

Abstract
AIM
Similar analgesics may have different analgesic potencies especially in patients in whom the pain system is sensitized. The aim was to investigate different opioid effects on experimental pain after the sensitized pain system was mimicked evoking hyperalgesia in healthy volunteers.
METHODS
Twenty-four healthy volunteers were randomized to treatment with morphine (30 mg orally) and oxycodone (15 mg orally) or placebo in a double-blind crossover study. Hyperalgesia was induced by oesophageal perfusion with acid and capsaicin. Several exploratory endpoints were studied using skin heat, muscle pressure and oesophageal mechanical, heat and electrical stimulation. Effects on pain from deeper structures were considered most important.
RESULTS
Different analgesic potencies were found. Oxycodone had a greater analgesic effect than morphine attenuating pain from: (i) heat stimulation of skin (P= 0.016); difference between the means of 0.39°C, 95% CI 0.22, 2.09. (ii) muscle pressure (P < 0.001); difference between the means of 11.93kPa, 95% CI 5.4, 18.5. (iii) oesophageal heat stimulation (P < 0.001); difference between the means of 38.54 cm2, 95% CI 15.37, 61.71 and (iv) oesophageal electrical stimulation (P= 0.016); difference between the means of 6.69mA, 95% CI 1.23, 12.13.
CONCLUSION
After sensitization of the pain system different analgesic potencies of morphine and oxycodone were found in response to skin, muscle and oesophageal pain stimulation, in which oxycodone had a greater effect. As similar differential analgesic potencies of the two opioids have been found in patients with chronic pain, the experimental hyperalgesia model bridged findings from studies in healthy volunteers to patients.
Keywords: experimental model, hyperalgesia, morphine, oxycodone, pain
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Previous studies using short-lasting experimental pain stimulations in healthy volunteers have shown differences in opioid effects regarding visceral pain stimulations. However, these differences can be more pronounced in patients due to a sensitized pain system. Therefore, the aim of the present study was to mimic the clinical situation by investigating opioid effects on experimental pain in healthy volunteers after experimentally evoked hyperalgesia.
WHAT THIS STUDY ADDS?
We now know that morphine and oxycodone exerts different effects in the sensitized pain system as we found a greater analgesic effect of oxycodone in response to skin, muscle and oesophageal pain stimulation. This supports clinicians' experiences that oxycodone can be superior to morphine in the treatment of some pain conditions. The evoked hyperalgesia bridged findings from studies in healthy volunteers to patients, and new fundamental knowledge on different analgesic effects in hyperalgesia was found.
Introduction
Chronic pain is most often associated with hyperalgesia which is difficult to treat [1, 2]. Different opioids are used in chronic pain treatment. However, the clinical significance of the potential effects of opiates on inflammatory processes is unclear [3]. Inflammation can effectuate changes in the pain systems [4–7]. These changes may include up-regulation of specific opioid receptors [8–11]. Animal studies have shown that opioid-binding to κ-receptor sites is increased in inflammation likely caused by up-regulation of κ-receptors in the nervous system [8, 12]. This may have clinical impact for some opioids, i.e. morphine and oxycodone which are thought to have different pharmacological profiles [13]. Hence, morphine binds to the µ-opioid receptor with stronger affinity and rodent experiments have indicated that oxycodone is a partial κ-agonist [14–18]. This could cause different analgesic potencies of morphine and oxycodone in diseases with inflammation.
Application of experimental pain models in a crossover study design with appropriate baseline recordings offers a unique opportunity of revealing analgesic effects [19]. It has been recommended to include pain models in various tissues, as opioid analgesia can exhibit tissue dissimilarities [20]. Moreover, different modalities activate distinct pain mechanisms, and make it possible to investigate on a mechanistic basis how analgesics work [20]. However, the effect on pain from deeper structures (muscle and viscera) is considered most important as opioid analgesia is more robust in deep pain [20]. In a previous experimental pain study in healthy volunteers morphine and oxycodone were comparable in analgesic potency in the modulation of skin and muscle pain, but oxycodone showed a greater analgesic potency on visceral pain [21]. Chronic pancreatitis is characterized by long lasting inflammation, hyperalgesia and pain [22]. Subsequently an experimental pain study was performed in patients with chronic pancreatitis where oxycodone was more potent than morphine in attenuating the same experimental skin, muscle and visceral pain in patients [23]. This supports the theory of different analgesic potencies of morphine and oxycodone when hyperalgesia is present.
Pain experienced and reported by healthy volunteers is different from clinical pain, and in the laboratory it is not possible to reproduce the pathophysiology and the full complexity of the pain experience in patients [4, 24]. On the other hand, hyperalgesia, being a hallmark of clinical pain, can be evoked experimentally in healthy volunteers by sensitizing the oesophagus with capsaicin or acid [25–27]. Moreover, local sensitization of the oesophagus can evoke generalized hyperalgesia reported as increased sensitivity in other organs and tissues than the affected one [27–29]. However, experimental pain models with hyperalgesia are more difficult to control than simple and short lasting pain stimuli, but may initiate some of the inflammatory processes and changes in the pain system that are seen in the clinic [29, 30].
We hypothesized that an experimental human translational pain model including hyperalgesia in volunteers could mimic the sensitized pain system in patients providing knowledge on the potential analgesic effects of opiates in inflammation. Such a model could bridge findings from experimental pain studies in healthy volunteers to those in patients. The aims were: (i) to investigate whether morphine and oxycodone, compared with placebo, had differential analgesic potencies after experimentally induced hyperalgesia and (ii) to validate whether this effect varied between tissues.
Methods
The study was conducted according to the Declaration of Helsinki. The Ethics Committee for the Region of Northern Jutland (N-20070025) and the Danish Medicines Agency (2612-3463) approved the study. The study was conducted according to the rules of Good Clinical Practice (GCP) and was monitored by the GCP-unit, Aarhus University Hospital, Denmark.
Subjects and study design
The reliability and sensitivity of the experimental tests have been evaluated previously [19]. In a similar study 24 healthy volunteers have been sufficient to reveal different analgesic effects of morphine and oxycodone [21]. Thus 24 healthy Caucasian (12 females), opioid-naïve volunteers (mean age 27.8, range 20–48 years) participated in this randomized, placebo-controlled, double-blind, three-way crossover study of Latin square design. The study was carried out in the Research Laboratory at ‘Mech-Sense’, Department of Gastroenterology, Aalborg Hospital, Denmark. The subjects were informed about the possible risks of the study, gave their informed consent and were paid for participation. Medical examinations prior to the study were normal and all females used contraceptives and a pregnancy test was negative in all study periods. All volunteers were trained and familiarized with all pain stimulations on a screening day, which took place at least 1 week before the first study day. Each volunteer fasted for at least 4 h prior to the study. The subjects were monitored with an electrocardiogram during the entire study.
When the subject arrived at the laboratory, blood pressure and pulse were registered and a probe designed for multimodal stimulation of the oesophagus was inserted through the mouth. A bag was mounted on the end of the probe and placed 8 cm proximal to the lower oesophageal sphincter. The probe was taped to the chin. The probe has been described in detail previously [26, 31]. The skin and muscular pain stimulation devices were positioned. Figure 1 illustrates the study design. Pain recordings were obtained before (baseline, to obtain values before any drug was given) and 30, 60 and 90 min after induction of hyperalgesia and drug administration. Pain was assessed from skin, muscle and oesophageal stimulations following the same order for all stimulation series as illustrated in Figure 1. Subjects were blinded to all thresholds measured. This schedule should ensure sufficient testing throughout the opioid absorption phase and the beginning of the elimination phase without extending the test period too much [32, 33]. The degrees of four known adverse effects, nausea, dizziness, itching and sweating, were registered as (1) none, (2) slight, (3) heavy or (4) intolerable and specifically asked for after each pain test battery. Moreover, the degree of sedation was evaluated after each pain test battery by the finger tapping test. If the subject was sedated or otherwise feeling unwell during the study, blood pressure and pulse were measured again to ensure that everything was normal before the study continued. If the subject felt dizzy, nauseated or otherwise indisposed at the end of the study they were observed in the laboratory until the adverse effect ceased and they felt ready to leave.
