

Abstract
Immune cytokine interferon-γ (IFN-γ) plays a crucial role in immune-mediated demyelination diseases such as multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Our previous studies have shown that enforced expression of IFN-γ in the central nervous system (CNS) inhibits developmental myelination or remyelination in EAE demyelinated lesions. While many of the cellular actions of IFN-γ result from its activation of the signal transducer and activator of transcription 1 (STAT1) pathway, recent studies have shown that STAT1-independent pathways regulate some facets of IFN-γ biology. In this study, we dissected the role of STAT1-dependent and STAT1-independent pathways in IFN-γ-induced hypomyelination using a genetic approach. We found that the induction of the STAT1-dependent, IFN-γ responsive genes in response to this cytokine was abolished in the CNS of STAT1 null mice. Moreover, STAT1 deletion diminished oligodendrocyte loss, the reduction of myelinated axons and inflammatory response in the CNS of transgenic mice that ectopically express IFN-γ in the CNS. Nevertheless, IFN-γ-induced reduction of myelin sheath thickness in the CNS of these mice was not altered by STAT1 deletion. Collectively, these data demonstrated that both STAT1-dependent and STAT1-independent pathways are involved in the detrimental effects of IFN-γ on the myelination process.
Keywords: oligodendrocyte, myelin, multiple sclerosis, cytokine, signal transduction
Introduction
Interferon-γ (IFN -γ), a pleiotropic cytokine secreted by activated T-lymphocytes and natural killer cells, is critical for immune defense and autoimmunity (Boehm et al., 1997; Schroder et al., 2004). The biological effects of IFN-γ are elicited through activation of intracellular signal transduction pathways. The best characterized pathway is the Janus kinases (JAKs) – signal transducer and activator of transcription 1 (STAT1) pathway (Gysemans et al., 2008). IFN-γ exerts its effects on cells by binding to its cell surface receptors IFNγR1 and IFNγR2, resulting in oligomerization of the receptors. The receptor oligomerization leads to activation of JAK1 and JAK2 which facilitate trans-phosphorylation of the JAKs and the receptors. STAT1 is then recruited to the receptors and phosphorylated on tyrosine 701. Phosphorylated STAT1 undergoes dimerization, translocates to the nucleus and regulates gene expression by binding to γ-activated sequence (GAS) elements in the promoters of IFN-γ-responsive genes. It is known that many aspects of IFN-γ biology are through the STAT1 pathway. Nevertheless, evidence is accumulating that some facets of IFN-γ biology are through STAT1-independent pathways (Ramana et al. 2002; Shresta et al., 2005; Gough et al., 2008). In addition to STAT1, phosphatidylinositol 3-kinase (PI3-K), proto-oncogene c-Cbl and myeloid differentiation primary response gene 88 (MyD88) are recruited to the ligated IFNγR where they are activated (Alsayed et al., 2000; Nguyen et al., 2001; Sun and Ding, 2006). Moreover, phosphorylated JAKs activate Src family non-receptor tyrosine kinases c-Src, Fyn and Lyn in the presence of IFN-γ (Uddin et al., 1997; Chang et al., 2002; Qing and Stark, 2004).
IFN-γ is regarded as a major pro-inflammatory cytokine involved in myelin damage and repair in immune-mediated demyelination diseases such as multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) (Popko et al., 1997; Sredni-Kenigsbuch, 2002; Lees and Cross, 2007). Nevertheless, the precise role of the STAT1 pathway or STAT1-independent pathways in this cytokine’s effects on immune-mediated demyelination diseases remains unclear. Thus, investigation of the role of these pathways in the detrimental effects of IFN-γ in immune-mediated demyelination diseases is warranted.
In a previous study, we showed that the presence of IFN-γ in the CNS during development induces cerebellar medulloblastoma. Importantly, we demonstrated that the STAT1 pathway is required for IFN-γ-induced medulloblastoma formation, but that STAT1-independent pathways also play an important role in an IFN-γ-mediated disruption of granule cell migration (Lin et al., 2004). In this report, we show that STAT1 deletion diminishes the induction of STAT1 target genes, oligodendrocyte loss, and the reduction of myelinated axons in the CNS of transgenic mice that ectopically express IFN-γ in the CNS, but does not alleviate IFN-γ-induced reduction of myelin sheath thickness. Furthermore, we demonstrate that inflammatory response induced by IFN-γ in the CNS is dependent on the STAT1 pathway.
