

Abstract
AIMS
Cannabinoid receptor-1 (CB1) antagonists suppress appetite and induce weight loss. Direct antagonism of CB1 receptors on platelets might be an additional benefit for CB1 antagonists, but the role of CB1 receptors in platelets is controversial. We tested the hypothesis that the endocannabinoid, 2-arachidonyl glycerol (2-AG), induces platelet aggregation by a COX-mediated mechanism rather than through CB1 receptor activation, in blood obtained from healthy volunteers and patients with coronary artery disease receiving low dose aspirin.
METHODS
Aggregatory responses to the cannabinoids 2-AG and Δ9-THC were examined in blood sampled from healthy volunteers (n= 8) and patients (n= 12) with coronary artery disease receiving aspirin using whole blood aggregometry. The effects of CB1 (AM251) and CB2 (AM630) antagonists, as well as fatty acid amide hydrolase (FAAH) and monoacyl glycerol lipase (MAGL) inhibitors and aspirin on 2-AG-induced aggregation were also assessed.
RESULTS
AM251 (100 nm–30 µm) had no effect on platelet aggregation induced by either ADP (P= 0.90) or thrombin (P= 0.86). 2-AG, but not Δ9-THC, induced aggregation. 2-AG-induced aggregation was unaffected by AM251 and AM630 but was abolished by aspirin (P < 0.001) and by the MAGL inhibitor, URB602 (P < 0.001). Moreover, the aggregatory response to 2-AG was depressed (by >75%, P < 0.001) in blood from patients with coronary artery disease receiving aspirin compared with that from healthy volunteers.
CONCLUSIONS
2-AG-mediated activation of platelets is via metabolism to arachidonic acid by MAGL, and not through direct action on CB1 or CB2 receptors, at least in the acute phase.
Keywords: 2-AG, AM251, antithrombotic, endocannabinoid system, platelets, rimonabant
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
There are conflicting views in the literature as to whether cannabinoids have an impact on platelet activity and to what extent cannabinoid receptors are involved. This is an important issue to resolve because platelet effects of putative therapeutic cannabinoid inhibitors and stimulators will have an impact on their potential benefits and safety.
WHAT THIS STUDY ADDS
The data presented in this manuscript clearly show that the endocannabinoid 2-arrachidonyl glycerol can activate platelet activity, but that the effects are mediated through an aspirin-sensitive pathway that is not affected by cannabinoid receptor antagonists or FAAH inhibition, but is abolished by MAGL inhibition. The findings question the role of cannabinoid receptors in platelet function and suggest that platelet function is unlikely to be directly affected by cannabinoid receptor antagonists, at least in the acute phase.
Introduction
Human cannabinoid receptors, CB1 and CB2, bind the psychoactive compound, Δ9-tetrahydrocannabinol (Δ9-THC), found in Cannabis sativa[1, 2], as well as endogenous cannabinoid entities (endocannabinoids). In the last decade, the endocannabinoid system has been recognized to be important in controlling many physiological and pathological processes and has become an area of interest for novel drug development.
Endocannabinoids are derivatives of arachidonic acid (AA) and belong to the eicosanoid family. The two main endocannabinoids, 2-arachidonoyl glycerol (2-AG) and anandamide, are synthesized in response to increasing intracellular calcium concentrations by diacylglycerol lipase [3]. Metabolism is relatively non-specific and occurs through hydrolyzing enzymes, notably fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) [4]. Hydrolysis of 2-AG produces AA, which is the main substrate for cyclooxygenase (COX) enzymes that catalyze the conversion of AA into prostaglandin-H2 in the first step of the synthesis of prostaglandins and thromboxanes [5].
CB1 receptors are predominantly localized in central and peripheral nerves and mediate retrograde synaptic inhibition of endogenous cannabinoids [6]. However, they are also present in other cell types and cellular fragments, including immune cells and platelets [7]. CB2 receptors mediate inflammation and are found in many inflammatory cell types [8].
The physiological actions of endocannabinoids are diverse [9–13] but are believed to include modulation of platelet function. Platelets express CB1 and CB2 receptors and possess the metabolic pathways to both synthesize and degrade endocannabinoids [7]. However, data regarding the impact of CB receptor activation are contradictory: some studies have reported platelet activation by Δ9-THC [7, 14], anandamide [15] and 2-AG [16], whilst another has demonstrated that Δ9-THC inhibits platelet function [17]. Likewise, the mechanism by which endocannabinoids induce activation is unclear: anandamide-induced aggregation has been shown to be antagonized by a CB1 antagonist in one study [15], but not in another [18], where platelet activation was shown to be dependent on metabolism of anandamide to arachidonic acid.
CB1 receptor knockout mice have reduced bodyweight, reduced fat mass and reduced appetite [19]. In rat [20] and human [21, 22] studies, pharmacological CB1 antagonism using AM251 or SR141716 reduces food intake and leads to an increase in energy expenditure. These findings led to the clinical development of the CB1 receptor antagonist, SR141716, rimonabant, as an appetite suppressant that acts through inhibition of tonic CB1 receptor activity [23].