Figure 1.
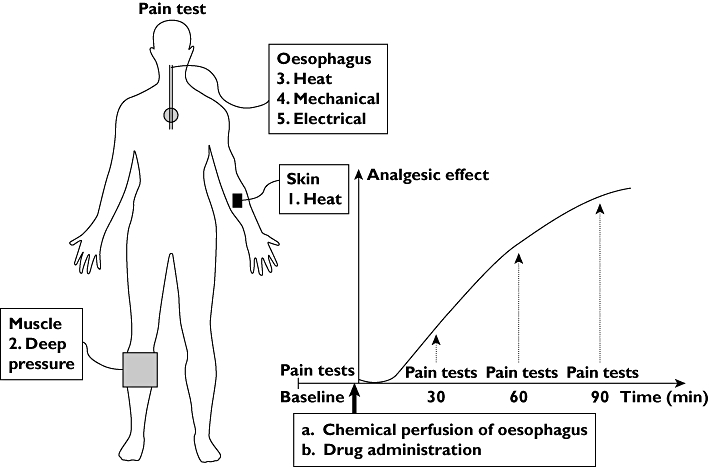
Flow-chart for the study protocol. A schematic illustration of the pain tests is shown on the left. Pain tests were performed in the order illustrated by the numbers. The order was similar for all stimulation series and between days and consisted of heat stimulation of the skin, mechanical stimulation of the muscle and heat, mechanical and electrical stimulation of the oesophagus. The graph (right) shows a schematic illustration of the analgesic effect. Pain tests were performed at baseline after which hyperalgesia was induced and the drug administered. Pain tests were performed 30, 60 and 90 min after baseline. Drug administration included either placebo, morphine (30 mg) or oxycodone (15 mg) in randomized order
The experiment was repeated three times with at least 1 week ‘wash-out’ intervals. The randomized order should deal with periodic effects e.g. learning effects, carry-over and interference effects. Hence bias from such effects would be evenly distributed in the three treatment periods.
Medication
Each subject was investigated in three separate periods and received 30 mg of morphine (morphine oral liquid mixture 2 mg ml−1, Hospital Pharmacies, Denmark) or 15 mg oxycodone (Oxynorm® oral liquid mixture 10 mg ml−1, Norpharma A/S, Hørsholm, Denmark) or placebo in randomized order (all masked in colour by 5 ml orange juice concentrate and diluted with water to a total of 20 ml liquid). The drugs were given immediately after baseline pain recordings. The colour of the mixture was masked in the orange juice and the mixture was infused through a catheter inside the probe ending in the distal oesophagus. The mixture was handled by a pharmacist not otherwise involved in the study to ensure blinding of all other investigators.
The dose of morphine was twice that of oxycodone. The rationale was that the greater oral bioavailability of immediate release oxycodone compared with morphine, and more rapid onset of analgesia could result in superior effects of oxycodone [34]. Thus, we chose a relatively high dose of morphine to be sure that differences in bioavailability did not favour oxycodone. Furthermore, the two doses have previously showed comparable analgesic potencies in experimental skin and muscle pain [21].
Skin stimulation
The cutaneous heat pain tolerance threshold was determined by a computer driven heat pain device (TSA-II NeuroSensory Analyzer, Medoc Ltd, Ramat Yishai, Israel). A thermode with a surface of 25 × 50 mm was applied to the volar surface of the left forearm at 10 cm from the elbow. The temperature increased from 32°C to a maximum of 52°C at a rate of 1°C s−1 until the threshold was reached. Three consecutive temperature measurements in °C were noted and the average computed. The threshold was defined as the highest stimulus intensity the volunteer could accept. The subjects were told to stop the stimulation by a click on a button when the pain tolerance threshold (PTT) was reached.
Muscle stimulation
Muscle stimulation was done by an electronic cuff algometer consisting of a pneumatic tourniquet cuff, a computer controlled air compressor, and an electronic 10 cm visual analogue scale (VAS) (Aalborg University, Denmark). The cuff algometer has been described in detail previously [35, 36]. The 0 and 10 cm extremes on the VAS were defined as ‘no pain’ and as ‘the worst pain imaginable’. The computer continuously controlled the compression rate to ensure a linear increase in pressure. A tourniquet cuff was wrapped around the gastrocnemius muscle. In order to ensure reliable pressure readings the cuff was fitted tightly around the limb before each measurement. All recordings were made on the right limb with the subject in a supine position. In addition to the hand-held pressure release button the inflation could be terminated both mechanically and from the computer program. The cuff was automatically inflated (compression rate 0.50 kPa s−1). The subject was instructed to rate the pain intensity continuously on the VAS from the first sensation of pain being the pain detection threshold (PDT). The pressure continued increasing and the subject pressed the release button when the cuff pressure pain tolerance threshold (PTT) was reached.
Oesophageal stimulation
Before the study all subjects were instructed how to use the 0–10 electronic VAS, for the visceral stimulations, where 0 = no perception, 1 = vague perception of mild sensation, 2 = definite perception of mild sensation, 3 = vague perception of moderate sensation, 4 = definite perception of moderate sensation, 5 = the pain threshold (PDT), 6 = mild pain, 7 = moderate pain, 8 = pain of medium intensity, 9 = intense pain and 10 = unbearable pain. This scale has been described in detail previously [37] and it has proved to be reliable in experimental oesophageal pain in both healthy volunteers and patients [19, 38–40].
The subjects were instructed to score the evoked pain and to differentiate this from unpleasantness in the throat. The subjects were asked to quantify the referred pain area, evoked at the maximal pain intensity by drawing the area on transparent paper, which was placed over the skin in the region where the pain was felt. The size of the referred pain area was later calculated and digitized (Trust, 1200 wireless tablet, Trust International BV, Dordrecht, the Netherlands). The visceral stimulations were stopped at moderate pain (VAS = 7) for the mechanical and the pain detection threshold (VAS = 5) for thermal and electrical stimuli stimulations. This was done to prevent severe autonomic reactions leading to vomiting of the probe. All data were displayed online (Openlab, GMC, Hornslet, Denmark) and stored for later analysis.
Mechanical stimulation
For mechanical stimulation the oesophageal bag was distended at a constant infusion rate of 15 ml min−1 until ‘moderate pain’ intensity ratings (VAS = 7) were reached. The volumes (ml) in the bag at ‘moderate pain’ were used for further analysis.
Thermal stimulation
A temperature probe (PR Electronics, Rønde, Denmark) monitored the fluid temperature continuously in the bag. The heat pain stimuli were performed by re-circulating water (60°C) in the bag. The infusion channels in the probe were attached to a specially designed pump system. OpenLab software (GMC Aps, Hornslet, Denmark) running on a computer was used to control an infusion/withdrawal syringe pump (PHD 2000, Harvard, Massachusetts, USA). The pump held two 140 ml syringes for infusion and withdrawal. The bag volume was kept constant by withdrawing the same amount of fluid as infused using the two large lumens. This was done to obtain a linear temperature increase of 1.5°C min−1. The temperature stimulus did not cause any mechanical stimulation since the amount of fluid in the bag did not change. Previous to this recirculation 4 ml of water was applied to the bag to ensure sufficient mucosa contact. The subject was instructed to rate the intensity continuously on the VAS from the first vague perception to the pain detection threshold. The temperature of the water in the bag at pain detection threshold was used for analysis.
Electrical stimulation
For electrical stimulation stainless steel electrodes were mounted on the probe 1 cm proximal to the bag. A computer-controlled constant current stimulator (University of Aalborg, Denmark) delivered a 25 ms train-of-five, 1 ms square-wave impulse (perceived as a single stimulus), to the oesophagus. Initially the pain detection threshold was found with increasing pain stimulations. The current intensity was increased in 1 mA in increments with random sham stimulations having the same or lower intensity until the pain detection threshold was found and the corresponding intensity in mA was used for analysis.
