

Abstract
Adenomatous polyposis coli (Apc) is critical for Wnt signaling and cell migration. The current study examined Apc expression during lung development, injury and repair. Apc was first detectable in smooth muscle layers in early lung morphogenesis, and was highly expressed in ciliated and neuroendocrine cells in the advanced stages. No Apc immunoreactivity was detected in Clara or basal cells, which function as stem/progenitor cell in adult lung. In ciliated cells, Apc is associated mainly with apical cytoplasmic domain. In response to naphthalene induced injury, Apcpositive cells underwent squamous metaplasia, accompanied by changes in Apc subcellular distribution. In conclusion, both spatial and temporal expression of Apc is dynamically regulated during lung development and injury repair. Differential expression of Apc in progenitor vs. non-progenitor cells suggests a functional role in cell type specification. Subcellular localization changes of Apc in response to naphthalene injury suggest a role in cell shape and cell migration.
Keywords: adenomatous polyposis coli, lung development, injury, repair, airway epithelium
INTRODUCTION
Cell renewal, which involves tissue embedded progenitor/stem cells, is critical for maintenance of tissue homeostasis and repair subsequent to injury. The canonical Wnt pathway mediated by β-catenin stabilization plays a key role in establishment and function of progenitor/stem cells. Activity of β-catenin is in turn controlled by Adenomatous Polyposis Coli (Apc), a 310 kDa protein. In the absence of a Wnt ligand, Apc inhibits Wnt signaling by promoting the phosphorylation of β-catenin in a cytoplasmic complex composed of Apc, Axin and glycogen synthase kinase-3β (Gsk3β). In this complex Apc is necessary for phosphorylation of β-catenin by Gsk3β and its subsequent degradation (Polakis, 1997). Deletion of Apc leads to accumulation of β-catenin and causes embryonic lethality before day 8 of gestation (Oshima et al., 1995). In the gut, high Apc levels correlate with loss of epithelial cell proliferative capacity. Low levels or absence of Apc are associated with the ability of cells to serve as functional tissue-embedded stem cells (Senda et al., 2007). The role of Apc in lung development and disease remains unknown.
Apc is a multifunctional protein that is found associated with several subcellular regions including cytoplasm, nucleus, plasma membrane, and microtubules (Nathke, 2004; Hanson and Miller, 2005; Langford et al., 2006a; Langford et al., 2006b; Aoki and Taketo, 2007). In addition to accumulation of β-catenin, loss of Apc leads to changes in cell migration, cell orientation, polarity and division (Etienne-Manneville and Hall, 2003; Dikovskaya et al., 2007; Kroboth et al., 2007). Cell shape determination may be mediated via association of membrane localized Apc with actin cytoskeleton (Rosin-Arbesfeld et al., 2001). In cell migration, Apc becomes localized to the growing plus-ends of microtubules in epithelial cell extensions (Nathke et al., 1996; Mimori-Kiyosue et al., 2000). The mechanism that controls the function and subcellular distribution of Apc appear to be dependent on Gsk3β. Phosphorylation of Apc by Gsk3β which promotes binding to β-catenin in the cytoplasmic destruction complex, also reduces Apc microtubule binding affinity and hence migration (Munemitsu et al., 1994). In contrast, phosphorylation of Gsk3β by a Cdc42-dependent mechanism promotes Apc microtubule interactions and thus leads to cell shape changes that accompany cellular migration (Zumbrunn et al., 2001; Etienne-Manneville and Hall, 2003).
Murine models of airway injury provide a useful in vivo tool to examine cell self-renewal, cell shape changes and migration. The airway epithelium is a functional landscape of complex and highly specialized cell types, the most abundant of which includes ciliated cells, Clara cells, basal cells, neuroendocrine cells, and goblet cells. The cytochrome P-450 activated Clara cell toxicant naphthalene (NAPH) selectively ablates Clara cells and triggers a repair mechanism. Within 24 hours, the NAPH-resistant epithelial cells, consisting primarily of columnar ciliated and some non-ciliated cells, undergo squamous metaplasia to cover the basement membrane of the denuded airway, a protective mechanism. During this period, dynamic changes in cell shape and cell migration are essential (Kida et al., 2008). Two to three days after NAPH-induced injury, cell proliferation increases to commence the repair of the injured epithelium. By seven to fourteen days, the normal cellular composition of the airway is re-established. The Clara cells repopulate the airway and the squamous or cuboidal epithelial cells again undergo changes in cell shape to re-establish the columnar phenotype of the airway epithelium. Cell lineage studies in this model of airway injury have revealed a subpopulation of Clara cells that is resistant to NAPH, which may act as tissue-embedded stem cells (Hong et al., 2001). These cells undergo self-renewal and have the capacity to generate progenitors of other lineages such as ciliated cells (Rawlins et al., 2009). In contrast, ciliated cells are post-mitotic and are thought to be incapable of undergoing mitosis (Rawlins et al., 2007). Neuroendocrine cells are also thought to proliferate and self-renew, but lack the capacity to generate other lineages. There is however evidence that basal cells may also function as stem cells (Hong et al., 2004; Rawlins and Hogan, 2006). In sum, NAPH induced injury response involves not only stem-cell mediated re-population of Clara cells (Giangreco et al., 2009), but also dynamic changes in cell shape and cell migration of the ciliated cells, providing a useful model for studying the underlying mechanisms.
In the current study, we examined the expression of Apc in embryonic and adult lungs, and found that the levels of Apc are cell type dependent and change dynamically as the lung develops. The pattern of Apc expression in the lung supports its function in regulating canonical Wnt signaling activity and cell proliferation potential. Furthermore, the subcellular distribution of Apc changes dynamically in response to NAPH-induced injury, correlating with cell shape changes and cell migration. In support of this, these changes are accompanied by corresponding changes in levels of phospho-Gsk3β. The cell-type specific distribution of Apc and its spatial and temporal relationship with β-catenin and Gsk3β suggests an important role for Apc in maintenance of tissue homeostasis during lung development and injury repair.
RESULTS
Apc expression during lung morphogenesis
Real time PCR analysis revealed that Apc is expressed throughout lung development (Figure 1, panel A). Apc protein was analyzed by western blot, using an anti-Apc polyclonal antibody (Materials & Methods). A protein extract from lung carcinoma H441 cells, transfected with a full-length Apc cDNA, was included as a positive control. As shown in Figure 1 (panel B), a strong band of 312 kDa, which is consistent with the predicted size of Apc, was present in the Apc-transfected H441 cells (lane 2). In protein extracts from un-transfected H441 cells, E18.5 and adult lungs, a band with similar mobility was also detected.
Figure 1. Apc expression in the mouse lung.