The controversy surrounding the role of CB1 receptors in mediating platelet activation requires clarification, given that any pro- or anti-aggregating effect of drugs acting at the CB1 receptor could have serious implications with respect to the safety or potential added benefit of CB1 receptor antagonism in patients with cardiovascular disease. We set out to test the hypothesis that the cannabinoids Δ9-THC and 2-AG cause platelet aggregation in whole blood in healthy volunteers and a group of coronary artery disease patients on long term, low dose aspirin. The mechanism by which 2-AG induces platelet aggregation was probed in vitro with the CB1 and CB2 receptor antagonists, the COX inhibitor, aspirin and inhibitors of FAAH and MAGL.
Methods
Ethics
This study was approved by the North of Scotland Research Ethics Committee (Ethics no. 06/S0901/46) and was conducted in accordance with the declaration of Helsinki and its amendments.
Healthy volunteers
Healthy non-smoking male volunteers aged 18–60 years, on no regular medications were recruited from local hospital staff. Volunteers were excluded if they had renal disease (creatinine >159 µm), diabetes (fasting blood glucose >6 mmol l−1, HbA1c >6.5%) or had participated in a clinical trial in the previous 3 months. A venous blood sample (100 ml) was taken from the antecubital fossa using a 21G butterfly needle.
Patients with coronary artery disease
Non-smoking male patients with a clinical history suggestive of coronary artery disease and who were undergoing routine coronary angiography were screened for suitability in the study. Patients were excluded if they had renal disease (creatinine >159 µm) or had participated in a clinical trial in the previous 3 months. As part of routine coronary angiography, an arterial sheath was inserted into their right radial or femoral artery. An arterial blood sample (100 ml) was taken from this site before catheterization. Demographics were recorded from patient medical notes and coronary disease status was defined as mild (<50% occlusion) moderate (50–75% occlusion) or severe (>75% occlusion); the number of major coronary vessels affected was also recorded.
Handling of blood samples
The first 5 ml of blood was discarded to remove blood affected by haemostasis in the needle. A 100 ml sample was drawn into two 50 ml syringes and then immediately transferred into tubes containing 3.8% trisodium citrate solution. Citrate was used as an anti-coagulant because of its negligible intrinsic effect on platelets [24]. A 9 ml sample was drawn at the same time into a lithium heparin-containing tube (Sarstedt Ltd, Leicester, UK) for biochemical screening (DCA 2000+ Analyser; Bayer, Newbury, UK). Renal status (Piccolo Renal Panel Plus; Abaxis, Darmstadt, Germany) and lipid status (Piccolo Lipid Panel Plus; Abaxis, Darmstadt, Germany) were recorded. Blood cell counts, including platelet count (Beckman Coulter ACT8: Beckman Coulter, High Wycombe, UK) were also obtained. Blood was promptly taken to the research laboratory after sampling and experiments were completed within 2 h of sampling. All sampling was carried out at approximately 09.00 h, ensuring minimization of previously observed circadian platelet variation [25], following a 12 h fast.
Preparation and treatment of platelets
Citrated blood for whole blood experiments was diluted 1:1 with 0.9% saline solution, as previously described [26], before 1 ml was added to a disposable aggregometry electrode and cuvette tube (LabMedics, Manchester, United Kingdom) containing a stirrer bar (LabMedics, Manchester, United Kingdom) and pre-warmed (37°C, 5 min). Aggregometry analysis was carried out using whole blood impedance aggregometry (37°C, stirrer speed 1000 rev min−1; Chrono-Log Model 700 Lumi-Aggregometer; LabMedics, Manchester, United Kingdom). Aggregation of platelets was determined by measuring the area under the curve (AUC) representing impedance change, from the baseline, over time.
Experimental protocols
Characterization of the responses of human blood to cannabinoid and non-cannabinoid platelet agonists
Preliminary studies were undertaken whereby blood samples from healthy volunteers were pre-incubated (10 min, 37°C), with a range of concentrations of AM251 (0.1–30 µm) prior to activation (6 min) with different agonists; 2-AG (200 µm), Δ9-THC (300 µm), ADP (20 µm; LabMedics, Manchester, UK) and thrombin (0.25 U ml−1; LabMedics, Manchester, UK). The potential of AM251 and AM630 themselves to induce aggregation alone was therefore also determined in these experiments.
Comparison of the aggregatory responses to 2-AG in healthy volunteers and CAD patients receiving aspirin
Aliquots (15 ml) of blood were transferred into seven tubes containing AM251, AM630 or DMSO (solvent control; final concentration, 0.5%). The final concentration for each drug was 30 µm, 3 µm and 300 nm. At time 10, 30, 60 and 120 min, 1 ml of blood from each tube was transferred to a cuvette within the aggregation chamber and 2-AG (100 µm) added; impedance was then recorded for the subsequent 6 min.