Induction of hyperalgesia
After all the baseline tests in all tissues the subjects underwent an oesophageal acid + capsaicin perfusion with a 100 ml solution consisting of 90 ml HCl 0.1 m (Bie & Berntsen, Rødovre, Denmark) and 1 mg capsaicin in 10 ml solvent (0.1 mg·ml−1 in polyoxyethylene sorbitan mono-oleate, Hospital Pharmacy, Aalborg Hospital, Denmark). The mixture was administered through a perfusion channel in the probe at a rate of 7 ml min−1. If the subject reached pain detection threshold (VAS = 5), the perfusion was stopped for 1 min and then continued until 100 ml was infused or until it became too unpleasant for the subject (pain intensity higher than PDT for more than 1 min). This method has been described previously [41].
Statistics
To eliminate errors relating to differences in baseline pain recordings, the change in stimulus intensity relative to baseline was used in the statistical analysis [19]. The results are expressed as differences between the means of the baseline corrected values and confidence intervals (95%) unless otherwise indicated. For multiple comparisons the overall effects were tested by two-way analysis of variance (anova) with the factors: 1) drug (with three levels: placebo, morphine and oxycodone) and 2) time (with three levels: 30, 60 and 90 min). This indicated whether there was a difference in the data obtained in the three drug groups (placebo, morphine and oxycodone) or in the data obtained at the three time points. When an overall significant difference was found, post hoc analyses were done based on a generalized linear model. To evaluate differences in drug effects, a total score over the 90 min was used in the post hoc analysis. For comparisons of side effects, Friedmans test was used. P < 0.05 was considered significant. The software package Sigma Stat vs. 3.0 (Systat Software, San Jose, California, USA) was used for the anova, whereas the software package STATA version 10.1 (StataCorp LP, College Station, Texas, USA) was used for the post hoc analysis.
Results
All volunteers completed the study. The results are shown in Table 1. The oesophageal perfusion with chemicals caused nausea and sweating in all volunteers. After administration of morphine two volunteers experienced nausea, sixteen sedation, two itching and one sweating. After administration of oxycodone six volunteers experienced nausea, sixteen sedation, six itching and four sweating. After administration of placebo one volunteer experienced nausea, two sedation, one itching and three sweating. None of the subjects reported any side effect as intolerable. There was no difference in the overall degree of side effects reported after morphine and oxycodone (P > 0.05). The finger tapping frequency increased over time in all three treatment arms. No difference was found in finger tapping frequency between placebo, morphine and oxycodone (F= 0.1, P= 0.9).
Table 1.
Results from all experimental pain stimulations given as mean ± CI
| Skin | Muscle | Oesophagus | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Heat | Pressure | Pressure | Pressure | Pressure | Heat | Heat | Electricity | Electricity | ||
| PTT (°C) | PDT (kPa) | PTT (kPa) | PTT (ml) | RPA (cm2) | PDT (°C) | RPA (cm2) | PDT (mA) | RPA (cm2) | ||
| Baseline | Placebo | 47.8 ± 0.8 | 24.1 ± 3.4 | 45.2 ± 7.1 | 27.2 ± 5.0 | 30.1 ± 21.2 | 48.0 ± 1.5 | 30.6 ± 15.4 | 16.5 ± 3.1 | 12.9 ± 7.7 |
| Morphine | 47.4 ± 0.8 | 23.3 ± 4.9 | 43.9 ± 7.3 | 29.6 ± 5.1 | 38.8 ± 27.9 | 50.1 ± 1.7 | 27.4 ± 11.5 | 16.7 ± 3.8 | 9.9 ± 8.5 | |
| Oxycodone | 47.8 ± 0.7 | 23.9 ± 4.6 | 45.2 ± 7.7 | 28.3 ± 5.1 | 22.4 ± 12.3 | 49.4 ± 1.8 | 32.1 ± 16.7 | 15.6 ± 3.1 | 10.7 ± 5.2 | |
| 30 min. | Placebo | 47.1 ± 0.9 | 23.8 ± 4.0 | 44.9 ± 6.9 | 26.9 ± 4.6 | 34.1 ± 23.3 | 48.1 ± 1.2 | 37.3 ± 19.1 | 12.0 ± 1.4 | 16.2 ± 8.9 |
| Morphine | 46.8 ± 0.8 | 25.4 ± 5.1 | 46.1 ± 7.5 | 29.8 ± 5.3 | 34.6 ± 20.8 | 48.2 ± 1.4 | 35.3 ± 19.8 | 14.2 ± 2.4 | 9.5 ± 6.3 | |
| Oxycodone | 47.6 ± 0.7 | 29.0 ± 6.3 | 51.8 ± 7.1 | 30.1 ± 5.0 | 24.8 ± 16.1 | 49.2 ± 1.4 | 21.4 ± 9.1 | 15.7 ± 2.5 | 10.7 ± 5.5 | |
| 60 min. | Placebo | 47.0 ± 0.7 | 25.9 ± 4.4 | 46.1 ± 7.1 | 29.0 ± 4.7 | 37.1 ± 21.4 | 48.3 ± 1.3 | 45.8 ± 23.7 | 12.0 ± 1.6 | 15.4 ± 7.9 |
| Morphine | 46.8 ± 0.8 | 28.6 ± 6.9 | 48.8 ± 7.7 | 31.7 ± 5.8 | 38.4 ± 18.7 | 49.2 ± 1.7 | 30.4 ± 11.2 | 14.1 ± 2.1 | 11.1 ± 6.7 | |
| Oxycodone | 47.9 ± 0.7 | 28.9 ± 5.9 | 53.2 ± 7.6 | 29.8 ± 6.1 | 25.5 ± 11.5 | 48.7 ± 1.6 | 24.2 ± 9.5 | 14.9 ± 2.1 | 11.7 ± 5.5 | |
| 90 min. | Placebo | 47.0 ± 0.9 | 27.2 ± 4.1 | 47.8 ± 7.0 | 30.9 ± 4.9 | 43.8 ± 25.2 | 49.3 ± 1.1 | 43.5 ± 22.3 | 12.1 ± 1.8 | 16.1 ± 9.1 |
| Morphine | 46.7 ± 0.9 | 29.1 ± 7.0 | 49.3 ± 7.7 | 33.0 ± 6.8 | 36.3 ± 18.3 | 49.9 ± 1.1 | 30.3 ± 10.1 | 14.0 ± 2.1 | 12.6 ± 6.1 | |
| Oxycodone | 47.6 ± 0.8 | 28.4 ± 6.0 | 53.9 ± 7.4 | 30.4 ± 6.0 | 24.9 ± 9.6 | 48.2 ± 1.4 | 24.1 ± 10.1 | 14.9 ± 2.6 | 10.6 ± 5.1 | |
| Statistical results | O>M = P | O = M>P | O>M>P | No difference | No difference | No difference | O>M = P | O>M = P | No difference | |
Data were obtained as pain detection thresholds (PDT), pain tolerance thresholds (PTT) or referred pain areas (RPA). The statistical results are summarized as better analgesic effect (>), equal effect (=) of placebo (P), morphine (M) and oxycodone (O).
Skin stimulation
Compared with placebo, administration of opioids increased PTT to heat stimulation (F= 9.5; P < 0.001) (Figure 2). In post hoc analysis oxycodone was found to be more effective than placebo (P < 0.001) with a difference between the means of 0.69°C (95% CI 1.12, 2.99) and more effective than morphine (P= 0.016) with a difference between the means of 0.39°C (95% CI 0.22, 2.09). The effect of morphine was not significantly different from placebo (P > 0.05).
Figure 2.
