

Abstract
Improvements in the treatment of primary biliary cirrhosis may depend upon dissection of mechanisms that determine recruitment of mononuclear cells to intralobular bile ducts, including the role of the chemokine-adhesion molecule CX3CL1 (fractalkine). We submit that there are unique interactions between intrahepatic biliary epithelial cells (BEC), endothelial cells (EC), liver sinusoidal endothelial cells (LSEC) and liver infiltrating mononuclear cells (LMC), and that such interactions will in part dictate the biliary-specific inflammatory response. To address this, we studied fresh explanted liver from pre-transplant patients with PBC and with inflammatory liver disease due to viral infection, designated as ‘disease controls’, and biopsy material from patients with a discrete liver tumor, ‘normal controls’. Using this clinical material, we isolated and stimulated BEC, EC, LSEC, and LMC with a panel of toll-like receptor (TLR) ligands. We also studied the interactions of these cell populations with LMC with respect to adhesion capability and production of TNF-α. Finally, we used fresh biopsy samples to evaluate mononuclear cells around intrahepatic biliary ductules using mAb specific to CD68 or CD154, markers for monocytes/macrophages and activated T cells respectively. We report herein that there are common properties of EC, LSEC and BEC, whether derived from PBC or viral hepatitis, but there are also significant differences, particularly in the potential in PBC for LMC to adhere to EC and BEC, and to produce TNF-α; such properties were associated with augmented CX3CL1 production by BEC from PBC liver. The processes defined herein suggest potential novel biotherapies for biliary specific inflammation.
Keywords: CX3CL1, biliary epithelial cells, endothelial cells, liver sinusoidal endothelial cells, toll-like receptors, liver infiltrating mononuclear cells, monocytes, TNF-α, primary biliary cirrhosis
Previous work in our laboratory on inflammatory chemotaxis in PBC has demonstrated participation of various chemokine ligands (1), and studies in other laboratories have implicated the chemokine CX3CL1 (fractalkine) in inflammatory liver disease (2, 3). Hence we set out to examine the contribution of CX3CL1 (4–9) to the bile duct destruction of PBC. Our previous findings in PBC indicated that CX3CL1 is elevated in serum concurrent with increased expression of the CX3CR1 receptor in liver mononuclear cells (LMC) (10), so leading us to posit that CX3CL1 could indeed be critical for the generation and persistence of the portal lymphocytic infiltration in PBC. We have herein taken advantage of our ability to isolate pure populations of multiple intrahepatic cell types including endothelial cells (EC), liver sinusoidal endothelial cells (LSEC) and BEC to directly study the interaction of CX3CL1-producing cells with LMC. We should note that several non-professional immunocompetent cells produce chemokines in response to ligands for toll-like receptors (TLR) (11, 12). Here we have evaluated CX3CL1 production from EC, LSEC and BEC exposed to TLR ligands and report that EC produced high amounts of CX3CL1 using one or another of several TLR ligands while LSEC never produced CX3CL1 with any ligand; BEC produced CX3CL1 on exposure to autologous LMC, TNF-α and a TLR3 ligand. In the process of simplifying the production system of CX3CL1 from BEC, we found that TLR3-stimulated BEC produced CX3CL1 after direct contact with TLR4-stimulated autologous monocytes. In summary, our data indicate that CX3CL1 and TNF-α, which are induced by TLR ligands, participate in processes that lead to disease-specific recruitment of lymphoid cells into the portal tracts of the liver and thereby to the characteristic chronic non-suppurative destructive cholangitis of PBC. This new knowledge on the mechanisms of lymphocyte homing and recruitment induced by innate immunity and, potentially, the ability to inhibit abnormal chemoattractant homing is a fertile area for future therapeutic intervention in PBC.
Materials and Methods
Subjects
There were 21 explanted livers studied, derived from 9 patients with PBC, 3 with hepatitis B virus infection, 7 with hepatitis C virus infection, and 2 with primary sclerosing cholangitis (PSC). All patients had end-stage liver cirrhosis without signs of other acute liver injury from an unrelated cause. The diagnosis of PBC was based on established criteria (13) and all such patients had a positive test for serum anti-mitochondrial antibodies (13). In addition, and for purposes of comparison in further nested substudies, we studied liver from an additional 5 non-cirrhotic PBC patients for analysis of liver infiltrating mononuclear cells, as well as ‘normal control’ liver from 4 patients undergoing resection of liver for isolated metastatic tumors, for concurrent analysis of intrahepatic biliary epithelial cells (BEC), endothelial cells (EC) and liver sinusoidal endothelial cells (LSEC). For studies on immunohistochemistry, fresh samples from wedge biopsies were available from 12 patients with PBC, 15 with hepatitis C and 7 with an hepatic neoplasm but normal surrounding liver. All such samples were studied after informed consent of the donor, and all experimental protocols were approved by the Research Ethics Committee of Kyushu University and the University of California at Davis.
Isolation of intrahepatic biliary epithelial cells (BEC), endothelial cells (EC), liver sinusoidal endothelial cells (LSEC) and liver-infiltrating mononuclear cells (LMC)
The isolation of nearly pure cell subpopulations from liver was achieved using methods previously described in detail (14). Liver specimens were first digested with 1 mg/ml of collagenase type I (Sigma-Aldrich, Tokyo, Japan). Cells from the digested tissue were gradient-separated to obtain LMC (15) that were cultured overnight, the adherent cell population was maintained in culture until there was full confluence, usually by day 14, and the non-adherent cell populations were stored in liquid nitrogen. Further, in a nested study, fresh LMC from non-cirrhotic PBC liver were obtained from liver biopsies (needle, n=3 and surgical, n=2) that were cut into smaller fragments and digested with collagenase type I for 20 minutes. Dissociated cells were filtered through a 150-μm mesh and separated by Ficoll centrifugation, and then immediately used for study of TNF-α production by LMC, as described below.
BEC were separated from adherent cells using CD326 (EpCAM) MicroBeads specific for epithelial cells as previously described (14). The cell phenotype was verified by immunohistochemistry with antibodies against cytokeratins 7 and 19 (Dako, Glostrup, Denmark); a cell purity exceeding 90% was deemed acceptable. The viability of all cells for each of the experiments of greater than 95% was established by trypan blue exclusion.