Effect of FAAH and MAGL inhibitors on responses to 2-AG in healthy volunteer whole blood and platelet rich plasma (PRP)
Whole blood
Blood (10 ml) from healthy volunteers was incubated with the FAAH inhibitor, URB597 (300 nm), the MAGL inhibitor, URB602 (300 µm), both URB597 + URB602 (300 nm and 300 µm, respectively) or vehicle control for 15 min (37°C, with occasional gentle swirling). In each case, the total final DMSO concentration was 1% v : v). Concentrations of URB597 and URB602 were derived from literature values for maximal inhibition of respective enzymes [27]. Aliquots (500 µl) of these samples were transferred to plastic cuvettes containing 500 µl saline. A further sample was treated for 30 min with aspirin (100 µm, 37°C) following saline dilution. Each sample was then treated with 2-AG (200 µm) and aggregation assessed for 6 min by the impedance method described above.
In experiments involving
PRP, citrated blood (40 ml) was centrifuged (130 g, 10 min, 20°C) and PRP was aspirated to a fresh tube, and kept at 37°C in a water bath, whilst platelet poor plasma was prepared from the remaining blood by centrifugation (13 000 g, 1 min, 20°C). PRP platelet count was normalized to 150 × 109 platelets l−1 by adding the appropriate volume of platelet poor plasma to PRP, which was then divided into glass cuvettes (500 µl) and treated with 2-AG (200 µm), whilst aggregation was assessed by standard turbidometric aggregometry.
Materials
Chemicals were of the purest analytical grade. 2-arachidonyl glycerol (2-AG), Δ9-tetrahydrocannbinol (Δ9-THC), 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide (AM251) and trisodium citrate were purchased from Sigma Aldrich (Dorset, UK). Adenosine diphosphate (ADP), thrombin and collagen were purchased from LabMedics (Manchester, UK). (6-iodio-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl) (4-methoxyphenyl) methanone (AM630; Axxora, Bingham, Nottingham, UK). 2-AG and Δ9-THC were evaporated from their stock solvent using a steady stream of nitrogen and reconstituted in DMSO. AM251 and AM630 were reconstituted in DMSO. All other chemicals were reconstituted in 0.9% saline solution. Rimonabant shares almost identical properties and structure with other CB1 selective antagonists, including AM251, which has similar selectivity for CB1 receptors and almost identical pharmacological actions [28, 29]. URB 597 and URB602 were obtained from Axxora (Cambridge, UK).
Statistics
The number of subjects used in each group is stated in the respective figure legends. All data on the graphs are expressed as the mean ± SEM. P values were obtained by the relevant statistical test as stated in the legend of each figure. P < 0.05 was considered to be statistically significant.
Results
Healthy volunteer and patient characteristics
Demographic and biochemical characteristics from the two groups studied are summarized in Table 1. Table 2 characterizes the patient population profiles and medications, including the severity of coronary artery disease in each patient.
Table 1.
Profiles of healthy volunteers and patients with coronary artery disease. Demographical measurements and biochemical results of two study groups
| Parameter | Healthy volunteers (n= 8) | Patients (n= 12) |
|---|---|---|
| Age (years) | 39 ± 4.3 | 65 ± 2.5*** |
| BMI (kg m−2) | 25 ± 1.6 | 29 ± 1 |
| BP (mmHg) | 127/79 ± 4/2 | 138/79 ± 4/3 |
| Total cholesterol (mm) | 4.8 ± 0.3 | 3.9 ± 0.2* |
| HDL cholesterol (mm)a | 1.2 ± 0.1 | 1.4 ± 0.1 |
| Triglycerides (mM) | 1.6 ± 0.4 | 1.4 ± 0.2 |
| Creatinine (µm) | 93.5 ± 7.0 | 108 ± 6.9 |
| Red blood cells (× 1012 cells l−1) | 4.8 ± 0.1 | 4.3 ± 0.1* |
| Haemoglobin (g l−1) | 13.0 ± 0.2 | 12.0 ± 0.4 |
| Haematocrit (%) | 42.7 ± 0.9 | 40.2 ± 1.2 |
| Platelets (× 109 cells l−1) | 209 ± 14 | 202 ± 16 |
Abbreviations and notes: calculated; BMI, body mass index; BP, blood pressure; HDL, high-density lipoprotein. Data expressed as mean ± SEM; Statistical analysis using two-tailed, unpaired t-test
P < 0.05;
P < 0.001).
Table 2.