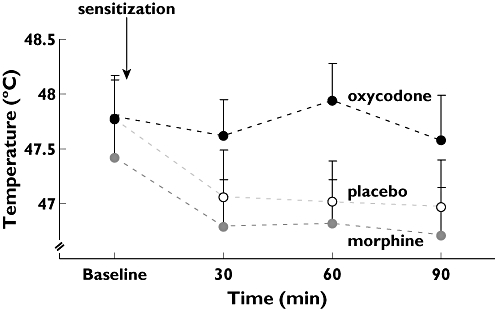
Results from painful skin stimulation. Stimulus intensities evoking skin pain tolerance threshold to heat stimulation at baseline, 30, 60 and 90 min after drug administration. Compared with placebo (white) and morphine (30 mg, grey) administration of oxycodone (15 mg, black) resulted in higher temperatures. The error bars represent SEM. To eliminate errors relating to differences in baseline pain recordings (illustrated by a lower baseline value for morphine in the figure), the change in stimulus intensity relative to baseline was used in the statistical analysis
Muscle stimulation
There was a difference in the effect of opioids and placebo regarding PDT (F= 3.8; P= 0.02). Post hoc analysis showed that morphine had a greater analgesic effect than placebo (P= 0.03) with a difference between the means of 9.32 kPa (95% CI 1.1, 17.6) and oxycodone had a greater analgesic effect than placebo (P= 0.012) with the difference between the means of 10.39 kPa (95% CI 2.3, 18.5). No difference was demonstrated between the effect of morphine and oxycodone (P > 0.05).
Changes in muscle PTT are illustrated in Figure 3. There was an overall effect of opioids (F= 21.7; P < 0.001). Post hoc analysis showed that morphine had a greater analgesic effect than placebo (P= 0.003) with a difference between the means of 9.74 kPa (95% CI 3.3, 16.1) and oxycodone had a greater analgesic effect than placebo (P < 0.001) with a difference between the means of 21.67 kPa (95% CI 15.2, 28.2). Oxycodone had a greater analgesic effect than morphine (P < 0.001) with a difference between the means of 11.93 kPa (95% CI 5.4, 18.5). Moreover, there was an overall effect of time on muscle PTT (F= 4.2; P= 0.016).
Figure 3.
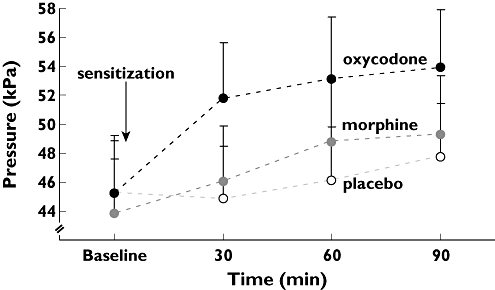
Results from painful muscle stimulation. The stimulus intensities evoking the muscular pain tolerance threshold (PTT) to pressure at baseline, 30, 60 and 90 min after drug administration are shown. Administration of both morphine (30 mg, grey) and oxycodone (15 mg, black) resulted in higher stimulus intensities to evoke PTT compared with placebo (white). Furthermore, oxycodone was superior to morphine. The error bars represent SEM. To eliminate errors relating to differences in baseline pain recordings (illustrated by a slightly lower baseline value for morphine in the figure), the change in stimulus intensity relative to baseline was used in the statistical analysis
Oesophageal stimulation
The visceral stimulation resulted in both local and referred pain. Figure 4 illustrates the average locations and sizes of the referred pain areas.
Figure 4.
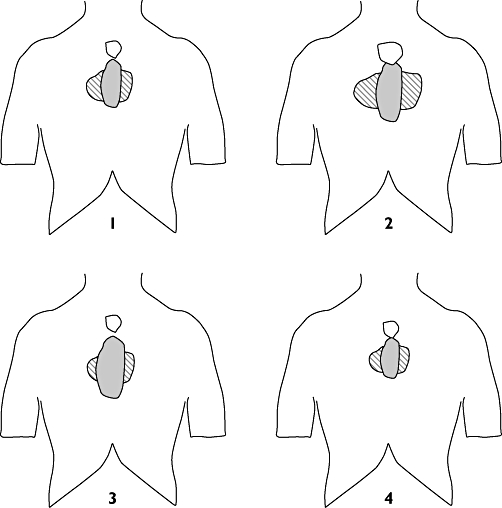
Illustration of referred pain areas to painful oesophageal stimulations. The mean of all reported referred pain areas is illustrated at (1) baseline; (2) after perfusion of the oesophagus with acid+capsaicin; (3) after perfusion of the oesophagus with acid + capsaicin and administration of morphine and (4) after perfusion of the oesophagus with acid + capsaicin and administration of oxycodone. The referred pain area was drawn immediately after the stimulation. The area reported after electrical stimulation (white) was often smaller than area reported after mechanical (grey) or thermal stimulation (hatched)
Mechanical stimulation
Regarding the tolerated volume, there was no difference between drugs (F= 0.1; P= 0.9). The referred pain areas were not affected after administration of opioids (F= 2.3; P= 0.1).
Thermal stimulation
There was no effect of opioids regarding temperature at PDT (F= 2.1; P= 0.122), but the referred pain area to heat in the oesophagus decreased (F= 13.7; P < 0.001). Post hoc analysis showed that oxycodone had a greater analgesic effect than placebo at decreasing the referred pain area (P < 0.001) with a difference in the means of 60.99 cm2 (95% CI 37.74, 84.24). Moreover, oxycodone had a greater analgesic effect than morphine (P < 0.001) with a difference in the means of 38.54 cm2 (95% CI 15.37, 61.71) (Figure 5). Morphine had no significant effect compared with placebo (P= 0.056).
Figure 5.
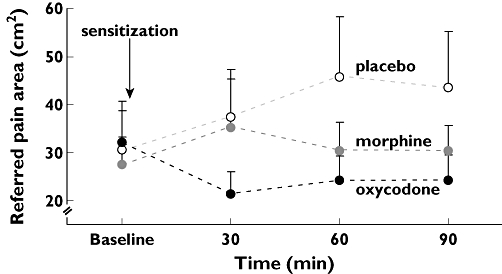
Results from painful heat stimulation of the oesophagus. The referred pain areas corresponding to pain detection thresholds to heat stimulation of the oesophagus at baseline, 30, 60 and 90 min after drug administration are shown. Morphine (30 mg, grey), oxycodone (15 mg, black) and placebo (white). The effect of oxycodone, but not morphine, was better than placebo. The error bars represent SEM. To eliminate errors relating to differences in baseline pain recordings, the change in stimulus intensity relative to baseline was used in the statistical analysis
Electrical stimulation
There was a significant effect of opioids on the stimulation intensity at PDT (F= 9.7; P < 0.001) (Figure 6). Post hoc analysis showed that oxycodone attenuated the pain response more than placebo (P < 0.001) with a difference in the means of 12.10 mA (95% CI 6.68, 17.53) and more than morphine (P= 0.016) with a difference in the means of 6.69 mA (95% CI 1.23, 12.13), Morphine showed no significant effect compared with placebo (P= 0.051). The referred pain areas to electrical stimulation in the oesophagus were not significantly affected by drugs (F= 1.7; P= 0.2).
Figure 6.
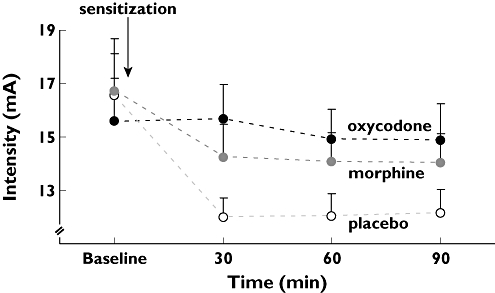
Results from painful electrical stimulation of the oesophagus. The stimulus intensities in pain detection thresholds to electrical stimulation of the oesophagus at baseline, 30, 60 and 90 min after drug administration are shown. Morphine (30 mg, grey), oxycodone (15 mg, black) and placebo (white). The effect of oxycodone, but not morphine, was better than placebo. The error bars represent SEM. To eliminate errors relating to differences in baseline pain recordings (illustrated by a slightly lower baseline value for oxycodone in the figure), the change in stimulus intensity relative to baseline was used in the statistical analysis
Discussion
The effects of morphine, oxycodone and placebo were compared in an experimental pain study after hyperalgesia was evoked in healthy volunteers using acid and capsaicin perfusion of the oesophagus. At the doses studied differences in analgesic potencies were found in various tissues. Oxycodone attenuated pain to heat stimulation of skin. Both opioids attenuated muscle pain, but oxycodone showed a greater analgesic effect than morphine. Oxycodone also showed greater analgesic effect compared with both morphine and placebo on pain from heat and electrical stimulation of the oesophagus.