EC were separated from adherent cells using CD31 microbeads specific for endothelial cells. Since LSEC do not express CD31 (16), but are positive for CD105 (17), these could be separated from both BEC and EC after separation of adherent cells using CD105 microbeads. LSEC were isolated using a density gradient (16–18). We confirmed that LSEC thus isolated were CD31 negative and CD105 positive. EC and LSEC were cultured with endothelial specific medium (HuMedia-EG2) (14). For the two cases of PSC the limited size of the liver specimens provided to us precluded isolation of EC and LSEC and thus limited the data available. For each cell population the yield of cells differed between samples, but all tissues were handled identically and the total number of cells used in each assay was standardized. Cells were studied in early cultures, at passages 4–6, to obviate the potential loss of phenotype after prolonged in vitro culture.
CX3CL1 production
To study CX3CL1 production, cells (1x105/400 μl in 24 well plates) were initially cultured for 48 hours in the presence of one or another of multiple TLR ligands; these included lipoteichoic acid (LTA), poly I:C, purified lipopolysaccharide (LPS), flagellin, CL-097, ODN2216 and ODN2006, all purchased from Invitrogen (San Diego, CA). The optimal concentrations were 2–10 μg/ml. Then BEC (1x105/200 μl in 24 well plates) and autologous LMC (2x106/200 μl in 24 well plates) were cultured for 24 hours to study CX3CL1 production in the presence of one or another of various ligands for TLR, and either IFN-γ or TNF-α, at final concentrations of 200U/ml or 0.1 μg/ml respectively. The supernatants from the cultured media with different TLR stimuli were analyzed for CX3CL1 production by sandwich ELISA kits (R&D Systems, Minneapolis, MN), using a combination of unlabeled and biotin- or enzyme-coupled monoclonal antibody to CX3CL1, for which the lower level of detectability was 200pg/ml.
In what we designate as ‘transwell’ experiments, BEC were grown in the bottom of wells, and LMC were added to the 24 well plate filter inserts with a pore size of 0.4 μmol/L (BD Biosciences, Bedford, MA). Blockage of cellular interactions was achieved with various antibodies including anti-CD154 (Ancell, Bayport, MN), anti-HLA class I, and anti-class II, HLA DP, DQ and DR (BD Biosciences, Bedford, MA). Each antibody was used at a predetermined optimal concentration of 5–40μg/ml. Briefly, anti-HLA class I, anti HLA DP, DQ and DR for BEC and anti-CD154 antibody for LMC were pre-incubated overnight, and cells were washed twice; BEC and LMC were co-cultured in the presence of poly I:C and TNF-α. In selected nested experiments, T cells, monocytes, NKT, NK or myeloid dendritic cells (mDCs) were separated as described and pre-treated with LPS (final concentration: 10μg/ml) for 24 hours, washed x3; poly I:C pre-treated BEC and 2x106/200μl of T cells, monocytes, NKT or NK cells, or mDCs were co-cultured in each well of a 24 well plate. In other selected nested experiments, BEC were pre-treated with poly I:C or poly I:C and LPS (final concentration:10μg/ml) for 24 hours, washed well, and then monocytes were added to the BEC preparation. Anti-TNF-α antibody or an irrelevant control antibody (final concentration, 10μg/ml) was used to confirm the role of TNF-α in the production of CX3CL1 from BEC.
Cell adhesion assay
Assays were performed for adhesion between EC, BEC or LSEC and LMC. Confluent monolayers of EC, BEC or LSEC were cultured in 24-well plates (1x105cells/well) and then overlaid with LMC (2x106/well) in the presence of poly I:C (final concentration 10μg/ml) and TNF-α (final concentration 0.1μg/ml) for 24 hours as described above. Non-adherent cells were removed by gentle rinsing and wells were washed 4 times. Adherent LMC were fixed and stained with Diff-Quick Stain, and counted as the number of adherent cells in ten random high power fields (HPF) as previously described (19).
Isolation of T cells, monocytes, NK cells, myeloid dendritic cells and NKT cells from LMC
T cells among LMC were separated using a Pan T cell isolation kit II. Non-T cells, i.e. B cells, NK cells, DCs, monocytes, granulocytes and erythroid cells, were indirectly magnetically labeled using a cocktail of biotin-conjugated antibodies against CD14, CD16, CD19, CD36, CD56, CD123, Glycophorin A and anti-biotin microbeads. Isolation of purified T cells was achieved by depletion of magnetically labeled cells by separation over a MACS column which was placed in the magnetic field of a MACS Separator; a purity of CD3+ T cells of >90% was confirmed by flow cytometry. Monocytes were separated with a monocyte isolation kit. Non-monocytes were indirectly magnetically labeled with a cocktail of biotin-conjugated monoclonal antibodies against CD3, CD7, CD16, CD19, CD56, CD123 and Glycophorin A, and anti-biotin microbeads. Isolation of monocytes was achieved by depletion of magnetically labeled cells; a purity of CD14+ monocytes of >90% was confirmed by flow cytometry. NK cells were separated with an NK isolation kit. Non-NK cells were indirectly magnetically labeled with a cocktail of biotin-conjugated antibodies against lineage-specific antigens, and anti-biotin microbeads. Isolation of NK cells was achieved by depletion of magnetically labeled cells; a purity of CD56+ NK cells of >90% was confirmed by flow cytometry. mDCs (CD1c+) were separated with an mDC isolation kit performed by two magnetic separation steps. In the first step, CD1c-expressing B cells were magnetically labeled with CD19 microbeads and, subsequently, depleted magnetically. In the second step, CD1c+ mDCs in the B cell-depleted flow-through fraction were indirectly magnetically labeled with CD1c-biotin and anti-biotin microbeads. Upon separation, the labeled CD1c+ mDCs were retained within the column and eluted after removing the column from the magnetic field. A purity of CD1c+ CD19- mDCs of >80% was confirmed by flow cytometry. NKT cells were separated with an NKT isolation kit. The isolation of NKT cells was performed in two magnetic separation steps. In the first step, NK cells and monocytes were indirectly magnetically labeled using a cocktail of biotin-conjugated antibodies and anti-biotin microbeads. The labeled cells were subsequently depleted by separation over a MACS Column. In the second step, CD3+CD56+ NKT cells were directly labeled with CD56 microbeads and isolated by positive selection from the pre-enriched NKT cell fraction. Upon separation, the labeled CD56+ cells were retained within the column and eluted after removing the column from the magnetic field. A purity of CD3+ CD56+ NKT cells of >80% was confirmed by flow cytometry.