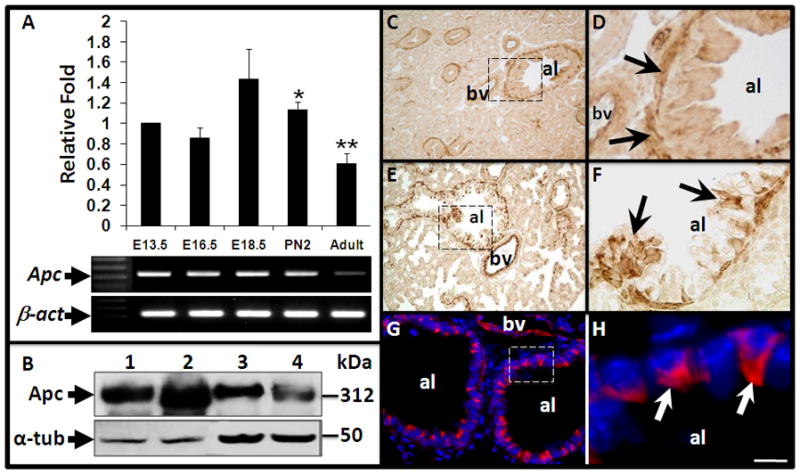
(A) Relative quantification of Apc mRNA by real-time PCR at various stages of mouse lung development. Data of each time point represent average from at least three individual lungs. The error bar represents standard deviation. Samples from E13.5 lungs served as “Calibrator”. *: p<0.05. **: p<0.01. (B) Western blot analysis of Apc protein in H441 cells and lung tissues. 1, H441 cells; 2, H441 cells transfected with Apc expression construct; 3, E18.5 lung; 4, adult lung. Note a band with predicted size of Apc was robustly increased in the transfected H441 cells (lane 2) as compared to H441 only (lane 1). Levels of α-tubulin (α-tub) in each sample were determined as loading control.
(C–H) Spatial distribution of Apc during lung development. Immunohistochemistry was performed to localize Apc protein in embryonic and adult lungs. (C) In E14.5 lung, extensive Apc staining is present in mesenchymal cells surrounding major airways. (D) High magnification of boxed area in panel C. (E) In E18.5 lung, Apc staining is present in epithelial cells and mesenchymal cells surrounding major airways and blood vessels. (F) High magnification of boxed area in panel E. Arrows indicate group of Apc positive epithelial cells along major airways. (G) In adult lung, extensive Apc staining is detectable in airway epithelial cells. (H) High magnification of boxed area in panel G. Nuclei were stained with DAPI. Arrows indicate Apc staining. al=airway lumen, bv=blood vessels. Scale bar: 60μm for Panels C and E; 20 μm for Panels D, F and G; 5μm for Panel H.
To determine the spatial distribution of Apc in the lung, we performed immunohistochemistry and immunofluorescence. In E14.5 embryonic lungs, high levels of Apc protein were detected in sub-epithelial mesenchymal cells surrounding major airways (Figure 1, Panels C and D). Apc is barely detectable in the epithelial cells. In E18.5 lungs, increasing number of Apcpositive cells were identified along the proximal airway (Figure 1, Panels E and F). In the adult lung, Apc expression was more robust and restricted to a subpopulation of airway epithelial cells in which Apc was predominantly localized to the apical cytoplasm (Figure 1, Panels G and H). Expression of Apc in the distal lung was not detectable by immunohistochemistry even though Apc mRNA was detected in isolated alveolar type II (ATII) cells by RT-PCR analysis (data not shown). Additionally, there was a reproducible level of Apc staining in the cells surrounding blood vessels at all stages (Figure 1, Panels C, D, E and G).
Cell-Type Specific Expression of Apc in adult mouse airways
The expression pattern of Apc in the sub-epithelial mesenchymal compartment appeared to overlap with that of smooth muscle cells. To validate this finding, we conducted double immunofluorescent staining with antibodies against Apc and α-smooth muscle actin (α-SMA), a marker for smooth muscle cells. In E15.5 lungs, Apc and α-SMA co-localized to the mesenchymal cells surrounding the large airways and blood vessels (Figure 2, Panels A–D). In the adult lung, Apc is mainly localized to the airway epithelial cells while its level in sub-epithelial smooth muscle cells is significantly decreased (Figure 2, Panels E–H).
Figure 2. Cell-type specific expression of Apc in mouse lung.
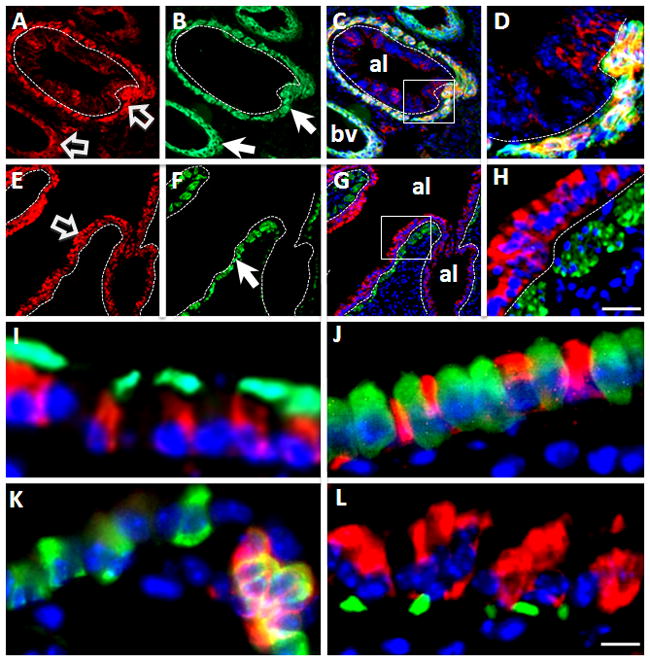
(A–H) Double immunofluorescent staining of Apc (red) and α-smooth muscle actin (α-SMA, green). (A–D) In E15.5 lung, Apc (red, panel A) and α-SMA (green, panel B) are co-localized in mesenchymal (smooth muscle) cells around large airway and blood vessels. Block arrows indicate Apc signal and arrows indicate α-SMA signal. High magnification of boxed area in merged image (panel C) is shown in panel D. (E–H) In adult lung, Apc is highly expressed in airway epithelial cells (block arrow), but weakly in smooth muscle cells as labeled by α-SMA staining (arrow, panel F). High magnification of boxed area of merged image (panel G) is shown in panel H. Nuclei were stained with DAPI. Dotted lines outline airway basement membrane. Scale bar, 30μm for Panels A–C and Panels E–G; 10μm for Panels D&H.