Materials and Methods
Mice breeding
Line 67 GFAP/tTA mice and line184 TRE/IFN-γ mice on the C57BL/6 background were mated with STAT1 null mice (strain 129S6/SvEv- STAT1tm1; kindly provided by Dr. H. Schreiber, The University of Chicago, Chicago, IL) (Meraz et al., 1996), and the resulting progeny were intercrossed to obtain GFAP/tTA; TRE/IFN-γ; STAT1 WT mice and GFAP/tTA; TRE/IFN-γ; STAT1 null mice (Lin et al., 2004). To prevent transcriptional activation of the TRE/IFN-γ transgene by tTA, 0.05 mg/ml doxycycline (Sigma, St. Louis, MO) was added to the drinking water and was provided ad libitum. All animal procedures were conducted in complete compliance with the National Institutes of Health’s (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Real-time PCR
RNA was isolated from the mouse brains using Trizol reagent (Invitrogen, Carlsbad, CA) and treated with DNaseI (Invitrogen, Carlsbad, CA) to eliminate genomic DNA. Reverse transcription was performed using Superscript First Strand Synthesis System for RT-PCR kit (Invitrogen, Carlsbad, CA). Real-time PCR was performed with iQ supermix (Bio-Rad, Hercules CA) on a Bio-Rad iQ real-time PCR detection system (Bio-Rad, Hercules CA). The primers and probes (Integrated DNA Technologies Inc., Coralville, IA) for real-time PCR were as follows: glyceraldehyde phosphate dehydrogenase (GAPDH) sense primer (CTCAACTACATGGTCTACATGTTCCA); GAPDH antisense primer (CCATTCTCGGCCTTGACTGT); GAPDH probe (TGACTCCACTCACGGCAAATTCAACG); major histocompatiblity complex (MHC)-I sense primer (ATTCCCCAAAGGCCCATGT); MHC-I antisense primer (GTCTCCACAAGCTCCATGTCC); MHC-I probe (TGCTGGGCCCTGGGCTTCTACC); IFN-γ-inducible transcription factor 1 (IRF-1) sense primer (ACCTACAGGTGTCACCCATGC); IRF-1 antisense primer (GCTGCCACTCAGACTGTTCAAA); IRF-1 probe (CCACCTCCGAAGCCGCAACAGACG); IFN-γ-inducible protein 10 (IP-10) sense primer (AGGGCCATAGGGAAGCTTGAAA); IP-10 antisense primer (CGGATTCAGACATCTCTGCTCATC); IP-10 probe (CCCTGCGAGCCTATCCTGCCCACG); myelin basic protein (MBP) sense primer (GCTCCCTGCCCCAGAAGT); MBP antisense primer (TGTCACAATGTTCTTGAAGAAATGG); MBP probe (AGCACGGCCGGACCCAAGATG); proteolipid protein (PLP) sense primer (CACTTACAACTTCGCCGTCCT); PLP antisense primer (GGGAGTTTCTATGGGAGCTCAGA); PLP probe (AACTCATGGGCCGAGGCACCAA); ceramide galactosyltransferase (CGT) sense primer ( TTATCGGAAATTCACAAGGATCAA); CGT antisense primer (TGGCGAAGAATGTAGTCTATCCAATA); CGT probe (CCGGCCACCCTGTCAATCGG); tumor necrosis factor-α (TNF-α) sense primer (GGCAGGTTCTGTCCCTTTCA); TNF-α antisense primer (ACCGCCTGGAGTTCTGGAA) TNF-α probe (CCCAAGGCGCCACATCTCCCT); inducible nitric oxide synthase (iNOs) sense primer (GCTGGGCTGTACAAACCTTCC); iNOs antisense primer (TTGAGGTCTAAAGGCTCCGG); iNOs probe (TGTCCGAAGCAAACATCACATTCAGATCC); CAATT enhancer-binding protein homologous protein/growth and DNA damage protein 153 (CHOP/GADD153) sense primer (CCACCACACCTGAAAGCAGAA); CHOP antisense primer (AGGTGCCCCCAATTTCATCT); CHOP probe (TGAGTCCCTGCCTTTCACCTTGGAGA); caspase-12 sense primer (ATGCTGACAGCTCCTCATGGA); caspase-12 antisense primer (TGAGAGCCAGACGTGTTCGT); caspase-12 probe (AGTCCAAGATACACTGAAGCTTTGTCCACGTGAT).
Immunohistochemistry
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in 0.1M PBS. The brains were removed, postfixed with paraformaldehyde, cryopreserved in 30% sucrose, embedded in OCT and frozen on dry ice. Frozen sections were cut in a cryostat at a thickness of 10μm. For immunohistochemistry, frozen sections were treated with −20°C acetone, blocked with PBS containing10% NGS and 0.1% Triton X-100 and incubated overnight with the primary antibody diluted in blocking solution. Appropriate fluorochrome- or enzyme-labeled secondary antibodies (Vector Laboratories, Burlingame, CA) were used for detection. An antibody against CC1 (APC7, 1:50; EMD Biosciences, Inc., La Jolla, CA) was used as a marker for mature oligodendrocytes. Antibody against MBP (1:1000; Sternberger Monoclonals, Lutherville, MA) was used to verify the degree of myelination. Antibody against CD3 (1:50, Santa Cruz Biotechnology) was used as a marker for T cells. Antibody against CD11b (1:50, Chemicon, Temecula, CA, USA) was used to determine the activation of microglia/macrophage. Fluorescent stained sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories). The stained sections were visualized and analyzed as previously described (Lin et al., 2006). We quantified CC1 positive cells by counting positive cells within the white matter of the spinal cord, the cerebellum, and the corpus callosum. Only those cells with nuclei observable by DAPI staining were counted.
Electron microscopy
For electron microscopy, we anesthetized and perfused mice with 0.1 M PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde. Brains were sliced into 1-mm sections. The section corresponding to the region of the fornix was trimmed, processed for electron microscopic analysis and oriented so that a cross-section of the corpus callosum was achieved. Thin sections were cut, stained with uranyl acetate and lead citrate and analyzed as previously described (Lin et al., 2006). We calculated the total percentage of myelinated axons. G-ratio was calculated as diameter of the axon divided by the diameter of axon and myelin. A minimum of 300 fibers per mouse, 3 mice per group was analyzed.
Statistics
Data are expressed as mean standard deviation. Multiple comparisons were statistically evaluated by one way ANOVA test using Sigmastat 3.1 software. Differences were considered statistically significant if p < 0.05.