TNF-α production
Cell populations (2x104/200μl in 96 well plates) were cultured for 48 hours in the presence of the TLR ligands described above at 10 μg/ml. Supernatants were analyzed for TNF-α production by sandwich ELISA kits (R&D Systems), using a combination of unlabeled and biotin- or enzyme-coupled monoclonal antibody to TNF-α. In all instances, known positive and known negative controls were used throughout and all assays were performed in triplicate.
Immunohistochemical stainings of liver specimens
Fresh liver specimens from 12 patients with PBC, 15 with hepatitis C infection and 7 with discrete intrahepatic tumors (unaffected non-tumor bearing liver) were fixed in 10% neutral-buffer formalin and snap frozen in OCT compound (Miles, Inc., Elkhart, IN). Deparaffinized and rehydrated sections, and frozen sections, were used for immunostaining for cell surface markers, CD68 (expressed particularly on monocytes/macrophages) and CD154 (expressed particularly on activated T cells). Endogenous peroxidase was blocked in normal goat serum diluted 1:10 (Vector Lab, Burlingame, CA) for 20 min; CD68 and CD154 were diluted 1:100 (Dako) and immunostaining was performed on coded sections and interpreted by a “blinded” qualified liver pathologist.
Statistical analysis
All experiments were performed in triplicate and data points shown are means of results of these triplicates. Comparisons between the points for data items were expressed as mean ± standard deviation (SD), and the significance of differences was determined by Student’s t test. All analyses were two-tailed and p values <0.05 were considered significant. Statistical analyses were performed using Intercooled Stata 8.0 (Stata Corp, College Station, TX).
Results
Production of CX3CL1 by populations of liver cells
We assessed the production of CX3CL1 by isolated populations of liver cells in PBC and control patients after stimulation by different TLR ligands. With endothelial cells, production was induced by LTA, poly I:C, LPS and flagellin, but not by CL-097, ODN2216 or ODN2006. Levels of CX3CL1 (ng/ml) in PBC versus non-PBC disease controls were for LTA 1.7±0.9 vs. 1.6±0.9 (n.s.), poly I:C 7.8±1.0 vs. 7.9±1.7 (n.s.), LPS 4.9±0.9 vs. 5.1±1.0 (n.s.) and flagellin 0.5±0.2 vs. 0.6±0.2 (Figure 1A). Levels of CX3CL1 (ng/ml) in normal liver controls included values of LTA 1.8±0.6, poly I:C 8.0±1.5, LPS 4.9±1.8 and flagellin 0.6±0.4 (Figure 1A); these differences were not significant. Although activated LSEC mediate CX3CL1 shedding and release of chemotactic peptides (20), with LSEC and BEC, neither of these cell types produced CX3CL1 after stimulation with any of the TLR ligands used (data not shown) in PBC, non-PBC disease controls and normal liver controls.
Figure 1.
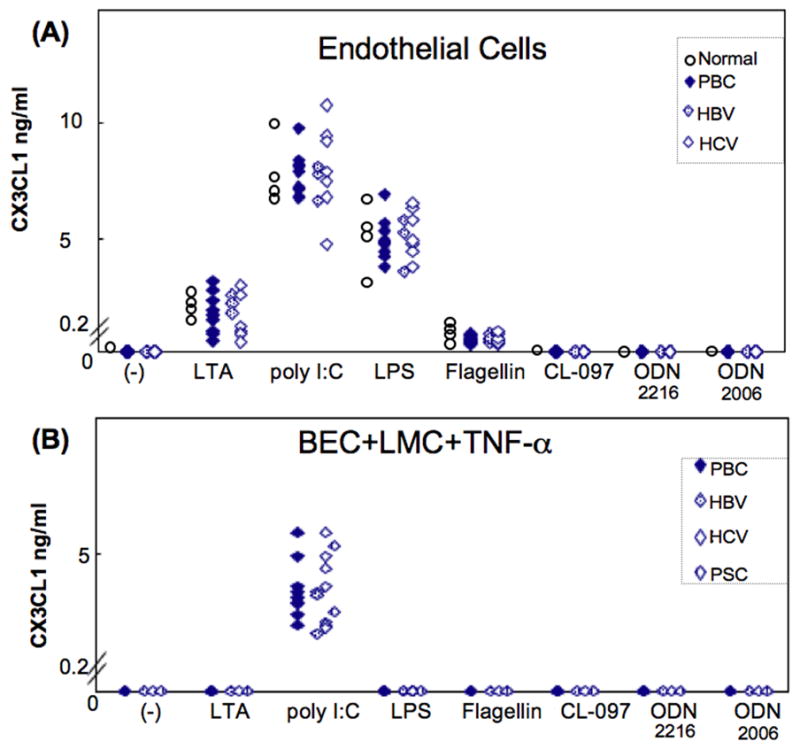
(A) CX3CL1 is produced by endothelial cells (EC) from PBC, normal livers (n=4) and disease control livers (3 HBV, 7 HCV) exposed to TLR ligands. Differences between PBC and controls were not significant. LSEC did not produce CX3CL1 with any TLR ligand (data not shown). (B) CX3CL1 is produced by BEC exposed to TLR ligands in the presence of autologous LMC and TNF-α. BEC production of CX3CL1 required the presence of LMC and TNF-α, and was elicited only with poly I:C. Differences between PBC and disease control livers were not significant. BEC did not produce CX3CL1 when exposed only to LMC or LMC with IFN-γ (data not shown). Open circle: normal. Black diamond: PBC. Dashed diamond: HBV. Open diamond: HCV. Hatched diamond: PSC.
Since previous reports demonstrated that BEC produce chemokines in co-culture with autologous LMC (1), and since TNF-α and IFN-γ enhance CX3CL1 production from mucosal endothelial cells (21), we used an LMC and BEC co-culture system with or without the addition of TNF-α or IFN-γ. No production of CX3CL1 by BEC with LMC was induced with any TLR ligands (data not shown). However, BEC in the presence of LMC, with TNF-α but not with IFN-γ, did together with poly I:C produce CX3CL1, 3.7±0.1 vs.3.5±1.2 (ng/mL); again, the difference between PBC and controls was not significant (Figure 1B). Thus the presence of TNF-α is critical for CX3CL1 production by BEC. The possibility that lymphocytes produced CX3CL1 (22) was excluded by irradiation of LMC which did not significantly alter the results (data not shown). Also, LMC without BEC never produced CX3CL1 whatever TLR ligand was used for stimulation, nor after addition of IFN-γ or TNF-α. In the case of non-diseased controls, we were unable to study CX3CL1 production from BEC with LMC and TNF-α, as sufficient LMC were not available.