(I–L). Cell type specific expression of Apc in airway epithelium of adult lung. (I) Double immunofluorescent staining of Apc (red) and β-tubulin IV (green). (J) Double immunofluorescent staining of Apc (red) and CC10 (green). (K) Double immunofluorescent staining of Apc (green) and PGP9.5 (red). (L) Double immunofluorescent staining of Apc (red) and P63 (green). Nuclei were stained with DAPI. Note that Apc co-localizes with β-tubulin IV (panel I) and PGP9.5 (panel K). Scale bar, 10μm.
To define the identity of epithelial cells that express Apc, we examined adult mouse lung by double immunostaining of Apc with the following epithelial cell markers; β-tubulin IV for ciliated cells (Fanucchi et al., 1997); CC10 for Clara cells (Singh et al., 1988); PGP9.5 for neuroendocrine cells (Poulsen et al., 2008) and P63 for basal cells (Chilosi and Doglioni, 2001). Apc was co-localized with β-tubulin IV (Figure 2, Panel I) and PGP9.5 (Figure 2, Panel K). Little if any co-localization was observed between Apc and CC10 (Figure 2, Panel J), or Apc and P63 (Figure 2, Panel L). These data indicate that in adult lung epithelium Apc is highly expressed in ciliated and neuroendocrine cells.
Co-localization of Apc with β-Catenin in the lung epithelium
In Wnt signaling, Apc plays a critical role in regulating β-catenin phosphorylation and degradation. We compared the distribution of Apc and total- and phosphorylated-β-catenin in embryonic and adult lungs by immunohistochemistry. In E14.5 lungs, Apc is present predominately in the sub-epithelial mesenchymal cells whereas total β-catenin was detected in almost every cell with a higher intensity in the epithelial compartment (Figure 3, Panels AC). In the adult lung, Apc is mainly located at the apical cytoplasm of sub-populations of epithelial cells. In contrast, β-catenin is primarily detected at the lateral junctions between each epithelial cell (Figure 3, Panels D–F). Interestingly, cytoplasmic accumulation of β-catenin is present mainly in epithelial cells with high levels of Apc. Therefore, accumulation of Apc correlates with cytoplasmic accumulation of total-β-catenin in lung epithelial cells.
Figure 3. Co-localization of Apc with β-catenin in the lung epithelium.
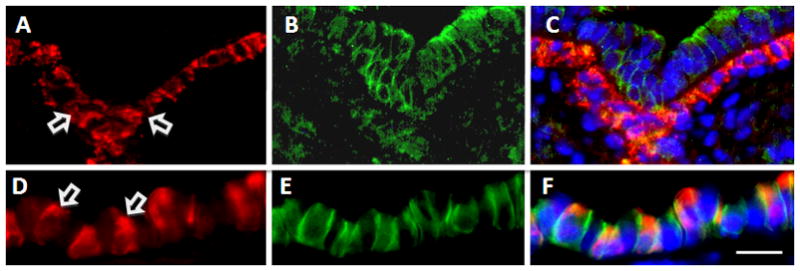
(A–F) Double immunofluorescent staining of Apc with total β-catenin. (A–C) In E14.5 lung, Apc is predominantly expressed in smooth muscle cells (A), whereas membranous β-catenin is present in every cell with a higher intensity in the epithelial compartment (B). (D–F) In adult lung, Apc is predominantly expressed in the cytoplasm of a subpopulation of airway epithelial cells (D). Cytoplasmic β-catenin was observed in the Apc positive cells (E and F). Block arrows in A and D indicate Apc signal. Scale bars, 20μm for Panels A–C; 10μm for Panels D–F.
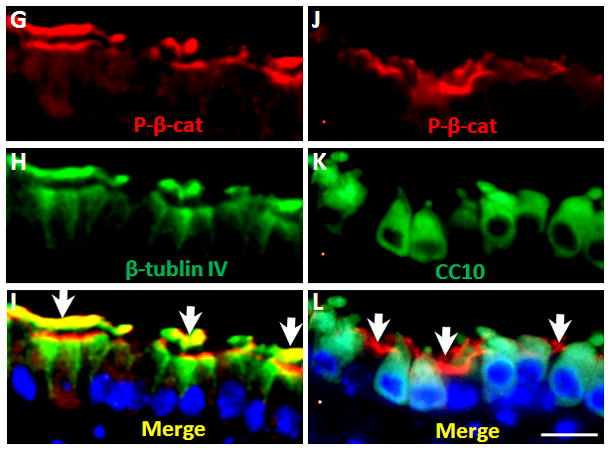
(G–L) Double immunofluorescent staining of phospho-β-catenin with β-tubulin IV (G–I) or CC10 (J–L) in adult lungs. Phospho-β-catenin (P-β-cat, red, G&I) was detected in β-tubulin IVpositive cells (H&I), but not in CC10positive cells (K&L). Arrows in G and J indicate P-β-cat signals. Scale bar, 10μm.
We next determined whether the phosphorylated-β-catenin (phospho-β-catenin) is spatially associated with Apc expression using an antibody against phospho-β-catenin. This antibody detects endogenous levels of β-catenin only when phosphorylated on residues serine 33, 37 or threonine 41. We found that phospho-β-catenin was co-localized with β-tubulin IV at the apical membrane of the ciliated cells (Figure 3, Panels G–I). Little, if any co-localization was observed with CC10 (Figure 3, Panels J–L). Both Apc and phospho-β-catenin are localized to the ciliated cells. This is consistent with their functional relationship in canonical Wnt signaling. The latter observations on co-localization of Apc with cytoplasmic total β-catenin and phospho-β-catenin indicate that the machinery that phosphorylates β-catenin is active in ciliated cells.
NAPH-induced changes in epithelial cell shape and cell migration are accompanied by alterations in subcellular distribution of Apc
In addition to its role in Wnt signaling, Apc regulates cytoskeletal networks and cell shape and migration. Both Wnt signaling and cell migration have been proposed to be critical in the lung epithelial response to NAPH injury (Park et al., 2006; Kida et al., 2008). We therefore examined expression and distribution of Apc in the NAPH-injury model. Figure 4 illustrates the effects of NAPH in adult mouse lung. Bronchioles of mice treated with corn oil (used as control) appeared normal with uniformly distributed Apcpositive, and CC10positive cells (Figure 4, Panels A & B). One day after NAPH administration, CC10positive cells were detached from the bronchiolar basement membrane and shed into the airway lumen. The remaining cells that had undergone squamous metaplasiato cover the injured airways were all Apcpositive (Figure 4, Panels C & D). Three days after injury, Apcpositive cells started to regain some cytoplasmic staining (Figure 4, Panels E & F, Please see below for details). 14 days after injury, CC10positive cells were detected again scattered among Apcpositive cells (Figure 4, Panels G & H).
Figure 4. Double immunofluorescent staining of Apc (red) with CC10 (green) in NAPH-injured lungs.