Patient characteristics and medications
| Patient | Age (years) | BMI (kg m−2) | BP (mmHg) | TChol : HDL ratio | Platelets (× 109 cells l−1) | CAD | Previous revascularization | T2DM | Antiplatelet therapy | Other drugs and dose (mg) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 68 | 28 | 142/84 | 3 | 85 | 3 VD | CABG | Y | ASA75, Clpd75 | Frus80, En2.5, Ator20, GTN |
| 2 | 75 | 36 | 103/58 | 5.3 | 265 | 3 VD | CABG | Y | ASA75, Clpd75 | Frus40, Ator80, IsoMn25, Los50, Metclp10, Omep20, Metf500, GTN |
| 3 | 70 | 24 | 129/73 | 2.9 | 205 | 2 VD | CABG | – | ASA75 | Ator80, Omep20, Ram5, Metp50 |
| 4 | 56 | 29 | 152/94 | 1.7 | 159 | N | – | – | ASA75 | GTN |
| 5 | 73 | 30 | 128/74 | 2.4 | 257 | 2 VD | Stent | – | ASA75 | Sim40, Adiz90, GTN |
| 6 | 61 | 25 | 151/75 | 3.0 | 217 | 2 VD | – | – | ASA75 | Sim40, Aten50, GTN |
| 7 | 64 | 30 | 136/82 | 3.2 | 179 | 1 VD | – | – | ASA75 | Sim40, Aten50, Per4, GTN |
| 8 | 65 | 28 | 150/90 | 2.6 | 190 | 1 VD | – | – | ASA75 | Ator10 |
| 9 | 65 | 36 | 135/75 | 3.9 | 188 | M | – | – | ASA75, Clpd75 | Ator80, Aten50, Los100, Omep15, Bfmt2.5, Aml10, GTN |
| 10 | 53 | 28 | 138/84 | 2.4 | 255 | N | – | – | ASA75 | Sim20, Adiz120, Per8, Bfmt2.5, GTN |
| 11 | 46 | 28 | 138/85 | 4.9 | 267 | 2 VD | – | – | ASA75 | Ator40, Bis2.5, GTN |
| 12 | 75 | 24 | 158/78 | 3.1 | 156 | 1 VD | – | Y | ASA 75 | Ator20, Cand4, GTN |
| Mean | 65 | 29 | 138/79 | 3.2 | 202 | |||||
| SEM | 2.5 | 1 | 4/3 | 0.3 | 16 |
ASA, aspirin; Adiz, adizem; Aml, amlodipine; Aten, atenolol; Ator, atorvastatin; BMI, body mass index; BP, blood pressure; Bfmt, bendroflumethiazide; Bis, bisoprolol; CABG, coronary artery bypass graft; CAD, coronary artery disease; Cand, candesartan; Clpd, clopidogrel; En, enalapril; Frus, furosemide; GTN, glyceryl trinitrate spray; HDL, high-density lipoprotein; IsoMn, isosorbide mononitrate; LAD, left anterior descending artery; Los, losartan; M, mild; Metclp, metoclopramide; Metf, metformin; Metp, metoprolol; N, normal; Omep, omeprazole; Per, perindopril; Ram, ramipril; Sim, smivastatin; Tchol, total cholesterol; T2DM, type 2 diabetes mellitus; VD, vessel disease.
Characterization of the response of blood from healthy volunteers to cannabinoid and non-cannabinoid agonists
The CB1/CB2 agonist, Δ9-THC (300 µm), had no pro-aggregatory effect in the absence or presence of either AM251 (100 nm–30 µm) or AM630 (100 nm–30 µm). 2-AG (200 µm) did induce aggregation, but this was not affected by pre-incubation with AM251 (Figure 1). Similarly, AM251 had no effect on aggregation induced by either ADP (20 µm; Figure 2A) or thrombin (0.25 U ml−1; Figure 2B). AM251 itself had no aggregatory effects beyond a small, aggregatory response also induced by the vehicle control. To account for this, all subsequent responses involved subtracting individual vehicle control values for both the agonist and antagonist from the final aggregatory response to give a true value of aggregation as the area under the curve over a 6 min period.
Figure 1.

The aggregatory effect of 2-AG (200 µm) on its own (control) and after pre-incubation with AM251 (100 nm–30 µm), in whole blood samples from healthy human volunteers (n= 6). It is important to note that data were standardized by subtracting vehicle-induced aggregation in each case and are expressed as the mean ± SEM; statistical analysis was performed using a one-way anova and no significance was found (P= 0.78)
Figure 2.

The aggregatory effect of (A) ADP (20 µm) and (B) thrombin (0.25 U ml−1) on their own (control) and after pre-incubation with AM251 (100 nM–30 µm), in whole blood samples from healthy human volunteers (n= 6). It is important to note that data were standardized by subtracting vehicle-induced aggregation in each case and are expressed as the mean ± SEM; statistical analysis was performed using a one-way anova and no significance was found [(A) P= 0.90; (B) P= 0.86]
The effects of AM251, AM630 and aspirin on 2-AG-induced platelet aggregation in blood from healthy volunteers and CAD patients on aspirin
Pre-incubation with AM251 and AM630 had no impact on subsequent aggregation induced by 2-AG (100 µm) at any of the incubation times tested over the 120 min incubation period in blood obtained from either normal, aspirin naïve volunteers, or from CAD patients. However, a significant difference in the aggregatory response to 2-AG (100 µm) was observed between healthy volunteers and patients (P < 0.0001) for all drug concentrations and incubations times for both AM251 and AM630 (Figure 3A,B, respectively).
Figure 3.