Production of CX3CL1 by BEC requires direct contact with LMC via CD40/CD154 interaction
BEC did not produce CX3CL1 on co-culture with poly I:C-pre-treated LMC in the presence of TNF-α, illustrated by representative data for one PBC liver (Figure 2A), and indicating that BEC but not LMC require poly I:C stimulation for production of CX3CL1. Such production decreased markedly when the BEC and LMC populations were separated by a filter in a transwell system (Figure 2B). We assessed the functional effects of CD40, HLA class I and HLA class II molecules on BEC by testing the capacity of blocking antibodies to CD154 and HLA molecules to suppress production of CX3CL1 by BEC. Production of CX3CL1 by BEC was significantly decreased when CD40 on BEC was blocked from interacting with CD154 on LMC: representative data are shown in Figure 2C. Having shown that LMC and TNF-α are critically required for production of CX3CL1 by BEC, we next examined in detail the role of LMC and TNF-α production.
Figure 2.

CX3CL1 is produced by BEC in the presence of TNF-α and poly I:C with autologous LMC. Representative data is shown from liver of one PBC patient. (A) Poly I:C pre-treatment of BEC plus autologous LMC in the presence of TNF-α enabled production by BEC of CX3CL1 with LMC whereas poly I:C treatment of LMC did not. Thus BEC but not LMC is required for poly I:C. (B) CX3CL1 production by BEC in the presence of TNF-α and poly I:C required direct cell contact with autologous LMC, since CX3CL1 production under transwell conditions with autologous LMC was minimal. (C) CX3CL1 production by BEC in the presence of TNF-α and poly I:C with autologous LMC was significantly inhibited by a mAb specific to CD154 (a-154) but not by mAbs specific to MHC class I or II (a-MHC I, II) (*p < 0.05). Thus interaction between CD40 from BEC and CD154 from LMC facilitates CX3CL1 production.
Assay for adherence of LMC to EC or LSEC
LMC in the presence of poly I:C and TNF-α adhered to EC and BEC and, notably, the number of such adherent LMC from PBC liver exceeded that for controls cases, 394±94 vs. 116±45 (p<0.01) for EC and 180±63 vs. 65±40 (p<0.01) for BEC. However, only very few LMC adhered to LSEC, whether from PBC liver 21±14 or controls 20±15 (p>0.05) (Figure 3).
Figure 3.
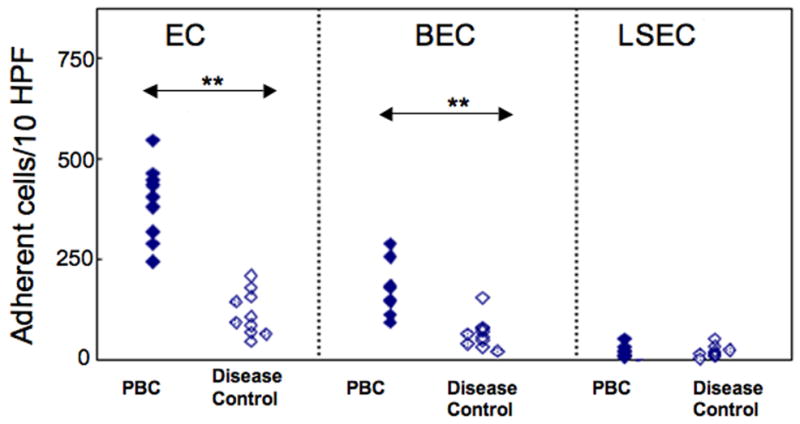
Autologous LMC adhesion assay using EC, BEC and LSEC. Adherent LMC were stained and counted in ten random high power fields. LMC from PBC livers adhered in greater numbers than did LMC from disease control livers, using either EC (**p<0.01) or BEC (**p<0.01) whereas LMC adhered only minimally to LSEC in all instances.
Production of TNF-α in the presence of LPS by LMC (T cell, Mo, NKT, NK and mDC)
The necessity of TNF-α for production by BEC of CX3CL1 led us to assess the source of available liver TNF-α. As shown in Figure 4, LMC produced TNF-α following stimulation with most TLR ligands, and values for PBC exceeded those for disease controls. The data for LTA were 751±163 vs. 547±138 pg/ml (p<0.05), for LPS 1699±253 vs. 1303±244 pg/ml (p<0.01) and for CL-097 956±188 vs. 726±154 pg/ml (p<0.05) (Figure 4). In the case of early non-cirrhotic PBC, only a limited quantity of LMC was available so that TNF-α production was measured only with or without LPS stimulation; here, TNF levels were 1825±334 pg/ml which did not differ significantly from cirrhotic PBC (p>0.05). There were however differences between non-cirrhotic PBC and cirrhotic disease controls (p<0.05) (Figure 4). We then determined which subpopulations of LPS-stimulated LMC produced TNF-α and, as shown in Figure 5, The data for PBC livers versus disease control livers were for monocytes 476±131 vs. 336±65 pg/ml (p<0.05), for NK cells 179±51 vs. 107±36 pg/ml (p<0.01) and for mDC 264±60 vs. 199±38 pg/ml (p<0.05). Thus the major but not exclusive source of TNF-α was monocytes, and production of TNF-α was relatively greater in PBC.
Figure 4.