Methodological considerations
A period effect could affect the results, but the randomization of the study ensured that any such effect was equally distributed in all treatment arms. Moreover, previous studies have demonstrated stable and reproducible pain recordings in multi-modal, multi-tissue experimental pain studies both within and between days [19, 41]. Therefore, a period effect was not believed to influence the analysis significantly in the present study. Another factor, sedation, could also potentially influence study outcome. Quante et al. investigated this issue after morphine administration in an experimental pain study in healthy volunteers and found no effect on sedation variables as the experimental pain was likely to increase arousal level thus counteracting morphine-induced sedation [42]. This could apply to the present study involving a rather intensive stimulation paradigm and the increased arousal was supported by an increase in finger tapping frequency in all three treatment arms. Thus, it is not likely that sedation influenced the study outcome.
In a previous study the current model of oesophageal sensitization was evaluated and evoked consistent hyperalgesia in healthy volunteers [41]. Therefore, further investigation of the hyperalgesia was not done in the present study. Hyperalgesia in chronic pain is often seen as a state of hypersensitivity of the central nervous system (CNS) that amplifies nociceptive input arising from damaged tissues [4]. Changes in the CNS after peripheral tissue injury have been shown in animal [43] and human studies [39, 44–47]. In healthy volunteers experimentally induced peripheral sensitization in muscle and viscera have also evoked both peripheral and central sensitization [26–28, 30, 48, 49]. Thus, shorter chemical perfusion of the oesophagus can induce generalized hyperalgesia with increased sensitivity in remote organs and other tissues and act as a translational bridge to the clinical situation.
When studying analgesic effects on experimental pain it is essential to choose the right dose, dosing regime and time point for testing the analgesia as the kinetic profile could affect the results [20]. A previous study demonstrated similar pharmacokinetic-pharmacodynamic relationships for somatic analgesia for morphine and oxycodone [50]. tmax is also comparable for the two opioids as for both morphine and oxycodone it was previously found to be 1 h [51, 52]. Moreover, after 30 min both morphine and oxycodone should be present at the CNS effect sites [50]. Therefore, it is not likely that the pharmacokinetic differences can fully explain the different analgesic effects between the opioids. A total score over 90 min was found to be the most appropriate method for the post hoc analysis as in pain treatment the analgesic effect is also evaluated over time and not at a specific time point.
The analgesic potency ratio of oral morphine : oxycodone has been widely discussed [34, 53]. However, it has been estimated and found to range from 1:1 to 2.2:1 [54–59]. The consensus in the pain management community is that oral oxycodone is 1.5 to 2 times as potent as oral morphine regarding analgesia [60, 61]. In the present study a high dose of morphine was chosen to secure that differences in bioavailability did not favour oxycodone analgesic efficacy. Furthermore, in a previous study the doses have showed comparable analgesic potencies in human experimental skin and muscle pain [21]. However, the analgesic potency ratio of oral morphine : oxycodone can be different from the non-analgesic pharmacodynamic ratio. Some clinical studies have suggested that oxycodone produces less severe side effects than morphine whereas a study in healthy volunteers suggested that oral morphine and oral oxycodone have a different relative potency (closer to 3:1) regarding non-analgesic pharmacodynamics, with oxycodone producing more side effects [61]. No significant difference in adverse event profiles was found in the present study. However there was a trend towards more reports of side effects after oxycodone than after morphine.
Opioid side effects could complicate blinding of the placebo administration. An inert placebo substance was included as the aim was to demonstrate differential effect of opioids and not to reveal analgesic efficacy. However, the inert placebo was to some degree masked by nausea and sweating caused by the chemical perfusion of oesophagus.
Numerous endpoints could limit the impact on interpretation of the findings by the multiple testing problem. The two-way anova was chosen for statistical analysis as it decreases the probability of type 1 errors in multiple testing. However, the aim was not to investigate whether all trials were positive. The exploratory endpoints are thought to provide potentially worthwhile information about the treatment that could serve to generate hypotheses for future studies. For this reason, endpoints that are prespecified in the design of a clinical trial as exploratory do not require any correction for multiplicity [62]. The unpleasantness of pain is associated with the limbic structures in the brain, an area where opioids traditionally are known to modulate the pain response. As deep pain is considered more unpleasant than skin type of pain, opioid analgesia is more robust in deep pain [20]. Therefore, the results from the deeper structures were considered most important. However, results from skin stimulation supported results from deeper structures and comparable results were demonstrated in all tissues which strengthen the conclusion.
Different effects of morphine vs. oxycodone
Skin
The skin heat stimulation had a narrow dynamic range of a few °C and might not be clinically relevant. However, it supports the other results and indicates different anti-hyperalgesic potencies of morphine and oxycodone as an induced generalized hyperalgesia (demonstrated in the placebo arm) was blocked by oxycodone, but not significantly by morphine. As the P value for the effect of morphine approached significance it could be hypothesized that an increased sample size would be necessary to demonstrate an effect of morphine in this experimental pain model of hyperalgesia. Previously, Staahl et al. demonstrated equal analgesic potencies of the same doses of morphine and oxycodone in the skin heat pain model [21]. These conflicting results could be due to the effect of the induced generalized hyperalgesia, as this was believed to affect both the pain system and the opioid system.
Muscle
An overall increase in tolerated pressure was found over time, most likely due to habituation to the tonic stimulation. Thus, no sensitization was found for this modality after the induction of generalized hyperalgesia. This could be due to the fact that nerves from the gastrocnemius muscle terminate in the lumbar and sacral part of the spinal cord, whereas nerves from the oesophagus and the arm are believed to terminate predominantly in the cervical and thoracic part of the spinal cord. Therefore, spinal sensitization caused by acid and capsaicin perfusion of the oesophagus will not necessarily affect incoming pain signals from the leg. However, the analgesic effect of morphine and oxycodone was demonstrated and oxycodone had greater analgesic effect than morphine (and placebo) in attenuating PTT. Opioids mainly attenuate pain intensities above the PDT [20]. This could explain why a differential effect was only found regarding PTT. Staahl et al. also demonstrated differential analgesic effects on experimental muscle stimulation in which oxycodone had a greater effect in patients with chronic pancreatitis [23]. Conversely, they could not demonstrate differential effects of morphine and oxycodone on pressure evoked muscle pain in healthy volunteers, when no sensitization was induced [21]. A significant effect of morphine was only demonstrated on this deep muscle pressure, which could be due to a higher sensitivity and reproducibility of this stimulation method.
Oesophagus
An analgesic effect of oxycodone using heat stimulation of the oesophagus was previously shown in healthy volunteers [21]. In the current study a new method ensuring linear rise in temperature was used, but no analgesic effect was demonstrated. This could be related to the faster change in temperature, which could cause uncertainty in pain assessments as it takes some time to rate the intensity. As individual differences in reaction time could not be excluded this could affect the accuracy of rating. Therefore, in future studies a slower temperature increase should be recommended. However, the heat evoked referred pain areas were smaller after oxycodone compared with placebo and morphine. This likely reflects the effects of the drug on spinal or supraspinal changes in the pain system after induction of hyperalgesia. The low baseline value for morphine on referred pain areas from heat stimulation of the oesophagus could not be the reason for lack of significance of morphine effect, as all results were baseline corrected for the same reason before analysis. However, as for the effect of morphine on skin heat pain, the P value for the effect of morphine on referred pain area from oesophageal heat stimulation approached significance, indicating an effect of morphine. However, in parallel with other results the analgesic effect of oxycodone was greater. In a previous study the opioids attenuated the response to oesophageal mechanical stimulation in healthy volunteers [21]. However, no effect was found in the present study. Acid perfusion causes oesophageal motility changes seen as increasing number and sizes of contractions [63]. This could lead to squeezing of the balloon as well as traction force exerted in the pharynx and therefore the data must be carefully interpreted.