Results
STAT1 deletion attenuates the expression of IFN-γ-responsive genes in transgenic mice expressing IFN-γ in the CNS
We have generated transgenic mice that allow for the temporally controlled delivery of IFN-γ to the CNS using the tetracycline-controllable system (Lin et al., 2004). The expression of the IFN-γ transgene is repressed in GFAP/tTA; TRE/IFN-γ double-transgenic mice treated with doxycycline solution from conception. CNS-specific expression of IFN-γ could be detected at approximately postnatal day 12 (P12) in double transgenic mice released from doxycycline on embryonic day 16 (E16). We have shown that the presence of IFN-γ in the CNS during development causes oligodendrocyte loss, hypomyelination and cerebellar medulloblastoma (Lin et al., 2004; 2005). We have also shown that the STAT1 pathway is necessary for IFN-γ-induced medulloblastoma formation, and that STAT1-independent pathways participate in an IFN-γ mediated disruption of granule cell migration (Lin et al, 2004). In this study, we pursued a genetic approach to dissect the role of the STAT1 pathway and STAT1-independent pathways in IFN-γ-induced oligodendrocyte loss and hypomyelination. The GFAP/tTA and TRE/IFN-γ mice were crossed to STAT1 null mice (Meraz et al., 1996), and the resulting progeny were intercrossed to obtain GFAP/tTA; TRE/IFN-γ; STAT1 WT mice and GFAP/tTA; TRE/IFN-γ; STAT1 null mice. As previously described, GFAP/tTA; TRE/IFN-γ; STAT1 WT mice released from doxycycline at E16 (IFN-γCNS+;STAT1 WT mice) showed severe tremor and ataxia, and ~80% of these mice died by P28 (Lin et al., 2004). In contrast, GFAP/tTA; TRE/IFN-γ; STAT1 null mice released from doxycycline at E16 (IFN-γCNS+;STAT1 null mice) showed a much milder phenotype. These animals displayed a minor tremor and ataxia, but they all survived at least 8 weeks. Furthermore, real-time PCR analysis showed that the upregulation of the STAT1-dependent, IFN-γ-responsive genes IRF-1 and IP-10 was abolished in the CNS of IFN-γ expressing mice on a STAT1 null background (Figure 1). Nevertheless, the presence of IFN-γ significantly increased the expression of MHC-I in the CNS of STAT1 null mice compared to control double transgenic mice on a STAT1 wild type or null background treated with doxycycline throughout gestation and postnatal development (IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice), although the increase was modest compared to IFN-γCNS+; STAT1 WT mice. It is known that IFN-γ regulates MHC-I expression through both the STAT1 pathway and STAT1-independent pathways (Boehm et al., 1997). Taken together, these data indicate IFN-γ regulates the expression of its responsive genes in the CNS through the STAT1 pathway or STAT1-independent pathways.
Figure 1. STAT1 deletion attenuated the expression of IFN-γ-responsive genes in transgenic mice expressing IFN-γ in the CNS.
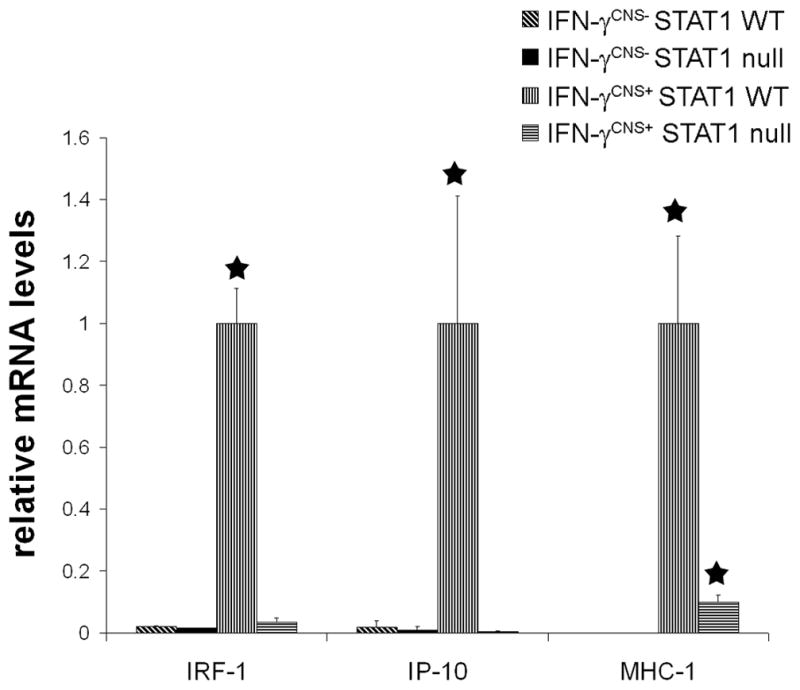
Real-time PCR analysis revealed that the presence of IFN-γ significantly increased the levels of IRF-1, IP-10 and MHC-1 mRNAs in the brain of 21-day-old mice on a STAT1 wild type background. The increased expression of STAT1-dependent, IFN-γ-responsive genes IRF-1 and IP-10 was abolished in the brain of IFN-γ-expressing mice on a STAT1 null background. Nevertheless, the level of MHC-1 mRNA in the brain of IFN-γ-expressing mice on a STAT1 null background was significantly elevated compared to control IFN-γCNS−;STAT1 WT mice or IFN-γCNS−; STAT1 null mice. mRNA levels are relative to the housekeeping gene GAPDH. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05.
STAT1 deletion diminished oligodendrocyte loss induced by IFN-γ
It is believed that the tremoring phenotype in mice that ectopically express IFN-γ in the CNS during development was primarily caused by oligodendrocyte loss and hypomyelination (Lin et al, 2005; 2008). Importantly, we did not find any evidence showing that STAT1 deletion significantly changed oligodendrocyte numbers or the myelination process in the CNS (Figure 2, 3 and 4). Additionally, western blot analysis showed that STAT1 deletion did not significantly alter the level of MBP protein in the CNS (data not shown). We next examine whether STAT1 deletion blocked oligodendrocyte loss in the CNS of IFN-γ-expressing mice. Oligodendrocytes, identified by CC1 immunostaining, were significantly reduced in the CNS white matter of 21-day-old IFN-γCNS+; STAT1 WT mice compared to control IFN-γCNS−;STAT1 WT mice or IFN-γCNS−; STAT1 null mice (Figure 2). Interestingly, oligodendrocyte numbers in the CNS white matter of IFN-γCNS+;STAT1 null mice were comparable to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice. Collectively, these data demonstrate that IFN-γ induces oligodendrocytes loss through the STAT1 pathway.
Figure 2. STAT1 deletion diminished oligodendrocyte loss induced by IFN-γ.