The aggregatory effect of 2-AG on its own (control) and after incubation with (A) AM251 (0.3, 3, 30 µm) and (B) AM630 (0.3, 3, 30 µm) at different time points (10–120 min), in whole blood samples from healthy human volunteers (n= 8; solid lines) and patients (n= 12; dotted lines). Data were standardized by subtracting vehicle-induced aggregation in each case and are expressed as the mean ± SEM; statistical analysis was performed using a two-way anova. Statistical significance was observed (***P < 0.001) in the difference between the average 2-AG-induced aggregation of healthy volunteers compared with patients for both drugs at all three concentrations of AM251 or AM630, as well as with the control. (A) Control ( ); AM251 30 µm (
); AM251 30 µm ( ); AM251 3 µm (
); AM251 3 µm ( ); AM251 300 nm (
); AM251 300 nm ( ); (B) Control (
); (B) Control ( ); AM630 30 µm (
); AM630 30 µm ( ); AM630 3 µm (
); AM630 3 µm ( ); AM630 300 nm (
); AM630 300 nm ( )
)
Sub-analysis of the data to interrogate whether patients on clopidogrel + aspirin (n= 3) responded differently to 2-AG than those on aspirin alone (n= 9) suggested that those on combined anti-platelet therapy were more responsive to 2-AG (AUC = 23.4 ± 6.0) than those on aspirin alone (AUC = 9.5 ± 7.1). The low number of patients on clopidogrel + aspirin precluded meaningful statistics on these data.
Since all CAD patients were on aspirin therapy at the time of blood sampling, while the healthy volunteers were free of aspirin, to confirm that these observed effects were due to the presence of aspirin we assessed in vitro the effect of pre-incubating blood samples from the healthy volunteers with aspirin on both arachidonic acid (AA) and 2-AG-induced aggregation. Pre-incubation of blood with aspirin (100 µm) was shown to inhibit markedly aggregation induced by both 2-AG and AA (Figure 4A,B, respectively). This was statistically significant for 2-AG concentrations 100 µm (P < 0.001) and 200 µm (P < 0.05) and for AA concentrations 100 µm (P < 0.05), 300 µm (P < 0.01) and 750 µm (P < 0.001).
Figure 4.
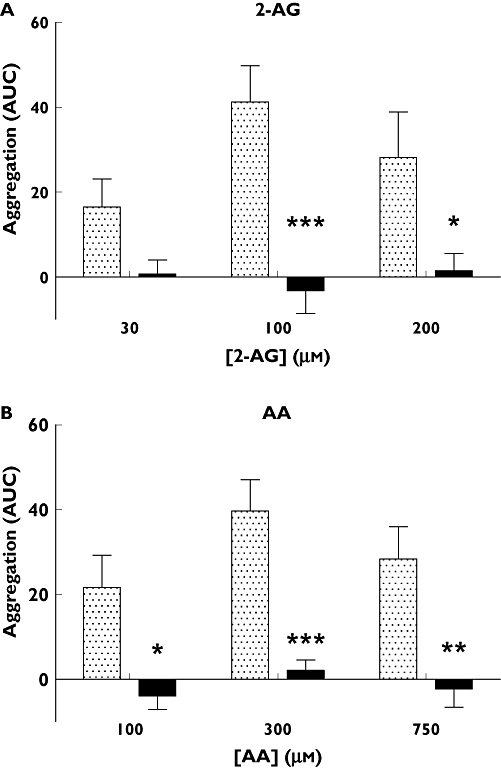
The inhibitory effect of aspirin (100 µm) on (A) 2-AG (30, 100 and 200 µm) and (B) AA (100, 300 and 750 µm) induced aggregation (n= 8). Data were standardized by subtracting vehicle-induced aggregation in each case and are expressed as the mean ± SEM; statistical analysis was performed using a two-way anova. 2-AG-induced aggregation was significantly inhibited by aspirin at concentrations of 100 µm (***P < 0.001) and 200 µm (*P < 0.05). AA-induced aggregation was significantly inhibited at all three concentrations, 100 µm (*P < 0.05), 300 µm (***P < 0.001) and 750 µm (**P < 0.01). Control ( ); + aspirin 100 µm (
); + aspirin 100 µm ( )
)
A final set of experiments conducted in blood from a separate cohort of healthy volunteers (n= 5) showed that a maximal concentration of the specific FAAH inhibitor, URB597, failed to have any significant effect on aggregation in response to 2-AG in either whole blood or PRP (Figure 5). However, the specific MAGL inhibitor, URB602, all but abolished aggregation (P < 0.001) in both whole blood and PRP. Similar inhibition was seen with preparations treated with both antagonists together (URB597 + URB602) and in those treated with aspirin (100 µm) as before.
Figure 5.

The inhibitory effect of URB597 (300 nm), URB602 (300 µm) and aspirin (100 µm) on 2-AG (200 µm)-induced aggregation in whole blood and platelet rich plasma. 2-AG-induced aggregation was inhibited by URB602, [URB597 + URB602] and aspirin in both whole blood and platelet rich plasma. ***P < 0.001 (two-way anova with Bonferoni post-test; n= 5). whole blood ( ); PRP (□)
); PRP (□)
Discussion
This study has demonstrated that, contrary to previous reports [16], 2-AG-induced platelet aggregation is mediated via activation of the COX pathway rather than through activation of CB1 receptors. The effect was found to be dependent on metabolism of 2-AG by MAGL, but not by FAAH. CB1 and CB2 antagonism had no effect on 2-AG-induced platelet aggregation in vitro, in blood from healthy volunteers and CAD patients, suggesting that endocannabinoids exert no tonic effect on platelets and any potential release of endocannabinoids as a consequence of platelet activation by either ADP or thrombin does not appear to involve CB1 or CB2 receptors. Δ9-THC was found to be completely inactive in platelets.