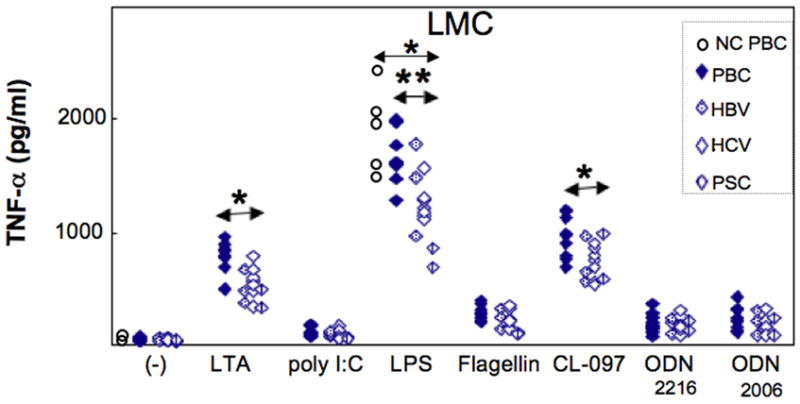
TNF-α production by LMC from PBC and control livers with different TLR ligand stimulation. Greater TNF-α responses were elicited by LTA, LPS and CL-097, and the TNF-α response for PBC and disease control livers was significantly greater for each of the above three TLR ligands LTA (p*<0.05), LPS (**p<0.01), CL-097 (*p<0.05). LMC from cirrhotic and non-cirrhotic PBC were compared and no differences were seen. Here, the open circle is non-cirrhotic PBC.
Figure 5.
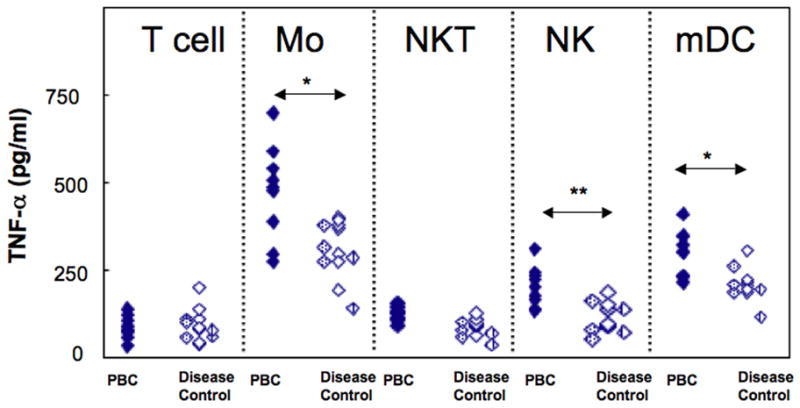
TNF-α production from different cell types under LPS stimulation using isolated cells from PBC and disease controls. From a starting population of LMC, separation was made of T cells, monocytes (Mo), NKT cells, NK cells and mDC. Monocytes elicited the highest TNF-α production among these populations, NK cells and mDC produced measurable TNF-α responses and T cell and NKT cell responses were minimal. TNF-α responses differed for PBC and disease control livers for Mo (*p<.05), NK cells (*p<0.01) and mDC (*p<0.05).
BEC pre-treated with Poly I:C produce CX3CL1 in presence of monocytes pre-treated with LPS
We have shown that direct contact of LMC, and TNF-α, were necessary for production by BEC of CX3CL1. Also, it is known that TLR4 ligands stimulate LMC to produce TNF-α. Accordingly we sought to ascertain which cell population among LMC is critical for CX3CL1 production by BEC. Our procedures included measurement of production of CX3CL1 by poly I:C pre-treated BEC, with LPS pre-treated MNCs of either T cells, monocytes, NKT, NK cells, or mDC (Figure 6). Even though NK and mDC did produce small amounts of TNF-α with LPS, production of CX3CL1 was rarely detectable when poly I:C-pre-treated BEC were co-cultured with LPS-pre-treated T cells, NKT or NK cells, or mDC: Figure 6A shows representative data for one PBC liver. On the other hand, CX3CL1 production was prolific when poly I:C pre-treated BEC were cultured with LPS pre-treated monocytes. Such production was not observed in the absence of LPS pre-treated monocytes, and the production was markedly inhibited after addition of anti-TNF-α (Figure 6B), indicating that LPS pre-treated monocytes provided the necessary direct contact, and TNF-α, for subsequent CX3CL1 production by BEC. Comparison of PBC and disease control livers showed that poly I:C pre-treated BEC from PBC livers produced relatively large amounts of CX3CL1 when cultured with LPS pre-treated monocytes, 2.1±0.5ng/ml vs. 1.3±0.4ng/ml p<0.01) (Figure 6C). Of note, in these experiments, only small amounts of CX3CL1 were produced from the two PSC livers.
Figure 6.
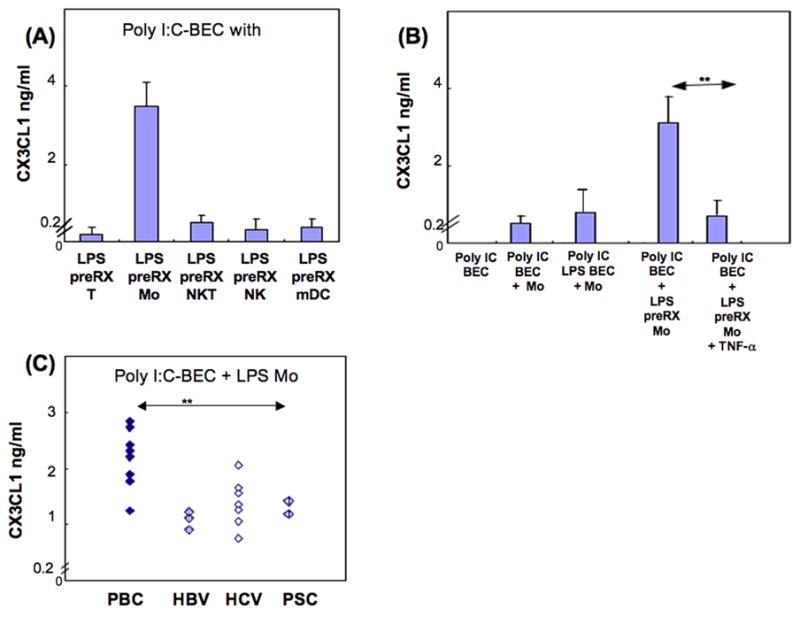
CXC3L1 production by BEC in the presence of monocytes: representative data from one PBC liver is shown in (A) and (B). (A) T cells, monocytes, NKT, NK or mDC were each pre-incubated with LPS for 24 hours and co-cultured with BEC that had been preincubated with poly I:C for 24 hours. Only monocytes showed augmented production of CX3CL1. (B) BEC pre-incubated with poly I:C or poly I:C and LPS were co-cultured with monocytes or LPS pre-incubated monocytes, as shown in 6A; anti-TNF-α abrogated the increased production by monocytes of CX3CL1 (**p < 0.01). (C) LPS pre-treatment of monocytes (Mo) induced a greater degree of CX3CL1 production by poly I:C pre-treated BEC from PBC livers than disease control livers (**p<0.01). Each of the TLR ligands was used at 10 μg/ml. Pre-treatment was performed for 24 hr and cells were washed twice before co-culture.