A. The presence of IFN-γ reduced the number of oligodendrocytes, detected by CC1 immunostaining (red fluorescence), in the corpus callosum of 21-day-old IFN-γCNS+; STAT1 WT mice but not IFN-γCNS+; STAT1 null mice. Blue fluorescence showed DAPI counterstain, n = 3, scale bar = 25μM. B. Quantitation of CC1-positive oligodendrocytes in the CNS white matter of 21-day-old mice. CC1 positive oligodendrocytes in the white matter of the spinal cord, the cerebellum, and the corpus callosum of IFN-γCNS+; STAT1 WT mice were significantly reduced compared to control IFN-γCNS−; STAT WT mice. In contrast, oligodendrocyte numbers in IFN-γCNS+; STAT1 null mice were comparable to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05.
Figure 3. STAT1 deletion attenuated hypomyelination induced by IFN-γ.

A. Real-time PCR analysis showed that the presence of IFN-γ significantly decreased myelin genes MBP, PLP, and CGT mRNA levels in the brain of 21-day-old IFN-γCNS+; STAT1 WT mice but not IFN-γCNS+; STAT1 null mice. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05. B, MBP immunostaining showed severe hypomyelination in the corpus callosum of 21-day-old IFN-γCNS+; STAT1 WT mice, and STAT1 deletion notably alleviated hypomyelination elicited by IFN-γ. N = 3 animals, scale bar = 50 μm.
Figure 4. STAT1 deletion attenuated hypomyelination induced by IFN-γ.
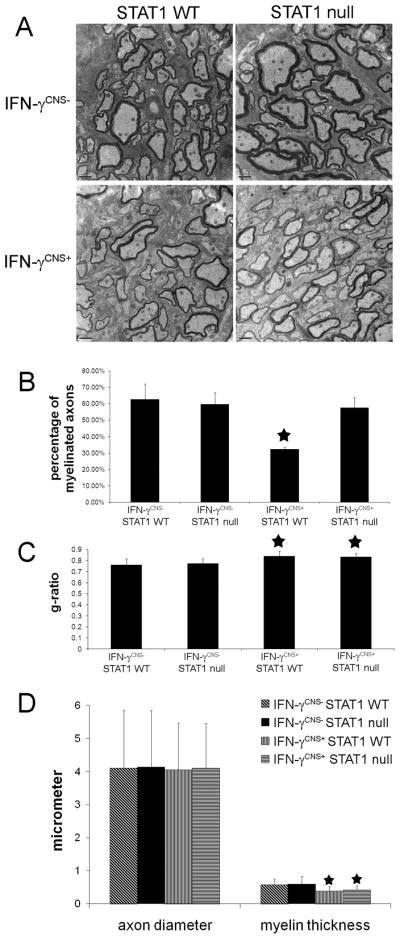
A. Electron micropscopy analysis showed that the presence of IFN-γ caused hypomyelination in the corpus callosum of 21-day-old mice on a STAT1 WT background and that STAT1 deletion ameliorated hypomyelination induced by IFN-γ in the CNS. N = 3 animals, scale bar = 200 nm. B. Electron micropscopy analysis revealed that the percentage of myelinated axons in the corpus callosum of 21-day-old IFN-γCNS+; STAT1 WT mice are significantly reduced compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice. Nevertheless, the percentage of myelinated axons in the corpus callosum of IFN-γCNS+; STAT1 null mice are comparable to control mice. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05. C. Electron microscopy analysis revealed that the presence of IFN-γ significantly increased the g-ratio in the corpus callosum of 21-day-old IFN-γCNS+; STAT1 WT mice and IFN-γCNS+; STAT1 null mice compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice. N = 300 axons, 100 axons per animals, error bars represent standard deviation, asterisk p < 0.05. D. Axonal diameter and myelin thickness in the corpus callosum of 21-day-old mice, N = 300 axons, 100 axons per animals, error bars represent standard deviation, asterisk p < 0.05.
STAT1 deletion attenuated hypomyelination induced by IFN-γ
We further determined the role of the STAT1 pathway in IFN-γ-induced hypomyelination. Real-time PCR analysis showed the presence of IFN-γ significantly decreased myelin genes MBP, PLP, and CGT mRNA levels in the brain of 21-day-old mice on aSTAT1 wild type background (Figure 3A). These mRNA levels were slightly lower in the brain of IFN-γCNS+; STAT1 null mice compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice, but the change, approximately 20%, were not statistically significant. Moreover, MBP immunostaining revealed severe hypomyelination in the corpus callosum of 21-day-old IFN-γCNS+;STAT1 WT mice (Figure 3B). While immunostaining for MBP was notably enhanced in the corpus callosum of 21-day-old IFN-γCNS+; STAT1 null mice compared to IFN-γCNS+; STAT1 WT mice, it was also notably decreased compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice.
We next quantified the myelin abnormalities in these mice by electron microscopy analysis. Ultrastructural examination revealed that the presence of IFN-γ significantly reduced the percentage of myelinated axons in the corpus callosum of 21-day-old mice on a STAT1 wild type background (32.30% ± 1.06%) compared to control IFN-γCNS−; STAT1 WT mice (62.60% ± 9.33% ) or IFN-γCNS−; STAT1 null mice (59.70% ± 7.11%). In contrast, the percentage of myelinated axons on IFN-γ expressing mice on a STAT1 null background (57.50% ± 6.19%) was comparable to age-matched control mice (Figure 4A and 4B). Moreover, we found that the presence of IFN-γ significantly reduced the thickness of myelin sheaths in mice on a STAT1 wild type background compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice (g-ratio, 0.841 ± 0.044 vs 0.760 ± 0.054 or 0.773 ± 0.043, p<0.05). Interestingly, the thickness of myelin sheaths in the CNS of IFN-γCNS+; STAT1 null mice was comparable to IFN-γCNS+; STAT1 WT mice (g-ratio, 0.833 ± 0.033 vs 0.841 ± 0.044, p > 0.05), which was significantly reduced compared to control mice (Figure 4C and 4D). Additional, electron microscopy analysis showed that neither STAT1 deletion nor the presence of IFN-γ significantly changed the diameter of axons in the CNS white matter (Figure 4D). Taken together, these data indicate that both the STAT1 pathway and STAT1-independent pathways are involved in the detrimental effects of IFN-γ on the myelination process. It is likely that the presence of IFN-γ reduces the numbers of oligodendrocyes and the percentage of myelinated axons in the CNS through the STAT1 pathway, but reduces the thickness of myelin sheaths through STAT1-independent pathways.