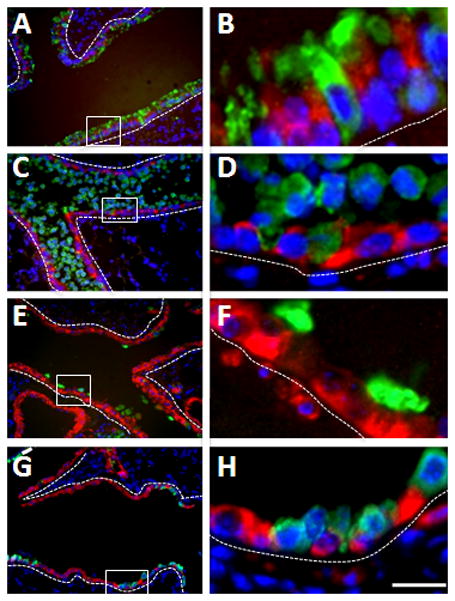
(A&B) Control mouse lung treated with corn oil (Day 0). (C&D) One day after NAPH injury. (E&F) Three days after NAPH injury. (G&H) Fourteen days after NAPH injury. Panels B, D, F and H are high magnification of boxed areas in Panels A, C, E, and G, respectively. Dotted lines outline airway epithelium. Scale bar, 40μm for Panels A, C, E, and G; 10μm for Panels B, D, F and H.
The latter observation demonstrated that in response to NAPH injury, Apcpositive epithelial cells undergo dynamic cell shape changes, a process that requires highly organized rearrangement of the cytoskeleton. Both Apc and β-catenin are involved in establishing and maintaining tissue architecture. The subcellular distribution of Apc with β-catenin during lung injury and repair was therefore further examined using confocal laser microscopy. As shown in Figure 5, in the normal adult lung, Apc is enriched in the apical and lateral cytoplasm of epithelial cells (ciliated cells) whereas β-catenin is localized in the membrane and cytoplasm (Figure 5, Panels A–C). Interestingly, 24 hours after NAPH administration, Apc distribution was no longer polarized (Figure 5, Panel D) whereas β-catenin was localized to the lateral membrane (Figure 5, Panel E) of surviving ciliated cells. As a result, there was a dramatic decrease of cytoplasmic Apc and β-catenin (Figure 5, Panel F; Figure 6). This subcellular distribution pattern persists for at least another 24 hours (data not shown). Cytoplasmic distribution of Apc and β-catenin was re-established 3 days subsequent to administration of NAPH (Figure 5, Panels G–I; Figure 6). The apical polarity pattern of Apc was restored completely14 days after exposure to NAPH (Figure 5, Panel J). The dynamic subcellular distribution of Apc and β-catenin suggests that the latter molecules play a role during NAPH-induced epithelial injury and repair, a process that involves dynamic cell shape change and cell migration (Kida et al., 2008).
Figure 5. Subcellular distribution of Apc and β-catenin after NAPH injury.
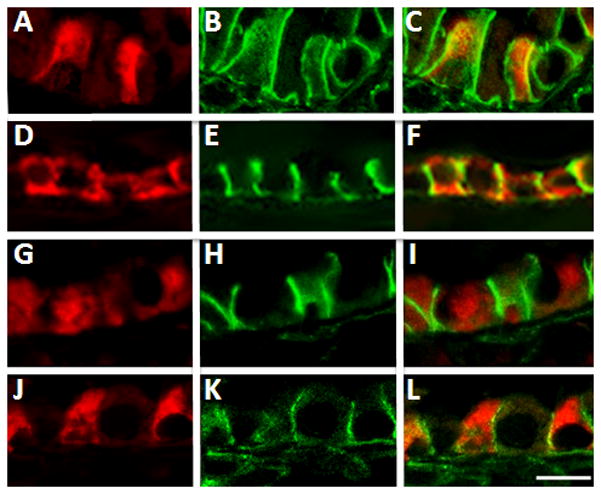
Double immunofluorescent staining was performed with antibodies against Apc (red) and total-β-catenin (green). (A–C) Control mouse lung treated with corn oil. (D–F) One day after NAPH injury. (G–I) Three days after NAPH injury. (J–L) Fourteen days after NAPH injury. Note, Apc subcellular distribution changed from a polarized (Day 0) to a non-polarized (Day 1) pattern in response to NAPH injury. The polarized Apc distribution pattern re-established by day 14. Scale bar, 5μm.
Figure 6. Western blot analysis for protein levels of Apc, phospho-β-catenin and phospho-Gsk3β in NAPH-injured lungs.
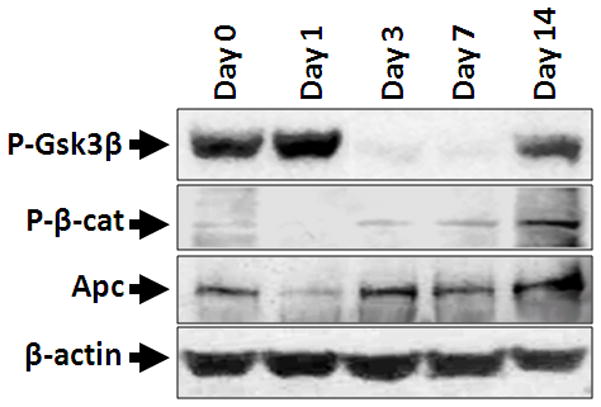
Protein levels of phospho-Gsk3β (P-Gsk3β), phospho-β-catenin (P-β-cat) and Apc were determined by western blot in mouse lungs treated with corn oil (Day 0) or NAPH (Day 1 to Day 14). Similar amount of total proteins were loaded as indicated by levels of β-actin.
NAPH-induced changes in Apc and β-catenin is accompanied by changes in Gsk3β
Phosphorylated Gsk3β (phospho-Gsk3β) controls Apc subcellular distribution and is essential for cell shape change and cell migration (Zumbrunn et al., 2001; Etienne-Manneville and Hall, 2003). We therefore examined the level of Gsk3β phosphorylation in the NAPH injured lungs. Twenty-four hours after NAPH injury, the Apcpositive epithelial cells responded by cell shape changes (squamous metaplasia) and loss of apical distribution of Apc (Figure 5, panels D). Correspondingly, the level of phospho-Gsk3β was robustly increased (Figure 6). Three days after NAPH injury, the cytoplasmic distribution of Apc was restored (Figure 5, panel G) and phospho-Gsk3β decreased to nearly undetectable levels. Interestingly, correlating to changes in Apc distribution and phospho-Gsk3β level, phospho-β-catenin levels decreased on day 1 and were re-established from day 3 of NAPH injury. Therefore, changes in Apc distribution and phospho-Gsk3β may have functional implications for the observed changes in cell shape and migration that constitute the airway epithelial cell response to NAPH-induced injury and onset of subsequent repair mechanisms.