Electrical stimulation is often the most reliable visceral stimulus as it is not affected by motility and has a high dynamic range [64]. Once more, the effect of morphine on electrically induced pain approached significance, indicating an effect which could be demonstrated with an increased sample size. Nevertheless, oxycodone had a greater effect than morphine in attenuating electrically induced pain by blocking the induced hyperalgesia. As the electrical stimulation bypasses the peripheral nerves the difference could be related to spinal and supraspinal changes in the pain system [5–7]. The referred pain area to electrical stimulation of the oesophagus increased after placebo indicating hyperalgesia. This hyperalgesia was also blocked by the opioids, but non-significantly. The lack of significance could be due to the high variance in how subjects reported the referred pain area after electrical stimulation.
The effect of morphine has previously been proved detectable in 24 healthy volunteers [21]. As the results only demonstrated a significant effect of morphine on muscle pressure, hyperalgesia might nevertheless have affected the variation and a larger sample size would have been more appropriate to demonstrate the effect of morphine. However, differential analgesic potencies of morphine and oxycodone were demonstrated in all tissues confirming that the sample size was sufficient for the main outcome of the study.
It has been demonstrated in animal studies that oxycodone may interact, at least in part, with a different population of opioid receptors or modulate µ-opioid receptor signalling in hyperalgesia in a subtly different way from other opioids [14, 65]. Moreover, the generalized hyperalgesia could lead to changes in the opioid system [66, 67]. For example a more pronounced up-regulation of κ-receptors in inflammation has been speculated [8]. Furthermore, animal studies have shown that binding to κ-receptor sites in the spinal cord are increased to a greater degree than binding to µ-receptors during peripheral inflammation [12]. It has been proposed from rodent experiments that oxycodone is a partial κ-agonist [14–18]. Therefore, it could be hypothesized that the differential analgesic potencies are caused by different pharmacological profiles of the opioids. Whether the present results are due to pharmacology and caused by different affinities for κ-opioid receptors cannot be concluded from this or any other human study as it would require a selective κ-antagonist suitable for human administration.
Translational pain research
Animal models may provide pharmacodynamic information during drug development, but have limitations mimicking human pain conditions. Hence, they are based on motor reflexes or behavioural responses, whereas human pain is a net result of complex sensory, affective and cognitive processing [68]. Research with patients offers a means to explore the actual pain states of interest. On the other hand, they suffer from difficulties in assessment of the pain, due to many confounding factors such as general malaise, fever, nausea, psychological status, etc [68]. Therefore, experimental pain models in healthy volunteers, controlling the pain stimulus and assessment, can act as a translational bridge from studies in animals to humans [24]. Traditional models are short lasting and have many limitations compared with the complex clinical pain conditions. However, experimental pain models can include hyperalgesia and be more consistent with phenomena observed in patients. They may reasonably explain many of the abnormal pain responses typical of chronic pain. This was demonstrated in the current translational study as the differential analgesic potency of morphine and oxycodone was comparable with the data previously found in our group on experimental pain stimulation in patients with chronic visceral pain [23]. Therefore, human experimental pain models of hyperalgesia may help to predict analgesic potency in a sensitized pain system. Models evoking controlled hyperalgesia have the advantages of experimental pain and reflect the clinical situation to a higher degree than acute models. Therefore, they may speed up development programmes within the industry and provide new fundamental knowledge on analgesics and pain mechanisms [24].
It is difficult to predict whether the demonstrated differential analgesic potencies have a significant impact on the clinical efficacy of the opioids. However, subtle differences in analgesic potencies as found in the present study are hard to elucidate in the clinical setting, but the results support the theory and practical experience about oxycodone being different from morphine regarding analgesia [1, 69, 70].
Acknowledgments
The study was supported by Norpharma (Mundipharma), the Research Initiative, Aarhus University Hospital, ‘Det Obelske Familie Fond’ and the Spar Nord Foundation.
Competing interests
C. S. has been employed by Grünenthal after completion of the study and manuscript. There are no other competing interests to declare.
REFERENCES
- 1.Riley J, Ross JR, Rutter D, Wells AU, Goller K, du Bois R, Welsh K. No pain relief from morphine? Individual variation in sensitivity to morphine and the need to switch to an alternative opioid in cancer patients. Support Care Cancer. 2006;14:56–64. doi: 10.1007/s00520-005-0843-2. [DOI] [PubMed] [Google Scholar]
- 2.Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10:287–333. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 3.De Schepper HU, Cremonini F, Park MI, Camilleri M. Opioids and the gut: pharmacology and current clinical experience. Neurogastroenterol Motil. 2004;16:383–94. doi: 10.1111/j.1365-2982.2004.00513.x. [DOI] [PubMed] [Google Scholar]
- 4.Curatolo M, Arendt-Nielsen L, Petersen-Felix S. Central hypersensitivity in chronic pain: mechanisms and clinical implications. Phys Med Rehabil Clin N Am. 2006;17:287–302. doi: 10.1016/j.pmr.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Cervero F. Visceral pain-central sensitisation. Gut. 2000;47(Suppl 4):iv56–7. doi: 10.1136/gut.47.suppl_4.iv56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anand P, Aziz Q, Willert R, van Oudenhove L. Peripheral and central mechanisms of visceral sensitization in man. Neurogastroenterol Motil. 2007;19:29–46. doi: 10.1111/j.1365-2982.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 7.Dimcevski G, Sami SA, Funch-Jensen P, Le Pera D, Valeriani M, Arendt-Nielsen L, Drewes AM. Pain in chronic pancreatitis: the role of reorganization in the central nervous system. Gastroenterology. 2007;132:1546–56. doi: 10.1053/j.gastro.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 8.Sengupta JN, Snider A, Su X, Gebhart GF. Effects of kappa opioids in the inflamed rat colon. Pain. 1999;79:175–85. doi: 10.1016/s0304-3959(98)00175-4. [DOI] [PubMed] [Google Scholar]
- 9.Pol O, Alameda F, Puig MM. Inflammation enhances mu-opioid receptor transcription and expression in mice intestine. Mol Pharmacol. 2001;60:894–9. doi: 10.1124/mol.60.5.894. [DOI] [PubMed] [Google Scholar]
- 10.Pol O, Palacio JR, Puig MM. The expression of delta- and kappa-opioid receptor is enhanced during intestinal inflammation in mice. J Pharmacol Exp Ther. 2003;306:455–62. doi: 10.1124/jpet.103.049346. [DOI] [PubMed] [Google Scholar]
- 11.Stein C. Peripheral mechanisms of opioid analgesia. Anesth Analg. 1993;76:182–91. doi: 10.1213/00000539-199301000-00031. [DOI] [PubMed] [Google Scholar]