STAT1 deletion diminished the inflammatory response induced by IFN-γ
IFN-γ possesses numerous immunomodulatory effects, including activation of microglia/macrophage, promotion of leukocyte adhesion, induction of many inflammatory mediators such as TNF-α and iNOs (Boehm et al., 1997; Popko et al, 1997). It has been shown enhanced expression of IFN-γ in the CNS induces T cell infiltration and microglia/macrophages activation, and upregulates the expression of TNF-α and iNOs (Corbin et al., 1996; LaFerla et al., 2000; Lin et al., 2008). Several lines of evidence have suggestedthat the inflammatory response induced by IFN-γ promotes oligodendrocyte loss and myelin abnormalities in the CNS (Popko et al., 1997; Buntinx et al., 2002). It is known that the STAT1 pathway is critical for the immunomodulatory function of IFN-γ (Gysemans et al., 2008). Therefore, we are interested in the role of the STAT1 pathway in IFN-γ induced inflammatory response in the CNS. CD3 immunostaining showed few infiltrated T cells in the CNS of 21-day-old IFN- γCNS+; STAT1 WT mice (Figure 5A). In contrast, T cells were undetectable in the CNS of IFN- γCNS+; STAT1 null mice or control IFN- γCNS−; STAT1 WT mice or IFN- γCNS−; STAT1 null mice. Moreover, we found that the presence of IFN-γ in the CNS dramatically activated microglia/macrophages and significantly stimulated the expression of TNF-α and iNOs in the CNS of 21-day-old mice on a STAT1 wild type background (Figure 5B and 5C). Nevertheless, STAT1 deletion diminished the activation of microglia/macrophages and the upregulation of TNF-α and iNOs. Thus, these data indicate that IFN-γ induces inflammatory response in the CNS through the STAT1 pathway.
Figure 5. STAT1 deletion diminished the inflammatory response induced by IFN-γ.

A, CD3 immunostaining showed few infiltrated T cells (arrow; CD3 positive T cells) in the spinal cord of 21-day-old IFN-γCNS+; STAT1 WT mice. T cells were undetectable in the CNS of IFN-γCNS−; STAT1 WT mice, IFN-γCNS−; STAT1 null mice or IFN-γCNS+;STAT1 null mice. N = 3 animals, scale bar = 25μm. B, CD11b immunostaining showed that the presence of IFN-γ activated microglia/macrophages in the spinal cord of 21-day-old IFN-γCNS+; STAT1 WT mice but not IFN-γCNS+;STAT1 null mice. N = 3 animals, scale bar = 25 μm. C, Real-time PCR analysis revealed significantly increased levels of TNF-α and iNOS mRNA in the brain of 21-day-old IFN-γCNS+; STAT1 WT mice compared to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice. Nevertheless, STAT1 deletion diminished the induction of TNF-α and iNOS mRNA in the brain of mice expressing IFN-γ in the CNS. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05.
STAT1 deletion diminished IFN-γ-induced endoplasmic reticulum (ER) stress response
ER stress, the stress of accumulating unfolded or misfolded proteins in the ER, activates an adaptive coordinated response, ER stress response, to limit further accumulation of unfolded proteins in the ER (Rutkowski and Kaufman, 2004; Ron and Walter 2007). In multicellular organisms, if these adaptive responses are not sufficient to resolve the folding problems in the ER, the ER stress will trigger an apoptotic program, such as activation of caspase-12 and CHOP, to eliminate the cells (Boyce and Yuan, 2006). We have shown that the presence of IFN-γ in the CNS during developmental myelination process causes oligodendrocyte death and hypomyelination through severe ER stress response in myelinating oligodendrocytes (Lin et al., 2005; 2008; 2009). Interestingly, we found that STAT1 deletion diminished the upregulation of the ER stress markers caspase-12 and CHOP in the CNS of IFN-γ-expressing mice (Figure 6). Thus, our data indicate that IFN-γ induces ER stress in oligodendrocytes and subsequent cell loss through the STAT1 pathway.
Figure 6. STAT1 deletion diminished IFN-γ-induced ER stress response.
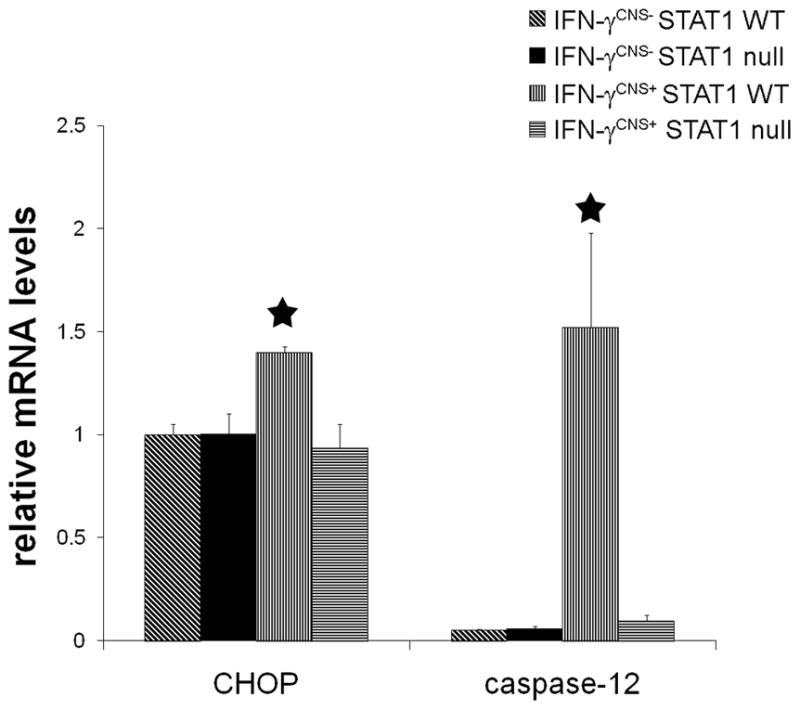
Real-time PCR analysis showed that the presence of IFN-γ significantly increased the expression of ER stress markers CHOP and caspase-12 in the brain of 21-day-old IFN-γCNS+; STAT1 WT mice. Nevertheless, the levels of CHOP and caspase-12 in the brain of IFN-γCNS+; STAT1 null mice were comparable to control IFN-γCNS−; STAT1 WT mice or IFN-γCNS−; STAT1 null mice.. N = 3 animals, error bars represent standard deviation, asterisk p < 0.05.
