr Opin Cell Biol. 1991;3:98–104. doi: 10.1016/0955-0674(91)90171-t. [DOI] [PubMed] [Google Scholar]
- Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, Ketterer DM, Pollice AA. Model for the genetic evolution of human solid tumors. Cancer Res. 1989;49:3344–54. [PubMed] [Google Scholar]
- Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038–42. doi: 10.1038/nature03958. [DOI] [PubMed] [Google Scholar]
- Silverman-Gavrila RV, Forer A. Effects of anti-myosin drugs on anaphase chromosome movement and cytokinesis in crane-fly primary spermatocytes. Cell Motil Cytoskeleton. 2001;50:180–97. doi: 10.1002/cm.10006. [DOI] [PubMed] [Google Scholar]
- Sluder G, Nordberg JJ. The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr Opin Cell Biol. 2004;16:49–54. doi: 10.1016/j.ceb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Takanishi DM, Jr, Hart J, Covarelli P, Chappell R, Michelassi F. Ploidy as a prognostic feature in colonic adenocarcinoma. Arch Surg. 1996;131:587–92. doi: 10.1001/archsurg.1996.01430180013002. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–72. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohtong R, Phattarasakul K, Jiraviriyakul A, Sutthiphongchai T. Dependence of metastatic cancer cell invasion on MLCK-catalyzed phosphorylation of myosin regulatory light chain. Prostate Cancer Prostatic Dis. 2003;6:212–6. doi: 10.1038/sj.pcan.4500663. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Watterson DM, Burgess WH. Immunoreactive levels of myosin light-chain kinase in normal and virus-transformed chicken embryo fibroblasts. Mol Cell Biol. 1984;4:2224–6. doi: 10.1128/mcb.4.10.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Hirohashi Y, Furuuchi K, Zhao H, Liu Q, Zhang H, Murali R, Berezov A, Du X, Li B, Greene MI. The centrosome in normal and transformed cells. DNA Cell Biol. 2004;23:475–89. doi: 10.1089/1044549041562276. [DOI] [PubMed] [Google Scholar]
- White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, Gollin SM. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2007;43:701–12. doi: 10.1016/j.oraloncology.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word RA, Casey ML, Kamm KE, Stull JT. Effects of cGMP on [Ca2+]i, myosin light chain phosphorylation, and contraction in human myometrium. Am J Physiol. 1991;260:C861–7. doi: 10.1152/ajpcell.1991.260.4.C861. [DOI] [PubMed] [Google Scholar]
- Xia D, Stull JT, Kamm KE. Myosin phosphatase targeting subunit 1 affects cell migration by regulating myosin phosphorylation and actin assembly. Exp Cell Res. 2005;304:506–17. doi: 10.1016/j.yexcr.2004.11.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.