

Abstract
Oxidative mechanisms of injury are important in many neurological disorders. Developing oligodendrocytes (pre-OLs) are particularly sensitive to oxidative stress-mediated injury. We previously demonstrated a novel function of phylloquinone (vitamin K1) and menaquinone 4 (MK-4; a major form of vitamin K2) in protecting pre-OLs and immature neurons against glutathione depletion-induced oxidative damage (Li et al. [2003] J. Neurosci. 23:5816–5826). Here we report that vitamin K at nanomolar concentrations prevents arachidonic acid-induced oxidative injury to pre-OLs through blocking the activation of 12-lipoxygenase (12-LOX). Arachidonic acid metabolism is a potential source for reactive oxygen species (ROS) generation during ischemia and reperfusion. Exposure of pre-OLs to arachidonic acid resulted in oxidative cell death in a concentration-dependent manner. Administration of vitamin K (K1 and MK-4) completely prevented the toxicity. Consistent with our previous findings, inhibitors of 12-LOX abolished ROS production and cell death, indicating that activation of 12-LOX is a key event in arachidonic acid-induced pre-OL death. Vitamin K1 and MK-4 significantly blocked 12-LOX activation and prevented ROS accumulation in pre-OLs challenged with arachidonic acid. However, vitamin K itself did not directly inhibit 12-LOX enzymatic activity when assayed with purified 12-LOX in vitro. These results suggest that vitamin K, or likely its metabolites, acts upstream of activation of 12-LOX in pre-OLs. In summary, our data indicate that vitamin K prevents oxidative cell death by blocking activation of 12-LOX and ROS generation.
Keywords: rat oligodendrocyte precursors, arachidonic acid, white matter injury, cerebral palsy, oxidative stress
Oxidative damage of cells is associated with many neurological disorders, including ischemia (Chong et al., 2004), neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases (Halliwell, 2006), and white matter injury such as periventricular leukomalacia (PVL; Haynes et al., 2005). Accumulating experimental data demonstrate that prevention of oxidative stress improves outcome in various animal models of neurodegenerative diseases and cerebral ischemic injury (Halliwell and Gutteridge, 2006). Therefore, development of novel antioxidants and elucidation of cellular pathways of oxidative stress in the central nervous system are of paramount importance in the development of effective therapeutics for neurological disorders.
PVL, the major pathology underlying the development of cerebral palsy in premature infants, is primarily a lesion involving damage to developing oligodendrocytes (pre-OLs; Volpe, 2001, 2003). Hypoxia/ischemia and maternal/fetal inflammation are considered two main causes of PVL. Oxidative stress resulting from generation of reactive oxygen/nitrogen species (ROS/RNS) is a well established sequela of ischemia (Vannucci and Hagberg, 2004; Blomgren and Hagberg, 2006) and inflammation (Rezaie and Dean, 2002) in the developing brain. Pre-OLs have been shown to be intrinsically sensitive to ROS- and RNS-induced damage (Back et al., 1998, 2007). Multiple lines of investigation strongly suggest that oxidative injury to pre-OLs plays an important role in the pathogenesis of PVL (Haynes et al., 2005). Oxidative damage to OLs was detected in human PVL cases (Haynes et al., 2003). Elevated oxidants and arachidonate metabolites were found in the cerebral spinal fluid of PVL patients (Inder et al., 2002) and in PVL white matter lesions (Back et al., 2005). Furthermore, administration of the antioxidant and glutathione precursor N-acetylcysteine ameliorated pre-OL degeneration and hypomyelination in an animal model of PVL (Paintlia et al., 2004).
Arachidonic acid is released upon hydrolysis of membrane phospholipids by phospholipase A2 in responding to various physiological or pathological stimuli (Piomelli, 1993). Increased arachidonic acid release occurs during brain ischemia as a result of the activation of phospholipases (Katsuki and Okuda, 1995). Once released, arachidonic acid is metabolized by three enzyme systems, cyclooxygenase, lipoxygenase, and epoxygenase, cyclooxygenase and lipoxygenase being the major metabolic enzymes. Metabolism of arachidonic acid by these enzymes produce free radicals and peroxides (Siesjo and Katsura, 1992; Paller and Jacob, 1994; Phillis et al., 2006). In addition to intracellular metabolism, arachidonic acid can also be liberated into the extracellular milieu upon phospholipase activation and act as a paracrine signal (Soliven et al., 1993; Takeda and Soliven, 1997). Cell culture studies demonstrate that arachidonic acid greatly potentiates glutathione depletion-induced oxidative toxicity in pre-OLs (Wang et al., 2004), in a glioma cell line (Higuchi et al., 2007), and in neurons (Li et al., 1997; Canals et al., 2003; Kramer et al., 2004; Kwon et al., 2005). The release and metabolism of arachidonic acid are also responsible for psychosine-initiated OL death and ROS generation (Giri et al., 2006). The cellular pathway of oxidative stress induced by arachidonic acid, glutathione depletion, or glutamate appears to involve the activation of a glial or neural 12-LOX (Li et al., 1997; Canals et al., 2003; Wang et al., 2004; Zhang et al., 2004, 2006; Kwon et al., 2005). LOXs catalyze the incorporation of molecular oxygen into specific positions of arachidonic acid, and, based on the position of oxygen insertion, are classified as 5-, 12-, or 15-LOX (Shimizu and Wolfe, 1990). 12-LOX is the major LOX found in the brain and generates predominately 12-hydroxyeicosatetraenoic acid (12-HETE; Hambrecht et al., 1987; Watanabe et al., 1993; Bendani et al., 1995). Genetic ablation or pharmacological inhibition of 12-LOX protects against ischemia/reperfusion-induced infarction in a mouse stroke model (Khanna et al., 2005; van Leyen et al., 2006).