DISCUSSION
Apc is an important regulator of cellular functions ranging from proliferation to migration and cell shape changes (Polakis, 1997; Etienne-Manneville and Hall, 2003; Wen et al., 2004; Langford et al., 2006a; Langford et al., 2006b). The current study examined the temporal and spatial expression of Apc during lung development and in a model of NAPH-induced airway injury and repair. The findings revealed three important properties of Apc with significant functional implications. First, during embryonic development, Apc expression was found to be dynamically regulated between the mesenchymal and endodermal cells of the murine lung. Second, within the endodermally-derived airway epithelium, Apc exhibited distinct cell type specificity, being present at high levels in post-mitotic ciliated cells. In contrast, Apc was either absent or undetectable in Clara and basal cells which are capable of self-renewal as well as generating other lineages in the airway cellular landscape. Third, epithelial Apc exhibits a distinct pattern of subcellular localization that is consistent with a role in regulating Wnt signaling and cell shape. This distribution becomes altered during cell shape changes that accompany the airway response to NAPH injury, and is reconstituted upon re-establishment of the normal airway epithelium.
Apc in lung morphogenesis
In adult animals, Apc is expressed in all tissues that have been tested, with high levels found in the brain, intestine and colon (Groden et al., 1991; Smith et al., 1993; Wong et al., 1996). Little to no information has been available on Apc expression in the lung. In the current study, we show that Apc expression changes dynamically during lung development. Immunohistochemical analysis revealed detectable Apc in the mesenchymal cells of murine lung as early as E13.5 (data not shown). Immunoreactivity was localized to subepithelial mesenchymal cells surrounding the nascent airway epithelium as well as the blood vessels. Double immunostaining with α-SMA antibody identified the mesenchymal Apcpositive cells as parabronchial smooth muscle. It is important to note that at this stage, epithelial expression of Apc could not a priori be ruled out, either because it may fall below the threshold of detectability, or because of the presence of distinct isoforms, not detectable by the specific antibody used. However, as embryonic lung development progressed, immunoreactive Apc became detectable and increased in distinct sub-populations of epithelial cells in E18.5 lungs. Further analysis identified the epithelial Apcpositive cells as ciliated and neuroendocrine cells. Concomitant with its rise in the epithelium, Apc in the parabronchial smooth muscle cells diminished significantly in the adult lung. Thus, Apc exhibits a distinct and dynamic pattern of expression in both the mesenchymal and endodermal compartments of the lung.
Absence of Apc in Clara cells
In the adult mouse lung, high levels of Apc were detectable only in ciliated and neuroendocrine cells of the airway epithelium. Double immunostaining with an antibody against the Clara cell-specific protein CC10 failed to reveal detectable Apc in this cell type, although, as mentioned, absence of immunoreactivity may be due to limited antigen availability or presence of distinct Apc isoforms. Nevertheless, the cell type-specific expression of Apc observed in the present study bears important functional implications, particularly as it relates to potential stem cell functions in the respiratory airways. In this regard, ciliated cells that are Apcpositive lack proliferative and self-renewal activities (Rawlins et al., 2007). In contrast, Clara cells which were found to be Apcnegative are known to serve as stem cells, undergoing not only self-renewal, but also serving as progenitors for derivation of other cell lineages in the repair and reconstitution of the normal airway epithelium, properties that define bona fide stem cells (Rawlins et al., 2009). The other cell type with the ability to undergo mitosis in response to injury is the neuroendocrine cell. However, while there is evidence for self-renewal, neuroendocrine cells are not known to generate non-neuroendocrine derivatives (Reynolds et al., 2000). This functional linkage between stem cells and absence of Apc has also been described in the gut epithelium (Senda et al., 2007), in which Apc contributes to gut organogenesis by supporting differentiation and restricting proliferation to the appropriate compartment. The post-mitotic enterocytes located in the upper half of the intestinal villi and the upper third of the colonic crypts exhibit intense immunoreactivity for Apc, whereas, those in the intestinal crypts and the lower third of the colonic crypts show faint or no Apc expression. The intestinal progenitor/stem cells reside near the bottom of the crypts and are Apcnegative. These cells proliferate to produce new enterocytes. Therefore, the functional link between absence of Apc and the ability of tissue-embedded stem cells to self-renew and to generate other cell types appears to be highly conserved in both the gut and the lung.
What is the role of Apc in ciliated cells?
In normal adult lung, the bulk of Apc is localized to the apical and lateral cytoplasm of columnar ciliated cells (Figure 2I). A similar intracellular distribution of Apc has been observed in enterocytes of the small intestine and colon (Miyashiro et al., 1995; Senda et al., 1996). By analogy to Apcpositive enterocytes, one functional consequence of high levels of Apc in the ciliated cells may be directly linked to their post-mitotic status and absence of proliferative capacity (Rawlins et al., 2007). Furthermore, double immunostaining for Apc and β-catenin showed that Apc and β-catenin are co-localized in the cytoplasm of ciliated cells, where phospho-β-catenin is detected. This correlation is consistent with the known function of Apc in regulating canonical Wnt signaling, which in turn controls cell proliferation (Munemitsu et al., 1995).
Apc likely participates in airway epithelial cell migration and cell shape changes
Apc regulates cell shape and cell migration (Etienne-Manneville and Hall, 2003; Wen et al., 2004; Langford et al., 2006a; Langford et al., 2006b; Kroboth et al., 2007). This discrete function of Apc is thought to be dependent on its subcellular distribution. Several studies have demonstrated that Apc exhibits high intracellular mobility, and shuttles between several subcellular destinations (Reinacher-Schick and Gumbiner, 2001; Bienz, 2002). While the best-characterized pool of Apc is within the destructive complex in the cytoplasm, biochemical fractions of full-length and mutant Apc have also been found that are tightly associated with the apical membrane of polarized epithelial cells (Reinacher-Schick and Gumbiner, 2001). Additionally, other studies have described concentrated Apc localization to the apical surface of intestinal epithelial cells (Senda et al., 1996) and cells within the developing Drosophila embryo (McCartney et al., 1999; Yu and Bienz, 1999). Whether the distribution pattern of Apc observed in the current study contributes to maintaining the structure and polarity of the ciliated cells remains to be determined.
In the NAPH-induced airway injury model, we observed dynamic alterations in the subcellular distribution of Apc in the ciliated cells that undergo changes in cell shape and migration. The level of Apc, which is high in uninjured ciliated cells, was reduced during the early (within 24–48 hours) phase of injury response, but was fully restored by the time epithelial cells regained their columnar cell phenotype on day 14 (Figure 5J). Thus in the airways, changes from columnar to squamous and back to columnar shape that signify pre-injury, injury and repair phases of the NAPH injury model respectively, are associated with alterations in intracellular localization of Apc. It is important to note that these observations are merely associative and whether such changes in Apc subcellular distribution are required for changes in ciliated cell shape and/or migration require further experimentation.