- 12.Iadarola MJ, Douglass J, Civelli O, Naranjo JR. Differential activation of spinal cord dynorphin and enkephalin neurons during hyperalgesia: evidence using cDNA hybridization. Brain Res. 1988;455:205–12. doi: 10.1016/0006-8993(88)90078-9. [DOI] [PubMed] [Google Scholar]
- 13.Riley J, Eisenberg E, Muller-Schwefe G, Drewes AM, Arendt-Nielsen L. Oxycodone: a review of its use in the management of pain. Curr Med Res Opin. 2008;24:175–92. doi: 10.1185/030079908x253708. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT. Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain. 2007;132:289–300. doi: 10.1016/j.pain.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 15.Ross FB, Smith MT. The intrinsic antinociceptive effects of oxycodone appear to be kappa-opioid receptor mediated. Pain. 1997;73:151–7. doi: 10.1016/S0304-3959(97)00093-6. [DOI] [PubMed] [Google Scholar]
- 16.Nozaki C, Saitoh A, Kamei J. Characterization of the antinociceptive effects of oxycodone in diabetic mice. Eur J Pharmacol. 2006;535:145–51. doi: 10.1016/j.ejphar.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Nozaki C, Saitoh A, Tamura N, Kamei J. Antinociceptive effect of oxycodone in diabetic mice. Eur J Pharmacol. 2005;524:75–9. doi: 10.1016/j.ejphar.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 18.Holtman JR, Wala EP. Characterization of the antinociceptive effect of oxycodone in male and female rats. Pharmacol Biochem Behav. 2006;83:100–8. doi: 10.1016/j.pbb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Staahl C, Reddy H, Andersen SD, Arendt-Nielsen L, Drewes AM. Multi-modal and tissue-differentiated experimental pain assessment: reproducibility of a new concept for assessment of analgesics. Basic Clin Pharmacol Toxicol. 2006;98:201–11. doi: 10.1111/j.1742-7843.2006.pto_211.x. [DOI] [PubMed] [Google Scholar]
- 20.Staahl C, Olesen AE, Andresen T, Arendt-Nielsen L, Drewes AM. Assessing analgesic actions of opioids by experimental pain models in healthy volunteers – an updated review. Br J Clin Pharmacol. 2009;68:149–68. doi: 10.1111/j.1365-2125.2009.03456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staahl C, Christrup LL, Andersen SD, Arendt-Nielsen L, Drewes AM. A comparative study of oxycodone and morphine in a multi-modal, tissue-differentiated experimental pain model. Pain. 2006;123:28–36. doi: 10.1016/j.pain.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Drewes AM, Krarup AL, Detlefsen S, Malmstrom ML, Dimcevski G, Funch-Jensen P. Pain in chronic pancreatitis: the role of neuropathic pain mechanisms. Gut. 2008;57:1616–27. doi: 10.1136/gut.2007.146621. [DOI] [PubMed] [Google Scholar]
- 23.Staahl C, Dimcevski G, Andersen SD, Thorsgaard N, Christrup LL, Arendt-Nielsen L, Drewes AM. Differential effect of opioids in patients with chronic pancreatitis: an experimental pain study. Scand J Gastroenterol. 2007;42:383–90. doi: 10.1080/00365520601014414. [DOI] [PubMed] [Google Scholar]
- 24.Arendt-Nielsen L, Curatolo M, Drewes A. Human experimental pain models in drug development: translational pain research. Curr Opin Investig Drugs. 2007;8:41–53. [PubMed] [Google Scholar]
- 25.Hammer J, Vogelsang H. Characterization of sensations induced by capsaicin in the upper gastrointestinal tract. Neurogastroenterol Motil. 2007;19:279–87. doi: 10.1111/j.1365-2982.2007.00900.x. [DOI] [PubMed] [Google Scholar]
- 26.Drewes AM, Schipper KP, Dimcevski G, Petersen P, Andersen OK, Gregersen H, Arendt-Nielsen L. Multi-modal induction and assessment of allodynia and hyperalgesia in the human oesophagus. Eur J Pain. 2003;7:539–49. doi: 10.1016/s1090-3801(03)00053-3. [DOI] [PubMed] [Google Scholar]
- 27.Willert RP, Delaney C, Kelly K, Sharma A, Aziz Q, Hobson AR. Exploring the neurophysiological basis of chest wall allodynia induced by experimental oesophageal acidification – evidence of central sensitization. Neurogastroenterol Motil. 2007;19:270–8. doi: 10.1111/j.1365-2982.2006.00890.x. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S, Woolf CJ, Hobson AR, Thompson DG, Aziz Q. Perceptual wind-up in the human oesophagus is enhanced by central sensitisation. Gut. 2006;55:920–5. doi: 10.1136/gut.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frokjaer JB, Andersen SD, Gale J, Arendt-Nielsen L, Gregersen H, Drewes AM. An experimental study of viscero-visceral hyperalgesia using an ultrasound-based multimodal sensory testing approach. Pain. 2005;119:191–200. doi: 10.1016/j.pain.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 30.Sami SA, Rossel P, Dimcevski G, Nielsen KD, Funch-Jensen P, Valeriani M, Arendt-Nielsen L, Drewes AM. Cortical changes to experimental sensitization of the human esophagus. Neuroscience. 2006;140:269–79. doi: 10.1016/j.neuroscience.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 31.Drewes AM, Schipper KP, Dimcevski G, Petersen P, Andersen OK, Gregersen H, Arendt-Nielsen L. Multimodal assessment of pain in the esophagus: a new experimental model. Am J Physiol Gastrointest Liver Physiol. 2002;283:G95–103. doi: 10.1152/ajpgi.00496.2001. [DOI] [PubMed] [Google Scholar]
- 32.Kalso E. Oxycodone. J Pain Symptom Manage. 2005;29:S47–56. doi: 10.1016/j.jpainsymman.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Christrup LL, Sjogren P, Jensen NH, Banning AM, Elbaek K, Ersboll AK. Steady-state kinetics and dynamics of morphine in cancer patients: is sedation related to the absorption rate of morphine? J Pain Symptom Manage. 1999;18:164–73. doi: 10.1016/s0885-3924(99)00068-8. [DOI] [PubMed] [Google Scholar]
- 34.Kalso E. How different is oxycodone from morphine? Pain. 2007;132:227–8. doi: 10.1016/j.pain.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 35.Jespersen A, Dreyer L, Kendall S, Graven-Nielsen T, Arendt-Nielsen L, Bliddal H, Danneskiold-Samsoe B. Computerized cuff pressure algometry: a new method to assess deep-tissue hypersensitivity in fibromyalgia. Pain. 2007;131:57–62. doi: 10.1016/j.pain.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Polianskis R, Graven-Nielsen T, Arendt-Nielsen L. Computer-controlled pneumatic pressure algometry – a new technique for quantitative sensory testing. Eur J Pain. 2001;5:267–77. doi: 10.1053/eujp.2001.0245. [DOI] [PubMed] [Google Scholar]
- 37.Drewes AM, Gregersen H, Arendt-Nielsen L. Experimental pain in gastroenterology: a reappraisal of human studies. Scand J Gastroenterol. 2003;38:1115–30. doi: 10.1080/00365520310004399. [DOI] [PubMed] [Google Scholar]
- 38.Frokjaer JB, Andersen SD, Ejskaer N, Funch-Jensen P, Arendt-Nielsen L, Gregersen H, Drewes AM. Gut sensations in diabetic autonomic neuropathy. Pain. 2007;131:320–9. doi: 10.1016/j.pain.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Drewes AM, Pedersen J, Reddy H, Rasmussen K, Funch-Jensen P, Arendt-Nielsen L, Gregersen H. Central sensitization in patients with non-cardiac chest pain: a clinical experimental study. Scand J Gastroenterol. 2006;41:640–9. doi: 10.1080/00365520500442559. [DOI] [PubMed] [Google Scholar]
- 40.Drewes AM, Reddy H, Pedersen J, Funch-Jensen P, Gregersen H, Arendt-Nielsen L. Multimodal pain stimulations in patients with grade B oesophagitis. Gut. 2006;55:926–32. doi: 10.1136/gut.2005.067769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olesen AE, Staahl C, Brock C, Arendt-Nielsen L, Drewes AM. Evoked human oesophageal hyperalgesia: a potential tool for analgesic evaluation? Basic Clin Pharmacol Toxicol. 2009;105:126–36. doi: 10.1111/j.1742-7843.2009.00422.x. [DOI] [PubMed] [Google Scholar]