Monocytes around BEC in the liver
Finally we investigated the presence of monocytes around bile ducts in the liver by immunohistochemistry. Comparing livers of patients with PBC and those with hepatitis C (disease controls), CD68+ monocytes/macrophages were enriched in PBC, predominantly in the portal area (Figure 7A), as were CD154+ activated T cells around biliary ductules (Figure 7B), indicative of greater invasion of CD68 and CD154+ cells into portal areas of liver in patients with PBC compared to those with hepatitis C. Actual cell counts are shown in Table 1.
Figure 7.
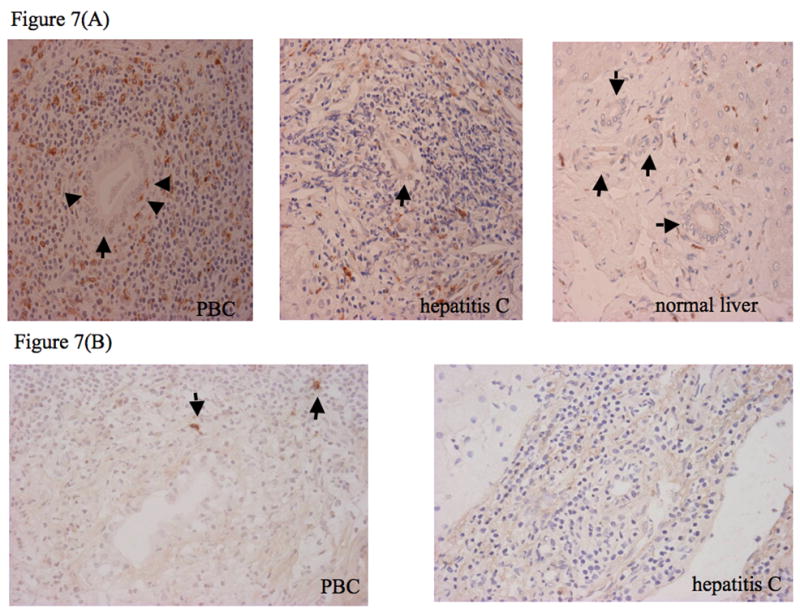
Immunohistochemical detection of CD68+ cells (mainly monocytes) and CD154+ cells (mainly activated T cells) in PBC liver, hepatitis C liver and normal liver. (A) Expression of CD68 on mononuclear cells around bile ducts. Mononuclear cells expressing CD68 (brown staining) are seen in the biliary epithelial layer and periductal tissue. In PBC, CD68+ cells are seen within the biliary epithelium (arrow heads) and are plentiful around the bile ducts (arrows), whereas in hepatitis C CD68+ cells are scattered, and in normal are rarely seen around bile ducts. (B) CD154 expression on mononuclear cells (arrows) around bile ducts is high in PBC liver, but weak on mononuclear cells around bile ducts in hepatitis C liver.
Table 1.
Immunohistochemical staining and counts of CD68+ and CD154− cells around bile ducts in liver sections from patients with PBC, chronic hepatitis C, and discrete liver tumor but otherwise normal liver.
| PBC (n=12) | Hepatitis C (n=15) | “Normal” liver (n=7) | |
|---|---|---|---|
| CD68 | 147.3+62.3* | 69.5+35.8 | 10.4+6.3 |
| CD154 | 3.4+0.8* | 1.0+0.9 | 0.5+0.4 |
In PBC, counts of CD68+ monocytes exceeded counts in hepatitis C and in “normal” liver (p<0.01, for both comparisons), as did counts of CD154+ activated T cells (p<0.01 for both comparisons).
Discussion
To facilitate understanding of the data herein, we have developed a schema, Figure 8, to reflect the chain of events amongst the liver subpopulations studied. We also note that the hypothesis that aberrant homing of T cell subsets are involved in the pathogenesis of PBC is based on earlier data in PSC (23). Samples from the study herein were primarily derived from end-stage disease, thus raising the issue of whether pathogenetic mechanisms that induce disease are overwhelmed by secondary immunological processes, including the contributions of fibrosis and extensive cholestasis. However, by reason of tissue access, this was a necessity. We should also note that prolonged culturing of EC, LSEC and BEC introduces the possibility of loss of cell differentiation which we attempted to obviate by studies of early cultures, passages 5–6. However, such reservations notwithstanding, we emphasize that the liver infiltrating mononuclear cells from PBC strongly induce production of CX3CL1 from BEC. In future studies these reservations could potentially be addressed by use of laser captured microdissection and in real-time analysis for study of site specific expression of messenger RNA from the relevant hepatic subpopulations. Indeed, a weakness of the data herein is the absence of completely normal non-diseased liver; such tissue is not readily available.
Figure 8.