The literature relating to the activity of endocannabinoids in platelets is controversial. Maccarrone et al. showed that high concentrations (1.3 mm) of anandamide caused platelet activation in human PRP [15]. The effect was unaffected by COX1 and FAAH inhibition, suggesting a cannabinoid receptor-dependent mechanism. Similar results were obtained in a later paper [16] with 2-AG, leading the authors to suggest that 2-AG-mediated aggregation is via activation of an as yet uncharacterized CB receptor. In contrast, Braud et al. showed similar anandamide-induced platelet aggregation, though at lower concentrations (3–10 µm), in washed platelets from rabbits, but activation was insensitive to CB1 antagonism and completely inhibited by COX1 and FAAH inhibition [18].
Our findings are in keeping with a previous report in human PRP [16], in that 2-AG induced platelet activation in whole blood in vitro. However, in stark contrast to the previous study, we found that the actions of 2-AG were not mediated by either CB1 or CB2 activation, but were abolished by the COX inhibitor, aspirin. The inference from these results is that 2-AG is metabolized in whole blood to generate arachidonic acid, the substrate for COX-mediated generation of the platelet activator, TXA2. The inability of AM251 or AM630 to inhibit 2-AG-mediated platelet aggregation throughout a 2 h incubation period infers that neither the antagonists themselves, nor any blood-derived metabolites that might infer inhibitory activity, were capable of inhibiting 2-AG-mediated aggregation. Whilst it is feasible that AM251 was ineffectual as a CB1 antagonist, we have evidence from in vivo studies in rats that the same batch of AM251 completely abolished the depressor responses to the CB1 agonist ACEA (unpublished observations).
In the present studies, we found that the CB1/CB2 agonist, Δ9-THC, at a concentration relevant to reported peak plasma concentrations observed post-marijuana inhalation [30], had no impact on platelet aggregation in blood from healthy volunteers (Figure 4). This is inconsistent with previous reports of Δ9-THC-mediated induction of GPIIb/IIIa exposure [7] and in conflict with anti-aggregatory effects of Δ9-THC reported elsewhere [17]. It is possible that by activating both CB1 and CB2 receptors, Δ9-THC could induce opposing responses mediated by the two receptors. However, the observation Δ9-THC failed to induce aggregation in the presence of either CB1 or CB2 receptor blockade does not support this notion and lends weight to the argument that CB1 (or indeed CB2) receptors play little or no part in platelet activation.
Several studies have shown that cannabinoids, notably 2-AG, can have a co-agonistic effect with normal physiological agonists such as ADP and thrombin [16] and it is recognized that, at least in the central nervous system, cannabinoid receptors mediate a tonic activity that is reversed by antagonists in the absence of exogenous cannabinoid agonists [23]. Our observations that AM251 alone did not have any effect on platelet activation induced by the physiological agonists, ADP or thrombin, suggests that there is no tonic activity associated with CB1 receptors in platelets.
The above studies in blood from healthy volunteers suggest that 2-AG-mediated activation is CB-receptor independent, but that the same is not necessarily true in patients with coronary artery disease, where changes in enzyme and receptor expression might alter the activation pathway(s) involved. Furthermore, all 12 CAD patients involved in the study were taking aspirin and three patients were also prescribed clopidogrel (Table 2). We found that the pro-aggregatory response was blunted in patients compared with healthy volunteers, most likely as a result of aspirin-mediated inhibition of the COX pathway, adding weight to the hypothesis that 2-AG-induced aggregation is via metabolism to arachidonic acid. No further inhibition was seen with either the CB1 or CB2 receptor antagonist, suggesting that cannabinoid receptors are not involved in the modest aggregation seen in response to 2-AG in patients. The observation that blood from the patients prescribed aspirin + clopidogrel appeared to be more responsive to 2-AG than that from patients on aspirin alone requires further exploration in a study specifically designed to test this hypothesis. However, the finding that blood from patients with coronary artery disease receiving aspirin showed reduced aggregatory responses led us to investigate whether or not this was due to COX inhibition. We therefore confirmed this in blood from healthy volunteers by studying the effects of COX inhibition in vitro with aspirin. We then went on to confirm that responses to 2-AG were abolished by the MAGL inhibitor, URB602, but not the FAAH inhibitor, URB597, adding conclusive evidence that metabolic conversion of 2-AG to AA by MAGL is a pre-requisite for 2-AG-mediated platelet activation.