Discussion
The presence of immune cytokine IFN-γ within the CNS in immune-mediated demyelinating disorders is believed to contribute to disease pathogenesis, however, the data concerning its roles in these disorders are at times contradictory (Popko et al., 1997; Steinman, 2001; Lees and Cross, 2007). IFN-γ is undetectable in the normal CNS, but becomes measurable during the symptomatic phase of MS and EAE (Popko et al, 1997; Imitola et al., 2005). Administration of IFN-γ to MS patients enhances inflammation in the CNS and exacerbates clinical symptoms, and treatment of patients with an IFN-γ antibody alleviates disability progression (Panitch et al., 1987; Skurkovich et al., 2001). Transgenic mice that ectopically express IFN-γ in the CNS during development display oligodendrocyte loss and myelin abnormalities (Corbin et al., 1996; LaFerla et al., 2000; Lin et al., 2005). A recent study has showed CNS delivery of IFN-γ specifically at the recovery stage of EAE impairs the clinical recovery of EAE and severely suppresses oligodendrocyte regeneration and remyelination (Lin et al., 2006). In contrast, mice with a mutation in either the gene encoding IFN-γ or IFNγR remain susceptible to EAE and in fact develop EAE with higher morbidity and mortality (Ferber et al., 1996; Willenborg et al., 1996). Moreover, it has been shown that CNS delivery of IFN-γ before EAE onset ameliorated the disease severity and prevented oligodendrocyte loss, demyelination and axonal damage (Furlan et al., 2001; Lin et al., 2007). Although evidence has suggested that IFN-γ exerts a direct effect on oligodendrocytes through the JAKs-STAT1 pathway (Agresti et al., 1998; Balabanov et al., 2006; Emery et al., 2006), the signal transduction pathways responsible for the paradoxical effects of IFN-γ in immune-mediated demyelination diseases remain unresolved. It is known that IFN-γ exerts its effects on cells through the STAT1 pathway or STAT1-independent pathways (Ramana et al. 2002; Gough et al., 2008). In this study, we demonstrate that both the STAT1 pathway and STAT1-independent pathways participate in the detrimental effects of IFN-γ on the myelination process. Using a genetic approach, we found that transgenic mice expressing IFN-γ in the CNS on a STAT1 null background display minor tremor and ataxia phenotypes, which are considerably milder compared to IFN-γ-expressing mice on a STAT1 wild type background. We further found that the upregulation of the STAT1-dependent, IFN-γ responsive genes was abolished in the CNS of IFN-γ-expressing mice on a STAT1 null background, but that the expression of other IFN-γ-responsive genes was increased in these mice. Moreover, we found that STAT1 deletion blocked oligodendrocyte loss and attenuated hypomyelination in response to IFN-γ. While STAT1 deletion diminished the reduction of myelinated axons in the CNS of IFN-γ-expressing mice, the reduction of the thickness of myelin sheaths in these mice was not affected by STAT1 deletion. Interestingly, moderate hypomyelination observed in the CNS of IFN-γ-expressing mice on a STAT1 null background correlated well with a mild behavioral phenotype exhibited by these mice.
While a recent report demonstrated that IFN-γ induced oligodendrocyte progenitor apoptosis through the STAT1 pathway in vitro (Wang et al., 2010), we did not find that the presence of IFN-γ starting on P12 significantly changed the numbers of PDGF-α receptor positive oligodendrocyte progenitor in the CNS of 21-day-old IFN-γ-expressing mice (data not shown). Since the proliferation of oligodendrocyte progenitors is largely complete and robust myelin is occurring in the CNS at P12, it is possible that the induction of IFN-γ is too late to alter the numbers of oligodendrocyte progenitor in these mice. Moreover, although we have shown that the numbers of apoptotic oligodendrocyte are significantly increased in the CNS of IFN-γ expressing mice on a C57BL/6; Swiss Webster hybrid background (Lin et al, 2005), we did not find any apoptotic oligodendrocytes in the CNS of IFN-γ-expressing mice on a C57BL/6; 129S6/SvEv hybrid background in this study. Nevertheless, the presence of IFN-γ significantly reduced oligodendrocyte numbers in the CNS of transgenic mice on a C57BL/6; Swiss Webster hybrid background (Lin et al, 2005) and on a C57BL/6; 129S6/SvEv hybrid background (Figure 2). It is known that microglia/macrophages are responsible for the removal of dead oligodendrocytes and myelin debris in various demyelination diseases (Glezer et al., 2007). Therefore, the lack of apoptotic oliogodendrocytes in the CNS of IFN-γ-expressing mice on a C57BL/6; 129S6/SvEv hybrid background likely reflects that IFN-γ-activated microglia/macrophages remove dead oligodendrocytes more efficiently in mice on a C57BL/6; 129S6/SvEv hybrid background than mice on a C57BL/6; Swiss Webster hybrid background.