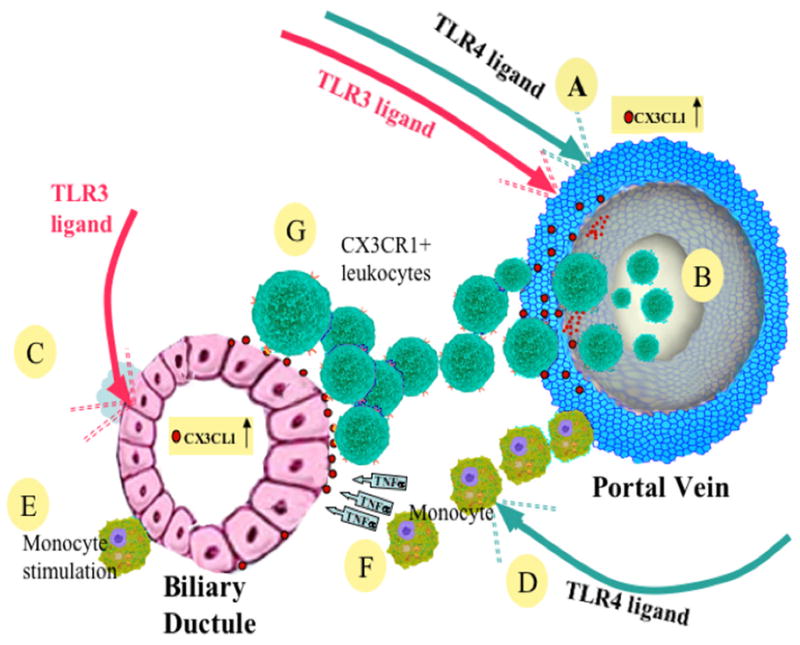
A diagrammatic representation of invasion of liver and destruction of bile ducts by CX3CR1+ cells. Shown are the pathways of activation by TLR 3 and 4 ligands; sources of CX3CR1 include vascular endothelial cells of the portal vein, the likely site of leukocyte entry. Upregulation of CX3CL1 on BEC is promoted by TLR 3 ligand, by activated monocytes via direct contact and by TNF-α secretion by monocytes. A chemotactically driven leukocyte-CX3CL1 interaction involving BEC promotes the destructive cholangitis of PBC. Events in detail are as follows. (A) Various TLR ligands particularly TLR ligands 3 (//in red) and 4 (//in green) stimulate vascular endothelial cells of the portal vein to produce CX3CL1. (B) CX3CR1+ leukocytes invade the liver from the portal vein. (C) TLR3 ligand stimulates BEC to upregulate CX3CL1. (D) TLR4 ligand activates resident and CX3CR1+ invading monocytes. (E) Monocytes stimulate BEC by direct contact to upregulate CX3CL1. (F) Monocytes stimulate BEC by secretion of TNF-α to upregulate CX3CL1. (G) CX3CR1+ leukocytes invade BEC by engaging CX3CL1, thereby causing cholangitis.
CX3CL1 is potentially involved in multiple other inflammatory processes. This has already been described not only in non-cholestatic disease, but also in lung inflammation with associated smoke injury (8, 24). Hence, the data herein is not necessarily specific for PBC. Our data should also be contrasted with studies in gut. Intestinal microvascular endothelial cells do not produce spontaneously CX3CL1, but can do so after stimulation with TNF-α or IFN-γ, or direct leukocyte contact, and this effect is significantly stronger using endothelial cells from patients with inflammatory bowel disease versus control endothelial cells (21). Interestingly, liver endothelial cells that are epithelial cell marker- negative and CD31+ adherent mononuclear cells, produced CX3CL1 upon TLR stimulation but production did not differ between livers from PBC patients and those from chronic viral hepatitis. Notably with LSEC (epithelial cell marker negative, CD31−and CD105+), adherent mononuclear cells failed to produce CX3CL1 under any form of stimulation, perhaps a reflection of the capacity of LSEC to induce antigen-specific tolerance within the liver (25). The CXCR3 ligands CXCL9 to CXCL11 are dominant on LSEC, whereas the CCR5 ligands are dominant on the portal vascular endothelium (26). Thus our findings suggest that CX3CR1+ cells invade the liver via the portal vascular endothelium. As noted in the data herein, we have demonstrated that EC, LSEC and BEC from disease controls behave similar to cell populations isolated from cirrhotic PBC and the other control populations studied in our CX3CL1 production assays, indicating that these liver cell subpopulations respond equally well against danger signals, i.e. TLR ligands, irrespective of changes related to fibrosis or in vitro culture artifacts.
In order for CX3CL1 to be produced by BEC, direct contact with autologous LMC is clearly required since this production was inhibited when the CD40-CD154 interaction was blocked, in line with a previous report that there was reduced production of chemokines from BEC exposed to activated liver macrophages after CD40-CD154 interaction had been blocked (27). These data take on added importance in light of the known capacity of biliary ductular cells in PBC to express CD40 (28). Furthermore TLR3 stimulation induces high levels of CD40 on BEC (1), and pretreatment of BEC with the TLR ligand poly I:C may increase the CD40-CD154 dependent interaction between BEC and LMC.
We particularly examined differences between LMC that were derived from patients with PBC versus those with an inflammatory disease of another causation, chronic viral hepatitis. Since CX3CL1 also functions as an adhesion molecule, we note that EC produced high levels of CX3CL1 compared to BEC, and that LMC from PBC patients attached to EC at higher frequencies than to BEC, whereas LMC from viral hepatitis patients showed only minimal attachment to EC or BEC. There were also significant differences in adhesion to either EC or BEC when LMC were compared from patients with PBC versus comparison cases. LMC after TLR4 stimulation produced TNF-α and, in this particular case, there were significantly higher levels of TNF-α produced from LMC from PBC compared to control cases.
There are clearly multiple interactions that occur in PBC and other inflammatory liver diseases with respect to cytokines, chemokines and their cognate receptors; such is the case for the murine models of PBC as well (29, 30). Within this context as well as the schema presented above, the immunobiology of CX3CL1 has been recently demonstrated to interact with multiple other receptors and molecules. Indeed, as examples, ADAM10, ADAM17 and MMP2, produced by activated hepatic stellate cells, may also lead to shedding of CX3CL1 (20). Thus, we report that the atypical chemokine-adhesion molecule CX3CL1 (fractalkine) is an important participant in PBC that leads to periductular accumulation of lymphoid cells. This conclusion should be tempered with our availability of clinical samples, primarily end-stage patients that may not mirror early events.
Acknowledgments
Financial support provided by Grant-in-Aid for Scientific Research (C) of Japan, 19590775 and National Institutes of Health grant DK39588.
List of Abbreviations
- PBC
primary biliary cirrhosis
- BEC
biliary epithelial cells
- DC
dendritic cells
- EC
endothelial cells
- LMC
liver infiltrating mononuclear cells
- LSEC
liver sinusoidal endothelial cells
- MHC
major histocompatibility complex
- mDC
myeloid dendritic cells
- NKT
natural killer T cells
- TLR
toll-like receptor
Contributor Information
Shinji Shimoda, Email: sshimoda@intmed1.med.kyushu-u.ac.jp.
Kenichi Harada, Email: kenichih@med.kanazawa-u.ac.jp.
Hiroaki Niiro, Email: hniiro@cancer.med.kyushu-u.ac.jp.