Activation of Gsk3β in injured airway epithelium
To validate the observations on subcellular changes in the distribution of Apc and their functional implications, we examined Gsk3β, a kinase that regulates Apc activity via phosphorylation. Activity of Gsk3β is also regulated by phosphorylation (Harwood, 2001). Unphosphorylated Gsk3β promotes binding of Apc to β-catenin and its subsequent degradation (Ha et al., 2004). Alternatively, when phosphorylated, Gsk3β catalyzes Apc interactions with microtubules and participates in regulating cell polarization and cell shape (Zumbrunn et al., 2001; Etienne-Manneville and Hall, 2003). Consistent with the latter functional relationships, levels of phospho-Gsk3β robustly increased 24 hours after NAPH treatment coincident with changes of subcellular localization of Apc (Figure 6). This is the period within which ciliated cells are observed to undergo squamous metaplasia accompanied by cell shape changes and migration. Accompanying this change was a reduction in phosphorylation of β-catenin, which leads to its stabilization and presumably activation of Wnt signaling (Figure 6). Three days after NAPH injury phospho-Gsk3β decreased and phospho-β-catenin increased (cessation of, or reduction in Wnt signaling) concomitant with re-establishment of cytoplasmic Apc distribution (Figures 5 & 6). Similar sequence of alterations have been documented during the repair phase of experimental models of smoke-induced airway injury (Liu et al., 2006).
Therefore, associated changes of Apc distribution and phospho-Gsk3β levels imply a role of Apc in ciliated cell shape change during NAPH injury. However, it can not be excluded that Apc distribution change may be secondary to the cell shape change. Further analysis involving Apc deletion will clarify the latter mechanism.
To summarize, Apc is expressed in both mesenchymal and endodermal compartments of the lung. Apc expression undergoes changes in cell-specificity during lung development. In the adult airways, high Apc levels occur in ciliated and neuroendocrine cells. In ciliated cells Apc may be involved in two distinct functions in regulating cell proliferation and cell shape and migration (Figure 7). The absence of Apc in airway epithelial cells may be functionally linked to their role as tissue-embedded stem cells. Clara cells appear to fulfill this definition, but the precise role of Apc in this regard awaits further experimentation.
Figure 7. Putative model for Apc functions in lung epithelial cells.
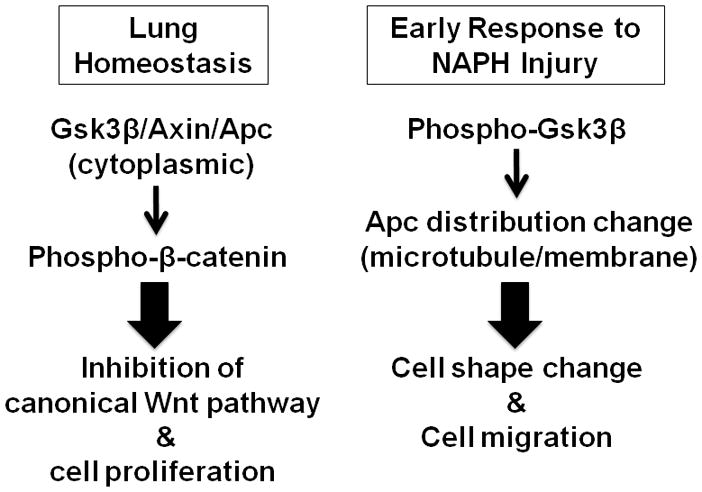
In normal lung homeostasis, Apc is mainly located in the cytoplasm and facilitates phosphorylation of β-catenin by Gsk3β and Ck1. This results in inhibition of canonical Wnt signaling, especially in fully differentiated cells such as ciliated cells.
Immediately after NAPH injury, levels of phospho-Gsk3β increase. Phospho-Gsk3β stimulates the interaction of Apc with microtubules and changes of Apc subcellular distribution. These changes may be the underlying mechanism for dynamic cell shape changes and cell migration during early response to NAPH injury in the lung.
EXPERIMENTAL PROCEDURES
Naphthalene Treatment and Tissue Collection
Female mice at 8 to 10 weeks of age were used. Naphthalene (NAPH, Sigma-Aldrich) or equivalent volumes of corn oil vehicle (Mazola, Cordova, TN) was administered by intraperitoneal injection at a dose of 250 mg/kg body weight. This dose of NAPH has been characterized to specifically ablate Clara cells with minimal inflammation within 2 days after injection (Van Winkle et al., 1995; Lawson et al., 2002). Naphthalene is concentrated in Clara cells that are enriched in P450 enzymes (CYP 2F2). Metabolism of NAPH generates toxic metabolites resulting in selective injury of Clara cells (Buckpitt et al., 2002). At 1, 3, 7 and 14days after NAPH injection, lungs were dissected for RNA or protein or perfused and fixed with 4% paraformaldehyde overnight for immunohistochemistry. Embryonic lungs at various stages of development were also collected for RT-PCR, western blot and immunohistochemistry.
All animals were maintained and housed in pathogen-free conditions according to a protocol approved by The University of Southern California Institutional Animal Care and Use Committee (IACUC) (Los Angeles, CA, USA).
Total RNA isolation and Polymerase Chain Reaction (PCR)
Relative levels of Apc mRNA in mouse lungs and cells were assessed by real-time RT-PCR as described previously (Li et al., 2009; Xing et al., 2010). Briefly, total RNA from lungs at various developmental stages and cell lines was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA). Two μg of total RNA were reverse transcribed with SuperScript II First-Strand Synthesis System for RT-PCR kit(Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Real time PCR was performed using a LightCycler (Roche Diagnostics, Mannheim, Germany). Sequences of the primers and resultant PCR product sizes were: (1) Mouse Apc: forward: 5′-ACCAGGACAAAAACCCAATG-3′ and reverse: 5′-TTCATTGCATGCCTATGCTC-3′ (105bp); (2) Mouse β-actin: forward: 5′-CCAACCGTGAAAAGATGACC-3′ and reverse: 5′-CCAGAGGCATACAGGGACAG-3′ (96bp). Specificity of each PCR reaction was verified by electrophoresis on 2% gel.
Relative quantification analysis was carried out with LightCycler software, Version 4 (Roche). The results were expressed as a normalized ratio (fold). β-actin was used as a reference gene for normalization (Strayer et al., 2002; Levesque et al., 2007). Samples from E13.5 lungs served as “Calibrator”.