It is interesting that STAT1 deletion did not alleviate the reduction of the thickness of myelin sheaths induced by IFN-γ. Clearly, this data demonstrated that STAT1-independent pathways activated by this cytokine interfered with the myelination process. Wang et al. (2004) have demonstrated that the presence of IFN-γ in the CNS increases not only phosphorylated STAT1, but also phosphorylated STAT3 and phosphorylated STAT5. Activation of STAT3 or STAT5 may contribute to the detrimental effects of IFN-γ on the myelination process in STAT1 null mice. On the other hand, it has been shown that phosphorylated JAKs activate Fyn in the presence of IFN-γ (Uddin et al., 1997). Several lines of evidence have shown that the Fyn pathway plays an important role in oligodendrocyte migration, differentiation and myelination (Sperber et al., 2001; Goto et al., 2008; Laursen et al., 2009). Moreover, we showed here that inflammatory response induced by IFN-γ in the CNS was abolished in STAT1 null mice. Take together, it is possible that dysregulation of the Fyn pathway in oligodendrocyte leads to the reduction of the thickness of myelin sheaths in CNS of IFN-γ expressing mice on a STAT1 null background. Nevertheless, the degree to which the STAT3 pathway, the STAT5 pathway or the Fyn pathway plays a role in IFN-γ-induced hypomyelination remains to be determined.
The pathological hallmarks of MS include inflammation, oligodendrocytes loss, demyelination, and axonal degeneration (Buntinx et al., 2002; Frohman et al., 2006). Regeneration of oligodendrocytes and subsequent remyelination is necessary to restore neurological function in MS patients (Franklin, 2002; Brück et al., 2003). IFN-γ is thought to be a major contributing factor to remyelinating oligodendrocyte apoptosis and remyelination failure in MS demyelinated lesions (Sun et al., 2004; Lin et al., 2006; Zawadzka and Franklin, 2007). Several lines of evidence have suggested that IFN-γ inhibits remyelination in the demyelinated lesions through enhancing inflammatory response (Popko et al., 1997; Renno et al., 1998; Buntinx et al., 2002; Sun et al., 2004). In addition, a recent study showed that the deleterious effects of IFN-γ on remyelination in the demyelinated lesions are mediated at least in part by ER stress in remyelinating oligodendrocytes (Lin et al., 2006). We showed here that the STAT1 deletion diminishes inflammatory response and ER stress in oligodendrocytes in the CNS of IFN-γ expressing mice. Moreover, IFN-γ-induced oligodendrocyte loss and reduction of myelinated axons are abolished in STAT null mice. These data suggest that therapeutic strategies that block the STAT1 pathway could suppress inflammatory response and alleviates ER stress in remyelinating oligodendrocytes in MS demyelinated lesions, resulting in an enhanced oligodendrocyte regeneration and remyelination. Nevertheless, STAT1 null mice are highly susceptible to EAE and develop more severe EAE (Bettelli et al., 2004). Thus, the potential beneficial effects of STAT1 blockage on the remyelination process needs to be carefully evaluated in immune-mediated demyelination diseases.
In summary, we have demonstrated that both the STAT1 pathway and STAT1-independent pathways are important in the detrimental effects of IFN-γ on the myelination process in the CNS. The STAT1 pathway is necessary for IFN-γ-induced oligodendrocyte loss, the reduction of myelinated axons and inflammatory response, whereas STAT1-independent pathways are involved in IFN-γ-induced reduction of the thickness of myelin sheaths. Our work suggests that STAT1 blockage could promote oligodendrocyte regeneration and remyelination in immune-mediated demyelinated lesions.
Acknowledgments
Grant information: This work was supported by grants TA 3026-A-1 (to Dr. Wensheng Lin) from the National Multiple Sclerosis Society and NS 034939 (to Dr. Brian Popko) from the National Institutes of Health.
The authors are grateful to Dr. Brian Popko for the generous support for this study. All the mice are generated at Dr. Brian Popko’s laboratory at The University of Chicago. This work was supported by grants NS 034939 (to Dr. Brian Popko) from the National Institutes of Health and TA 3026-A-1 (to Dr. Wensheng Lin) from the National Multiple Sclerosis Society.