Our findings challenge the previous literature that suggests 2-AG-induced aggregation is cannabinoid receptor-dependent and metabolic breakdown-independent [16]. The conclusive impact of URB602 on 2-AG-induced aggregation (Figure 5) in both whole blood and PRP, combined with the inhibitory effect of aspirin, strongly suggests that the mechanism of action of 2-AG is not through CB1 or CB2 receptors, but via MAGL-mediated conversion of 2-AG to AA prior to activation of COX-1. This finding goes some way to supporting the findings of Braud et al. [18] in that an endocannabinoid is shown to require metabolic conversion for activity. It also might explain the lack of effect of Δ9-THC on platelets. The specificity of MAGL for 2-AG clearly does not extend to Δ9-THC, rendering the latter ineffectual in this setting despite the possibility of CB1 receptors on platelets. The differences between our data and that of a previous report [16] are not due to the use of whole blood aggregometry in our study as opposed to platelet rich plasma previously: inhibition of MAGL in both preparations had the same inhibitory effect.
Our finding that 2-AG mediates a pro-aggregatory effect via a CB1 receptor-independent pathway implies that CB1 receptor antagonism might not have any benefit with respect to reducing thrombotic potential in patients, at least in the acute phase. However, a recent study, measuring platelet-bound fibrinogen in unstimulated whole blood, suggests that CB1 receptor antagonism depresses platelet activation in a mouse model of type 2 diabetes mellitus [31]. Treatment with rimonabant reduced fibrinogen binding (reflecting reduced glycoprotein IIb/IIIa exposure), reduced thrombin-induced platelet aggregation and reduced ADP-stimulated expression of P-selectin, a platelet activation marker. These findings suggest that chronic treatment with a CB1 receptor antagonist may cause changes in platelet function, although it is as yet unclear whether this is a species-dependent phenomenon or one that is due to subtle effects exerted during protracted therapy.
Limitations of study
The patient group and healthy volunteers used in this study were not matched. The aim was not to compare directly the effects of AM251 in platelets in the two groups but to establish whether the same principles of activation applied in CAD patients, given that their pathophysiology might differ from a healthy group. The major differences between the healthy and patient groups include increased age and the polypharmacy related to high-risk patients undergoing interventional therapies for cardiac disease. Drug therapies in these patient groups include medications directly targeting platelet function, such as aspirin and clopidogrel, and other medications, such as statins and anti-hypertensives, which may cause unexpected changes in platelet function. These differences must be taken into consideration when directly comparing the results from the two groups in this study. In addition, platelet aggregometry, whether in whole blood or PRP is nevertheless some distance from in vivo measures of platelet function. Whilst aggregometry is an accepted measure for this kind of pharmacological study, direct correlations to in vivo findings should be viewed with caution [32].
Conclusion
In conclusion, 2-AG induces aggregation in human platelets. The mechanism of platelet activation is via metabolism by the enzyme MAGL, to arachidonic acid rather than through direct activation of CB1 or CB2 receptors. Whilst the clinical potential of endocannabinoid receptor antagonists as therapies to modulate cardiovascular risk is potentially exciting, the current study demonstrates that there is likely to be minimal acute effects of these compounds in terms of platelet function. There is some evidence in animal models, however, to suggest that long-term therapy may inhibit platelet function, and this warrants further study.
Acknowledgments
Support for this project is gratefully acknowledged from NHS Highland Endowments, Highlands & Islands Enterprise, Scottish Funding Council, European Regional Development Fund and the Coronary Thrombosis Trust (Award No. CTT40/08).
Competing interests
There are no competing interests to declare.
REFERENCES
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–56. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bisogno T. Endogenous cannabinoids: structure and metabolism. J Neuroendocrinol. 2008;20(Suppl 1):1–9. doi: 10.1111/j.1365-2826.2008.01676.x. [DOI] [PubMed] [Google Scholar]
- 5.Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008;118:18–35. doi: 10.1016/j.pharmthera.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Mackie K. Signaling via CNS cannabinoid receptors. Mol Cell Endocrinol. 2008;286:S60–5. doi: 10.1016/j.mce.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deusch E, Kress HG, Kraft B, Kozek-Langenecker SA. The procoagulatory effects of delta-9-tetrahydrocannabinol in human platelets. Anesth Analg. 2004;99:1127–30. doi: 10.1213/01.ANE.0000131505.03006.74. [DOI] [PubMed] [Google Scholar]
- 8.Docagne F, Mestre L, Loria F, Hernangomez M, Correa F, Guaza C. Therapeutic potential of CB2 targeting in multiple sclerosis. Expert Opin Ther Targets. 2008;12:185–95. doi: 10.1517/14728222.12.2.185. [DOI] [PubMed] [Google Scholar]