- 42.Quante M, Scharein E, Zimmermann R, Langer-Brauburger B, Bromm B. Dissociation of morphine analgesia and sedation evaluated by EEG measures in healthy volunteers. Arzneimittelforschung. 2004;54:143–51. doi: 10.1055/s-0031-1296951. [DOI] [PubMed] [Google Scholar]
- 43.Roza C, Laird JM, Cervero F. Spinal mechanisms underlying persistent pain and referred hyperalgesia in rats with an experimental ureteric stone. J Neurophysiol. 1998;79:1603–12. doi: 10.1152/jn.1998.79.4.1603. [DOI] [PubMed] [Google Scholar]
- 44.Wilder-Smith CH, Robert-Yap J. Abnormal endogenous pain modulation and somatic and visceral hypersensitivity in female patients with irritable bowel syndrome. World J Gastroenterol. 2007;13:3699–704. doi: 10.3748/wjg.v13.i27.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azpiroz F, Bouin M, Camilleri M, Mayer EA, Poitras P, Serra J, Spiller RC. Mechanisms of hypersensitivity in IBS and functional disorders. Neurogastroenterol Motil. 2007;19:62–88. doi: 10.1111/j.1365-2982.2006.00875.x. [DOI] [PubMed] [Google Scholar]
- 46.Verne GN, Robinson ME, Price DD. Hypersensitivity to visceral and cutaneous pain in the irritable bowel syndrome. Pain. 2001;93:7–14. doi: 10.1016/S0304-3959(01)00285-8. [DOI] [PubMed] [Google Scholar]
- 47.Chang L, Mayer EA, Johnson T, FitzGerald LZ, Naliboff B. Differences in somatic perception in female patients with irritable bowel syndrome with and without fibromyalgia. Pain. 2000;84:297–307. doi: 10.1016/s0304-3959(99)00215-8. [DOI] [PubMed] [Google Scholar]
- 48.Arendt-Nielsen L, Svensson P. Referred muscle pain: basic and clinical findings. Clin J Pain. 2001;17:11–9. doi: 10.1097/00002508-200103000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Brock C, Andresen T, Frokjaer JB, Gale J, Olesen AE, Arendt-Nielsen L, Drewes AM. Central pain mechanisms following combined acid and capsaicin perfusion of the human oesophagus. Eur J Pain. 2009 doi: 10.1016/j.ejpain.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 50.Staahl C, Upton R, Foster DJ, Christrup LL, Kristensen K, Hansen SH, Arendt-Nielsen L, Drewes AM. Pharmacokinetic-pharmacodynamic modeling of morphine and oxycodone concentrations and analgesic effect in a multimodal experimental pain model. J Clin Pharmacol. 2008;48:619–31. doi: 10.1177/0091270008314465. [DOI] [PubMed] [Google Scholar]
- 51.Collins SL, Faura CC, Moore RA, McQuay HJ. Peak plasma concentrations after oral morphine: a systematic review. J Pain Symptom Manage. 1998;16:388–402. doi: 10.1016/s0885-3924(98)00094-3. [DOI] [PubMed] [Google Scholar]
- 52.Poyhia R, Seppala T, Olkkola KT, Kalso E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br J Clin Pharmacol. 1992;33:617–21. doi: 10.1111/j.1365-2125.1992.tb04090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bostrom E, Hammarlund-Udenaes M, Simonsson US. Blood-brain barrier transport helps to explain discrepancies in in vivo potency between oxycodone and morphine. Anesthesiology. 2008;108:495–505. doi: 10.1097/ALN.0b013e318164cf9e. [DOI] [PubMed] [Google Scholar]
- 54.Glare PA, Walsh TD. Dose-ranging study of oxycodone for chronic pain in advanced cancer. J Clin Oncol. 1993;11:973–8. doi: 10.1200/JCO.1993.11.5.973. [DOI] [PubMed] [Google Scholar]
- 55.Kalso E, Vainio A. Morphine and oxycodone hydrochloride in the management of cancer pain. Clin Pharmacol Ther. 1990;47:639–46. doi: 10.1038/clpt.1990.85. [DOI] [PubMed] [Google Scholar]
- 56.Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, Babul N, Ford I. Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. J Clin Oncol. 1998;16:3222–9. doi: 10.1200/JCO.1998.16.10.3222. [DOI] [PubMed] [Google Scholar]
- 57.Heiskanen TE, Ruismaki PM, Seppala TA, Kalso EA. Morphine or oxycodone in cancer pain? Acta Oncol. 2000;39:941–7. doi: 10.1080/02841860050215927. [DOI] [PubMed] [Google Scholar]
- 58.Curtis GB, Johnson GH, Clark P, Taylor R, Brown J, O'Callaghan R, Shi M, Lacouture PG. Relative potency of controlled-release oxycodone and controlled-release morphine in a postoperative pain model. Eur J Clin Pharmacol. 1999;55:425–9. doi: 10.1007/s002280050651. [DOI] [PubMed] [Google Scholar]
- 59.Foley KM. The treatment of cancer pain. N Engl J Med. 1985;313:84–95. doi: 10.1056/NEJM198507113130205. [DOI] [PubMed] [Google Scholar]
- 60.Hanks GW, Conno F, Cherny N, Hanna M, Kalso E, McQuay HJ, Mercadante S, Meynadier J, Poulain P, Ripamonti C, Radbruch L, Casas JR, Sawe J, Twycross RG, Ventafridda V. Morphine and alternative opioids in cancer pain: the EAPC recommendations. Br J Cancer. 2001;84:587–93. doi: 10.1054/bjoc.2001.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zacny JP, Lichtor SA. Within-subject comparison of the psychopharmacological profiles of oral oxycodone and oral morphine in non-drug-abusing volunteers. Psychopharmacology (Berl) 2008;196:105–16. doi: 10.1007/s00213-007-0937-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turk DC, Dworkin RH, McDermott MP, Bellamy N, Burke LB, Chandler JM, Cleeland CS, Cowan P, Dimitrova R, Farrar JT, Hertz S, Heyse JF, Iyengar S, Jadad AR, Jay GW, Jermano JA, Katz NP, Manning DC, Martin S, Max MB, McGrath P, McQuay HJ, Quessy S, Rappaport BA, Revicki DA, Rothman M, Stauffer JW, Svensson O, White RE, Witter J. Analyzing multiple endpoints in clinical trials of pain treatments: IMMPACT recommendations. Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials. Pain. 2008;139:485–93. doi: 10.1016/j.pain.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 63.Drewes AM, Reddy H, Staahl C, Funch-Jensen P, Arendt-Nielsen L, Gregersen H, Lundbye-Christensen S. Statistical modeling of the response characteristics of mechanosensitive stimuli in the human esophagus. J Pain. 2005;6:455–62. doi: 10.1016/j.jpain.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 64.Staahl C, Drewes AM. Experimental human pain models: a review of standardised methods for preclinical testing of analgesics. Basic Clin Pharmacol Toxicol. 2004;95:97–111. doi: 10.1111/j.1742-7843.2004.950301.x. [DOI] [PubMed] [Google Scholar]
- 65.Virk MS, Williams JT. Agonist-specific regulation of mu-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol. 2008;73:1301–8. doi: 10.1124/mol.107.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stanfa L, Dickenson A. Spinal opioid systems in inflammation. Inflamm Res. 1995;44:231–41. doi: 10.1007/BF01782974. [DOI] [PubMed] [Google Scholar]
- 67.Riviere PJ. Peripheral kappa-opioid agonists for visceral pain. Br J Pharmacol. 2004;141:1331–4. doi: 10.1038/sj.bjp.0705763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langley CK, Aziz Q, Bountra C, Gordon N, Hawkins P, Jones A, Langley G, Nurmikko T, Tracey I. Volunteer studies in pain research – opportunities and challenges to replace animal experiments: the report and recommendations of a Focus on Alternatives workshop. Neuroimage. 2008;42:467–73. doi: 10.1016/j.neuroimage.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 69.Lenz H, Sandvik L, Qvigstad E, Bjerkelund CE, Raeder J. A comparison of intravenous oxycodone and intravenous morphine in patient-controlled postoperative analgesia after laparoscopic hysterectomy. Anesth Analg. 2009;109:1279–83. doi: 10.1213/ane.0b013e3181b0f0bb. [DOI] [PubMed] [Google Scholar]
- 70.Narabayashi M, Saijo Y, Takenoshita S, Chida M, Shimoyama N, Miura T, Tani K, Nishimura K, Onozawa Y, Hosokawa T, Kamoto T, Tsushima T. Opioid rotation from oral morphine to oral oxycodone in cancer patients with intolerable adverse effects: an open-label trial. Jpn J Clin Oncol. 2008;38:296–304. doi: 10.1093/jjco/hyn010. [DOI] [PubMed] [Google Scholar]