Akinobu Taketomi, Email: taketomi@surg2.med.kyushu-u.ac.jp.
Yoshihiko Maehara, Email: maehara@surg2.med.kyushu-u.ac.jp.
Koichi Tsuneyama, Email: ktsune@med.u-toyama.ac.jp.
Kentaro Kikuchi, Email: kentaro@med.teikyo-u.ac.jp.
Yasuni Nakanuma, Email: pbcpsc@kenroku.kanazawa-u.ac.jp.
Ian R. Mackay, Email: Ian.MacKay@med.monash.edu.au.
M. Eric Gershwin, Email: megershwin@ucdavis.edu.
Koichi Akashi, Email: akashi@med.kyushu-u.ac.jp.
References
- 1.Shimoda S, Harada K, Niiro H, Yoshizumi T, Soejima Y, Taketomi A, et al. Biliary epithelial cells and primary biliary cirrhosis: the role of liver-infiltrating mononuclear cells. Hepatology. 2008;47:958–965. doi: 10.1002/hep.22102. [DOI] [PubMed] [Google Scholar]
- 2.Efsen E, Grappone C, DeFranco RM, Milani S, Romanelli RG, Bonacchi A, et al. Up-regulated expression of fractalkine and its receptor CX3CR1 during liver injury in humans. J Hepatol. 2002;37:39–47. doi: 10.1016/s0168-8278(02)00065-x. [DOI] [PubMed] [Google Scholar]
- 3.Wasmuth HE, Zaldivar MM, Berres ML, Werth A, Scholten D, Hillebrandt S, et al. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48:208–215. doi: 10.1016/j.jhep.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Fraticelli P, Sironi M, Bianchi G, D’Ambrosio D, Albanesi C, Stoppacciaro A, et al. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–1181. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52:1392–1401. doi: 10.1002/art.21140. [DOI] [PubMed] [Google Scholar]
- 6.Sawai H, Park YW, He X, Goronzy JJ, Weyand CM. Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2007;56:3215–3225. doi: 10.1002/art.22919. [DOI] [PubMed] [Google Scholar]
- 7.Bolovan-Fritts CA, Trout RN, Spector SA. Human cytomegalovirus-specific CD4+-T-cell cytokine response induces fractalkine in endothelial cells. J Virol. 2004;78:13173–13181. doi: 10.1128/JVI.78.23.13173-13181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McComb JG, Ranganathan M, Liu XH, Pilewski JM, Ray P, Watkins SC, et al. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am J Pathol. 2008;173:949–961. doi: 10.2353/ajpath.2008.071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med. 2009;206:595–606. doi: 10.1084/jem.20081385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–516. doi: 10.1002/hep.20582. [DOI] [PubMed] [Google Scholar]
- 11.Shang L, Fukata M, Thirunarayanan N, Martin AP, Arnaboldi P, Maussang D, et al. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology. 2008;135:529–538. doi: 10.1053/j.gastro.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loos T, Dekeyzer L, Struyf S, Schutyser E, Gijsbers K, Gouwy M, et al. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest. 2006;86:902–916. doi: 10.1038/labinvest.3700453. [DOI] [PubMed] [Google Scholar]
- 13.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 14.Kamihira T, Shimoda S, Nakamura M, Yokoyama T, Takii Y, Kawano A, et al. Biliary epithelial cells regulate autoreactive T cells: implications for biliary-specific diseases. Hepatology. 2005;41:151–159. doi: 10.1002/hep.20494. [DOI] [PubMed] [Google Scholar]
- 15.Shimoda S, Van de Water J, Ansari A, Nakamura M, Ishibashi H, Coppel RL, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163:1275–1289. doi: 10.1016/S0002-9440(10)63487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karrar A, Broome U, Uzunel M, Qureshi AR, Sumitran-Holgersson S. Human liver sinusoidal endothelial cells induce apoptosis in activated T cells: a role in tolerance induction. Gut. 2007;56:243–252. doi: 10.1136/gut.2006.093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumitran-Holgersson S, Ge X, Karrar A, Xu B, Nava S, Broome U, et al. A novel mechanism of liver allograft rejection facilitated by antibodies to liver sinusoidal endothelial cells. Hepatology. 2004;40:1211–1221. doi: 10.1002/hep.20434. [DOI] [PubMed] [Google Scholar]
- 19.Binion DG, West GA, Volk EE, Drazba JA, Ziats NP, Petras RE, et al. Acquired increase in leucocyte binding by intestinal microvascular endothelium in inflammatory bowel disease. Lancet. 1998;352:1742–1746. doi: 10.1016/S0140-6736(98)05050-8. [DOI] [PubMed] [Google Scholar]
- 20.Bourd-Boittin K, Basset L, Bonnier D, L’Helgoualc’h A, Samson M, Theret N. Cx3cl1/Fractalkine Shedding by Human Hepatic Stellate Cells: Contribution to Chronic Inflammation in the Liver. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sans M, Danese S, de la Motte C, de Souza HS, Rivera-Reyes BM, West GA, et al. Enhanced recruitment of CX3CR1+ T cells by mucosal endothelial cell-derived fractalkine in inflammatory bowel disease. Gastroenterology. 2007;132:139–153. doi: 10.1053/j.gastro.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- 23.Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med. 2004;200:1511–1517. doi: 10.1084/jem.20041035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Quement C, Guenon I, Gillon JY, Valenca S, Cayron-Elizondo V, Lagente V, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol. 2008;154:1206–1215. doi: 10.1038/bjp.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 26.Heydtmann M, Adams DH. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology. 2009;49:676–688. doi: 10.1002/hep.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alabraba EB, Lai V, Boon L, Wigmore SJ, Adams DH, Afford SC. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology. 2008;47:552–562. doi: 10.1002/hep.22011. [DOI] [PubMed] [Google Scholar]
- 28.Afford SC, Ahmed-Choudhury J, Randhawa S, Russell C, Youster J, Crosby HA, et al. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. Faseb J. 2001;15:2345–2354. doi: 10.1096/fj.01-0088com. [DOI] [PubMed] [Google Scholar]
- 29.Hsu W, Zhang W, Tsuneyama K, Moritoki Y, Ridgway WM, Ansari AA, et al. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(-/-) mice. Hepatology. 2009;49:133–140. doi: 10.1002/hep.22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]