Western Blot Analysis
Total protein was extracted with RIPA buffer (Sigma, St. Louis, MO) from embryonic and adult mice lung tissues, H441 cells, and H441 cells transfected with full-length Apc expression construct (Addgene plasmid 16507) (Morin et al., 1997). Protein concentration was determined by a BCA Protein Assay kit (Thermo Scientific, IL). 15–30μg of protein was loaded onto 3–8% NuPAGE gels and transferred to Immobilon-P membranes (Millipore Corp.). Membranes were then blocked with 5% milk in Tris-buffered saline and incubated with primary antibodies overnight. The secondary antibodies, goat-anti-rabbit or goat-anti-mouse IgG-HRP (Pierce), were applied at a 1:1000 dilution for 30 min. Membranes were reacted with chemiluminescence reagent ECL (Amersham Biosciences) and exposed to photographic film.
Immunohistochemistry
Immunohistochemistry was performed as described previously with some modifications (Li et al., 2008; Li et al., 2009). Briefly, routinely prepared histological sections were deparaffinized and endogenous peroxidase activity was quenched using 3% hydrogen peroxide. After blocking with normal horse serum, sections were incubated with a primary antibody at 4°C overnight. Impress-anti-rabbit or anti-goat IgG (Vector Laboratories) was applied for 50 min at room temperature. Staining was visualized by a Peroxidase Substrate Kit DAB (Vector Laboratories).
For immunofluorescence staining, the sections were incubated with primary antibodies overnight at 4°C. After the washing steps, the sections were reacted with a mixture of Cy3 anti-rabbit IgG or fluorescein anti-mouse or anti-goat IgG (Jackson ImmunoResearch Laboratories, INC) for 1 hr in the dark at room temperature. After thorough rinses with phosphate buffered saline (PBS, pH 7.0) containing 0.1% Triton X-100, the sections were mounted with VECTASHELD mounting medium containing DAPI (to visualize nuclei).
Primary antibodies that were used are: Apc, rabbit polyclonal antibody (Anaspec, San Jose, CA); α-smooth muscle-specific actin (α-SMA), mouse monoclonal antibody (Sigma, St. Louis, MO), β-tubulin IV, mouse monoclonal antibody (Biogenex); CC10, goat polyclonal antibody (Santa Cruz); P63, mouse monoclonal antibody (Santa Cruz Biotechnology); PGP9.5, rabbit polyclonal antibody (Anaspec, San Jose, CA); total-β-catenin, mouse monoclonal antibody (Transduction Laboratories); phospho(ser33/37/thr41)-β-catenin, rabbit polyclonal antibody (Santa Cruz Biotechnology); and phospho(ser9)-Gsk3β, rabbit monoclonal antibody (Cell Signaling Technology).
Acknowledgments
Grant support: This work was supported by NIH/NHLBI (HL075334, HL060231, & HL071945) and generous funds from the Hastings Foundation.
We thank Dr. Bert Vogelstein for the Apc expression construct, and Linyan Hu and Hongyan Chen for technical support. ZB holds the Ralph Edgington Chair in Medicine.
References
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- Bienz M. The subcellular destinations of APC proteins. Nat Rev Mol Cell Biol. 2002;3:328–338. doi: 10.1038/nrm806. [DOI] [PubMed] [Google Scholar]
- Buckpitt A, Boland B, Isbell M, Morin D, Shultz M, Baldwin R, Chan K, Karlsson A, Lin C, Taff A, West J, Fanucchi M, Van Winkle L, Plopper C. Naphthalene-induced respiratory tract toxicity: metabolic mechanisms of toxicity. Drug Metab Rev. 2002;34:791–820. doi: 10.1081/dmr-120015694. [DOI] [PubMed] [Google Scholar]
- Chilosi M, Doglioni C. Constitutive p63 expression in airway basal cells. A molecular target in diffuse lung diseases. Sarcoidosis Vasc Diffuse Lung Dis. 2001;18:23–26. [PubMed] [Google Scholar]
- Dikovskaya D, Schiffmann D, Newton IP, Oakley A, Kroboth K, Sansom O, Jamieson TJ, Meniel V, Clarke A, Nathke IS. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol. 2007;176:183–195. doi: 10.1083/jcb.200610099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Fanucchi MV, Murphy ME, Buckpitt AR, Philpot RM, Plopper CG. Pulmonary cytochrome P450 monooxygenase and Clara cell differentiation in mice. Am J Respir Cell Mol Biol. 1997;17:302–314. doi: 10.1165/ajrcmb.17.3.2774. [DOI] [PubMed] [Google Scholar]
- Giangreco A, Arwert EN, Rosewell IR, Snyder J, Watt FM, Stripp BR. Stem cells are dispensable for lung homeostasis but restore airways after injury. Proc Natl Acad Sci U S A. 2009;106:9286–9291. doi: 10.1073/pnas.0900668106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Hanson CA, Miller JR. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene. 2005;361:1–12. doi: 10.1016/j.gene.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Harwood AJ. Regulation of GSK-3: a cellular multiprocessor. Cell. 2001;105:821–824. doi: 10.1016/s0092-8674(01)00412-3. [DOI] [PubMed] [Google Scholar]
- Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am J Respir Cell Mol Biol. 2001;24:671–681. doi: 10.1165/ajrcmb.24.6.4498. [DOI] [PubMed] [Google Scholar]
- Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol. 2004;164:577–588. doi: 10.1016/S0002-9440(10)63147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida H, Mucenski ML, Thitoff AR, Le Cras TD, Park KS, Ikegami M, Muller W, Whitsett JA. GP130-STAT3 regulates epithelial cell migration and is required for repair of the bronchiolar epithelium. Am J Pathol. 2008;172:1542–1554. doi: 10.2353/ajpath.2008.071052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroboth K, Newton IP, Kita K, Dikovskaya D, Zumbrunn J, Waterman-Storer CM, Nathke IS. Lack of adenomatous polyposis coli protein correlates with a decrease in cell migration and overall changes in microtubule stability. Mol Biol Cell. 2007;18:910–918. doi: 10.1091/mbc.E06-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford KJ, Askham JM, Lee T, Adams M, Morrison EE. Examination of actin and microtubule dependent APC localisations in living mammalian cells. BMC Cell Biol. 2006a;7:3. doi: 10.1186/1471-2121-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford KJ, Lee T, Askham JM, Morrison EE. Adenomatous polyposis coli localization is both cell type and cell context dependent. Cell Motil Cytoskeleton. 2006b;63:483–492. doi: 10.1002/cm.20139. [DOI] [PubMed] [Google Scholar]