References
- Agresti C, Bernardo A, Del Russo N, Marziali G, Battistini A, Aloisi F, Levi G, Coccia EM. Synergistic stimulation of MHC class I and IRF-1 gene expression by IFN-gamma and TNF-alpha in oligodendrocytes. Eur J Neurosci. 1998;10:2975–2983. doi: 10.1111/j.1460-9568.1998.00313.x. [DOI] [PubMed] [Google Scholar]
- Alsayed Y, Uddin S, Ahmad S, Majchrzak B, Druker BJ, Fish EN, Platanias LC. IFN- gamma activates the C3G/Rap1 signaling pathway. J Immunol. 2000;164:1800–1806. doi: 10.4049/jimmunol.164.4.1800. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Strand K, Kemper A, Lee JY, Popko B. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J Neurosci. 2006;26:5143–5152. doi: 10.1523/JNEUROSCI.0737-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- Brück W, Kuhlmann T, Stadelmann C. Remyelination in multiple sclerosis. J Neurol Sci. 2003;206:181–185. doi: 10.1016/s0022-510x(02)00191-0. [DOI] [PubMed] [Google Scholar]
- Buntinx M, Stinissen P, Steels P, Ameloot M, Raus J. Immune-mediated oligodendrocyte injury in multiple sclerosis: molecular mechanisms and therapeutic interventions. Crit Rev Immunol. 2002;22:391–424. [PubMed] [Google Scholar]
- Chang YJ, Holtzman MJ, Chen CC. Interferon-gamma-induced epithelial ICAM-1 expression and monocyte adhesion. Involvement of protein kinase C-dependent c-Src tyrosine kinase activation pathway. J Biol Chem. 2002;277:7118–7126. doi: 10.1074/jbc.M109924200. [DOI] [PubMed] [Google Scholar]
- Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci. 1996;7:354–370. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]
- Emery B, Butzkueven H, Snell C, Binder M, Kilpatrick TJ. Oligodendrocytes exhibit selective expression of suppressor of cytokine signaling genes and signal transducer and activator of transcription 1 independent inhibition of interferon-gamma-induced toxicity in response to leukemia inhibitory factor. Neuroscience. 2006;137:463–472. doi: 10.1016/j.neuroscience.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Furlan R, Brambilla E, Ruffini F, Poliani PL, Bergami A, Marconi PC, Franciotta DM, Penna G, Comi G, Adorini L, Martino G. Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J Immunol. 2001;167:1821–1829. doi: 10.4049/jimmunol.167.3.1821. [DOI] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147:867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Goto J, Tezuka T, Nakazawa T, Sagara H, Yamamoto T. Loss of Fyn tyrosine kinase on the C57BL/6 genetic background causes hydrocephalus with defects in oligodendrocyte development. Mol Cell Neurosci. 2008;38:203–212. doi: 10.1016/j.mcn.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19:383–394. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Gysemans C, Callewaert H, Overbergh L, Mathieu C. Cytokine signalling in the beta-cell: a dual role for IFNgamma. Biochem Soc Trans. 2008;36:328–333. doi: 10.1042/BST0360328. [DOI] [PubMed] [Google Scholar]
- Imitola J, Chitnis T, Khoury SJ. Cytokines in multiple sclerosis: from bench to bedside. Pharmacol Ther. 2005;106:163–177. doi: 10.1016/j.pharmthera.2004.11.007. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Sugarman MC, Lane TE, Leissring MA. Regional hypomyelination and dysplasia in transgenic mice with astrocyte-directed expression of interferon-gamma. J Mol Neurosci. 2000;15:45–59. doi: 10.1385/JMN:15:1:45. [DOI] [PubMed] [Google Scholar]
- Laursen LS, Chan CW, ffrench-Constant C. An integrin-contactin complex regulates CNS myelination by differential Fyn phosphorylation. J Neurosci. 2009;29:9174–9185. doi: 10.1523/JNEUROSCI.5942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JR, Cross AH. A little stress is good: IFN-gamma, demyelination, and multiple sclerosis. J Clin Invest. 2007;117:297–299. doi: 10.1172/JCI31254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129:1306–1318. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- Lin W, Kemper A, McCarthy KD, Pytel P, Wang JP, Campbell IL, Utset MF, Popko B. Interferon-gamma induced medulloblastoma in the developing cerebellum. J Neurosci. 2004;24:10074–10083. doi: 10.1523/JNEUROSCI.2604-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12:379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361–33368. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- Popko B, Corbin JG, Baerwald KD, Dupree J, Garcia AM. The effects of interferon- gamma on the central nervous system. Mol Neurobiol. 1997;14:19–35. doi: 10.1007/BF02740619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- Renno T, Taupin V, Bourbonnière L, Verge G, Tran E, De Simone R, Krakowski M, Rodriguez M, Peterson A, Owens T. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol Cell Neurosci. 1998;12:376–389. doi: 10.1006/mcne.1998.0725. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20– 28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Shresta S, Sharar KL, Prigozhin DM, Snider HM, Beatty PR, Harris E. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary dengue virus infection in mice. J Immunol. 2005;175:3946–3954. doi: 10.4049/jimmunol.175.6.3946. [DOI] [PubMed] [Google Scholar]
- Skurkovich S, Boiko A, Beliaeva I, Buglak A, Alekseeva T, Smirnova N, Kulakova O, Tchechonin V, Gurova O, Deomina T, Favorova OO, Skurkovic B, Gusev E. Randomized study of antibodies to IFN-gamma and TNF-alpha in secondary progressive multiple sclerosis. Mult Scler. 2001;7:277–284. doi: 10.1177/135245850100700502. [DOI] [PubMed] [Google Scholar]
- Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sredni-Kenigsbuch D. TH1/TH2 cytokines in the central nervous system. Int J Neurosci. 2002;112:665–703. doi: 10.1080/00207450290025725. [DOI] [PubMed] [Google Scholar]
- Steinman L. Blockade of gamma interferon might be beneficial in MS. Mult Scler. 2001;7:275–276. doi: 10.1177/135245850100700501. [DOI] [PubMed] [Google Scholar]
- Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- Sun D, Newman TA, Perry VH, Weller RO. Cytokine-induced enhancement of autoimmune inflammation in the brain and spinal cord: implications for multiple sclerosis. Neuropathol Appl Neurobiol. 2004;30:374–384. doi: 10.1111/j.1365-2990.2003.00546.x. [DOI] [PubMed] [Google Scholar]
- Uddin S, Sher DA, Alsayed Y, Pons S, Colamonici OR, Fish EN, White MF, Platanias LC. Interaction of p59fyn with interferon-activated Jak kinases. Biochem Biophys Res Commun. 1997;235:83–88. doi: 10.1006/bbrc.1997.6741. [DOI] [PubMed] [Google Scholar]
- Wang J, Lin W, Popko B, Campbell IL. Inducible production of interferon-gamma in the developing brain causes cerebellar dysplasia with activation of the Sonic hedgehog pathway. Mol Cell Neurosci. 2004;27:489–496. doi: 10.1016/j.mcn.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ren Z, Tao D, Tilwalli S, Goswami R, Balabanov R. STAT1/IRF-1 signaling pathway mediates the injurious effect of interferon-gamma on oligodendrocyte progenitor cells. Glia. 2010;58:195–208. doi: 10.1002/glia.20912. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- Zawadzka M, Franklin RJ. Myelin regeneration in demyelinating disorders: new developments in biology and clinical pathology. Curr Opin Neurol. 2007;20:294–298. doi: 10.1097/WCO.0b013e32813aee7f. [DOI] [PubMed] [Google Scholar]