- 9.Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J. 2005;7:E625–54. doi: 10.1208/aapsj070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valverde O, Karsak M, Zimmer A. Analysis of the endocannabinoid system by using CB1 cannabinoid receptor knockout mice. Handb Exp Pharmacol. 2005;168:117–45. doi: 10.1007/3-540-26573-2_4. [DOI] [PubMed] [Google Scholar]
- 11.Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci. 2005;8:585–9. doi: 10.1038/nn1457. [DOI] [PubMed] [Google Scholar]
- 12.Bonz A, Laser M, Kullmer S, Kniesch S, Babin-Ebell J, Popp V, Ertl G, Wagner JA. Cannabinoids acting on CB1 receptors decrease contractile performance in human atrial muscle. J Cardiovasc Pharmacol. 2003;41:657–64. doi: 10.1097/00005344-200304000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol. 1999;276:H2085–93. doi: 10.1152/ajpheart.1999.276.6.H2085. [DOI] [PubMed] [Google Scholar]
- 14.Levy R, Schurr A, Nathan I, Dvilanski A, Livne A. Impairment of ADP-induced platelet aggregation by hashish components. Thromb Haemost. 1976;36:634–40. [PubMed] [Google Scholar]
- 15.Maccarrone M, Bari M, Menichelli A, Del Principe D, Agro AF. Anandamide activates human platelets through a pathway independent of the arachidonate cascade. FEBS Lett. 1999;447:277–82. doi: 10.1016/s0014-5793(99)00308-7. [DOI] [PubMed] [Google Scholar]
- 16.Maccarrone M, Bari M, Menichelli A, Giuliani E, Del Principe D, Finazzi-Agro A. Human platelets bind and degrade 2-arachidonoylglycerol, which activates these cells through a cannabinoid receptor. Eur J Biochem. 2001;268:819–25. doi: 10.1046/j.1432-1327.2001.01942.x. [DOI] [PubMed] [Google Scholar]
- 17.Formukong EA, Evans AT, Evans FJ. The inhibitory effects of cannabinoids, the active constituents of Cannabis sativa L. on human and rabbit platelet aggregation. J Pharm Pharmacol. 1989;41:705–9. doi: 10.1111/j.2042-7158.1989.tb06345.x. [DOI] [PubMed] [Google Scholar]
- 18.Braud S, Bon C, Touqui L, Mounier C. Activation of rabbit blood platelets by anandamide through its cleavage into arachidonic acid. FEBS Lett. 2000;471:12–6. doi: 10.1016/s0014-5793(00)01359-4. [DOI] [PubMed] [Google Scholar]
- 19.Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thöne-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423–31. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams CM, Kirkham TC. Reversal of delta 9-THC hyperphagia by SR141716 and naloxone but not dexfenfluramine. Pharmacol Biochem Behav. 2002;71:333–40. doi: 10.1016/s0091-3057(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 21.Scheen AJ, Van Gaal LG, Despres JP, Pi-Sunyer X, Golay A, Hanotin C. Rimonabant improves cardiometabolic risk profile in obese or overweight subjects: overview of RIO studies. Rev Med Suisse. 2006;2:1916–23. [PubMed] [Google Scholar]
- 22.Van Gaal LF, Michiels JJ. Obesity, health issues, and cardiovascular disease. Semin Vasc Med. 2005;5:1–2. doi: 10.1055/s-2005-871736. [DOI] [PubMed] [Google Scholar]
- 23.Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–24. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 24.Golanski J, Pietrucha T, Baj Z, Greger J, Watala C. Molecular insights into the anticoagulant-induced spontaneous activation of platelets in whole blood – various anticoagulants are not equal. Thromb Res. 1996;83:199–216. doi: 10.1016/0049-3848(96)00129-6. [DOI] [PubMed] [Google Scholar]
- 25.Dalby MC, Davidson SJ, Burman JF, Davies SW. Diurnal variation in platelet aggregation with the PFA-100 platelet function analyser. Platelets. 2000;11:320–4. doi: 10.1080/09537100050144731. [DOI] [PubMed] [Google Scholar]
- 26.Serebruany V, Mckenzie M, Meister A, Fuzaylov S, Gurbel P, Atar D, Gattis W, O'Connor C. Whole blood impedance aggregometry for the assessment of platelet function in patients with congestive heart failure (EPCOT trial) Eur J Heart Fail. 2002;4:461–7. doi: 10.1016/s1388-9842(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 27.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–12. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 28.Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005;168:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- 29.Pertwee RG. GPR55: a new member of the cannabinoid receptor clan? Br J Pharmacol. 2007;152:984–6. doi: 10.1038/sj.bjp.0707464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azorlosa JL, Heishman SJ, Stitzer ML, Mahaffey JM. Marijuana smoking: effect of varying delta 9-tetrahydrocannabinol content and number of puffs. J Pharmacol Exp Ther. 1992;261:114–22. [PubMed] [Google Scholar]
- 31.Schafer A, Pfrang J, Neumuller J, Fiedler S, Ertl G, Bauersachs J. The cannabinoid receptor-1 antagonist rimonabant inhibits platelet activation and reduces pro-inflammatory chemokines and leukocytes in zucker rats. Br J Pharmacol. 2008;154:1047–54. doi: 10.1038/bjp.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santilli F, Rocca B, De Cristofaro R, Lattanzio S, Pietrangelo L, Habib A, Pettinella C, Recchiuti A, Ferrante E, Ciabattoni G, Davì G, Patrono C. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: implications for aspirin “resistance. J Am Coll Cardiol. 2009;53:667–77. doi: 10.1016/j.jacc.2008.10.047. [DOI] [PubMed] [Google Scholar]