- Lawson GW, Van Winkle LS, Toskala E, Senior RM, Parks WC, Plopper CG. Mouse strain modulates the role of the ciliated cell in acute tracheobronchial airway injury-distal airways. Am J Pathol. 2002;160:315–327. doi: 10.1016/S0002-9440(10)64375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque BM, Zhou S, Shan L, Johnston P, Kong Y, Degan S, Sunday ME. NPAS1 regulates branching morphogenesis in embryonic lung. Am J Respir Cell Mol Biol. 2007;36:427–434. doi: 10.1165/rcmb.2006-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Li A, Li M, Xing Y, Chen H, Hu L, Tiozzo C, Anderson S, Taketo MM, Minoo P. Stabilized beta-catenin in lung epithelial cells changes cell fate and leads to tracheal and bronchial polyposis. Dev Biol. 2009;334:97–108. doi: 10.1016/j.ydbio.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Li C, Liu YH, Xing Y, Hu L, Borok Z, Kwong KY, Minoo P. Mesodermal deletion of transforming growth factor-beta receptor II disrupts lung epithelial morphogenesis: cross-talk between TGF-beta and Sonic hedgehog pathways. J Biol Chem. 2008;283:36257–36264. doi: 10.1074/jbc.M806786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MG, Li NP, Wu RL, Ma Y, Hong YZ, Tian D, Zhu M. Dynamic changes of adenomatous polyposis coli protein and glycogen synthase kinase 3beta in the repair of the injured airway epithelial cells in smoking mice. Sheng Li Xue Bao. 2006;58:255–261. [PubMed] [Google Scholar]
- McCartney BM, Dierick HA, Kirkpatrick C, Moline MM, Baas A, Peifer M, Bejsovec A. Drosophila APC2 is a cytoskeletally-associated protein that regulates wingless signaling in the embryonic epidermis. J Cell Biol. 1999;146:1303–1318. doi: 10.1083/jcb.146.6.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J Cell Biol. 2000;148:505–518. doi: 10.1083/jcb.148.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro I, Senda T, Matsumine A, Baeg GH, Kuroda T, Shimano T, Miura S, Noda T, Kobayashi S, Monden M, et al. Subcellular localization of the APC protein: immunoelectron microscopic study of the association of the APC protein with catenin. Oncogene. 1995;11:89–96. [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Souza B, Muller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–3681. [PubMed] [Google Scholar]
- Nathke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–366. doi: 10.1146/annurev.cellbio.20.012103.094541. [DOI] [PubMed] [Google Scholar]
- Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Wells JM, Zorn AM, Wert SE, Laubach VE, Fernandez LG, Whitsett JA. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. 2006;34:151–157. doi: 10.1165/rcmb.2005-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta. 1997;1332:F127–147. doi: 10.1016/s0304-419x(97)00008-5. [DOI] [PubMed] [Google Scholar]
- Poulsen TT, Biologist XN, Poulsen HS, Linnoila RI. Acute damage by naphthalene triggers expression of the neuroendocrine marker PGP9.5 in airway epithelial cells. Toxicol Lett. 2008 doi: 10.1016/j.toxlet.2008.06.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins EL, Hogan BL. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006;133:2455–2465. doi: 10.1242/dev.02407. [DOI] [PubMed] [Google Scholar]
- Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4:525–534. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins EL, Ostrowski LE, Randell SH, Hogan BL. Lung development and repair: contribution of the ciliated lineage. Proc Natl Acad Sci U S A. 2007;104:410–417. doi: 10.1073/pnas.0610770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinacher-Schick A, Gumbiner BM. Apical membrane localization of the adenomatous polyposis coli tumor suppressor protein and subcellular distribution of the beta-catenin destruction complex in polarized epithelial cells. J Cell Biol. 2001;152:491–502. doi: 10.1083/jcb.152.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SD, Hong KU, Giangreco A, Mango GW, Guron C, Morimoto Y, Stripp BR. Conditional clara cell ablation reveals a self-renewing progenitor function of pulmonary neuroendocrine cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1256–1263. doi: 10.1152/ajplung.2000.278.6.L1256. [DOI] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R, Ihrke G, Bienz M. Actin-dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. EMBO J. 2001;20:5929–5939. doi: 10.1093/emboj/20.21.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senda T, Iizuka-Kogo A, Onouchi T, Shimomura A. Adenomatous polyposis coli (APC) plays multiple roles in the intestinal and colorectal epithelia. Med Mol Morphol. 2007;40:68–81. doi: 10.1007/s00795-006-0352-5. [DOI] [PubMed] [Google Scholar]
- Senda T, Miyashiro I, Matsumine A, Baeg GH, Monden T, Kobayashil S, Monden M, Toyoshima K, Akiyama T. The tumor suppressor protein APC colocalizes with beta-catenin in the colon epithelial cells. Biochem Biophys Res Commun. 1996;223:329–334. doi: 10.1006/bbrc.1996.0894. [DOI] [PubMed] [Google Scholar]
- Singh G, Singh J, Katyal SL, Brown WE, Kramps JA, Paradis IL, Dauber JH, Macpherson TA, Squeglia N. Identification, cellular localization, isolation, and characterization of human Clara cell-specific 10 KD protein. J Histochem Cytochem. 1988;36:73–80. doi: 10.1177/36.1.3275712. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Johnson KA, Bryan TM, Hill DE, Markowitz S, Willson JK, Paraskeva C, Petersen GM, Hamilton SR, Vogelstein B, et al. The APC gene product in normal and tumor cells. Proc Natl Acad Sci U S A. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strayer M, Savani RC, Gonzales LW, Zaman A, Cui Z, Veszelovszky E, Wood E, Ho YS, Ballard PL. Human surfactant protein B promoter in transgenic mice: temporal, spatial, and stimulus-responsive regulation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L394–404. doi: 10.1152/ajplung.00188.2001. [DOI] [PubMed] [Google Scholar]
- Van Winkle LS, Buckpitt AR, Nishio SJ, Isaac JM, Plopper CG. Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am J Physiol. 1995;269:L800–818. doi: 10.1152/ajplung.1995.269.6.L800. [DOI] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- Wong MH, Hermiston ML, Syder AJ, Gordon JI. Forced expression of the tumor suppressor adenomatosis polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc Natl Acad Sci U S A. 1996;93:9588–9593. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Li C, Li A, Sridurongrit S, Tiozzo C, Bellusci S, Borok Z, Kaartinen V, Minoo P. Signaling via Alk5 controls the ontogeny of lung Clara cells. Development. 2010;137:825–833. doi: 10.1242/dev.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Bienz M. Ubiquitous expression of a Drosophila adenomatous polyposis coli homolog and its localization in cortical actin caps. Mech Dev. 1999;84:69–73. doi: 10.1016/s0925-4773(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11:44–49. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]