Previously, we reported that phylloquinone (vitamin K1) and menaquinone 4 (MK-4; a vitamin K2) protect pre-OLs and immature neurons against glutathione depletion-induced oxidative injury and generation of ROS (Li et al., 2003). However, the mechanisms by which K1 and MK-4 prevent ROS generation and oxidative injury in pre-OLs remain undefined. 12-LOX has been shown to play an essential role in oxidative death of pre-OLs (Wang et al., 2004). In this study, we examined whether vitamin K prevents arachidonic acid-induced pre-OL death by blocking 12-LOX activation.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), Hank’s balanced salt solution (HBSS), Earle’s balanced salt solution (EBSS), penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was from Hyclone (Logan, UT). Human platelet-derived growth factor (PDGF) and basic fibroblast growth factor (b-FGF) were from Pepro-Tech (Rocky Hill, NJ). 2,3,5-Trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-1,4-benzoquinone (AA861) and 12-HETE were purchased from BioMol Research Laboratory (Plymouth, PA). The selective 12-LOX inhibitor N-benzyl-N-hydroxy-5-phenylpentamide (BHPP; also named BMD-122) was a generous gift from Dr. Lawrence J. Marnett (Vanderbilt University, Nashville, TN). Unless specified otherwise, all other reagents were from Sigma (St. Louis, MO).
Primary Cell Cultures
Primary pre-OL cultures were prepared from the forebrains of 1–2-day-old Sprague-Dawley rat pups by using a differential detachment method (McCarthy and de Vellis, 1980; Li et al., 2005; Chen et al., 2007). Briefly, forebrains free of meninges were digested with HBSS containing 0.01% trypsin and 10 μg/ml DNase and triturated with DMEM containing 20% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin. Dissociated cells were plated onto poly-D-lysine-coated 75-cm2 flasks and fed every other day for 7–10 days. After a 1-hr preshake to remove microglia, the flasks were shaken overnight at 200 rpm to separate pre-OLs from the astrocyte layer (Li et al., 2005). The suspension was plated onto uncoated petri dishes for 1 hr to remove further the residual contaminating microglia/astrocytes. Pre-OLs were plated into poly-L-ornithine-coated 96-well or 24-well plates and maintained in a serum-free basal defined medium (BDM containing 0.1% bovine serum albumin, 50 μg/ml human apotransferrin, 50 μg/ml insulin, 30 nM sodium selenite, 10 nM D-biotin, and 10 nM hydrocortisone in DMEM supplemented with PDGF 10 ng/ml and bFGF 10 ng/ml for 5–9 days at 37°C in a humid atmosphere of 5% CO2 and 95% air. The cell cultures were primarily OL progenitors and precursors [ , myelin basic protein−] and are therefore referred to as pre-OLs. Contamination by astrocytes and microglia was normally less than about 2–3%.
Cell Treatment
To induce oxidative stress, unless specified otherwise, freshly prepared arachidonic acid (stock concentration 100 mM) in anhydrous dimethylsulfoxide (DMSO) was added to cells in their growth medium at concentrations specified in the figure legends. Vitamin K1 and MK-4 were also made fresh in anhydrous DMSO as a ×1,000 working solution and added at the same time as arachidonic acid. In experiments in which OLs were exposed to vitamin K, the same volumes of the vehicle DMSO were also added to corresponding controls. The final concentration of DMSO in the culture medium was ≤0.1% and had no effect on cell viability, proliferation, or morphology as determined by Alamar Blue viability assay, cell counting, and phase-contrast image analysis. For glutathione depletion experiments, cells were washed three times with EBSS containing 1 mg/ml BSA and then exposed to growth medium containing no cystine. Omission of cystine, the precursor for glutathione synthesis, in the culture medium resulted in depletion of intracellular glutathione and oxidative death (Oka et al., 1993; Yonezawa et al., 1996; Back et al., 1998).
Cell Survival Determination
Cell survival was determined after treatment for 20–24 hr using Alamar Blue (Southern Biotechnology, Birmingham, AL), a tetrazolium dye that is reduced by living cells to a colored product. This assay is similar in principle to the 3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability assay and has been previously validated as an accurate measure of survival of OLs in our culture system (Back et al., 1999). All results of cell death assays were also confirmed by visual inspection under a phase-contrast light microscope. Briefly, culture medium in 96-well plates was aspirated, and cells were incubated with 200 μl of assay solution prepared by diluting ×100 stock solution of Alamar Blue into EBSS for 2 hr at 37°C. The fluorescence of the assay solution, reflecting cell viability, was measured with a fluorescence plate reader (FluoroCount; Packard Instrument Co.) using an excitation wavelength of 560 nm and an emission wavelength of 590 nm. All survival assays were performed in triplicate, and cell survival was expressed as mean ± SD.
Intracellular Free Radical Accumulation Measurement and Imaging
Intracellular free radical generation was evaluated with dichlorohydrofluorescin diacetate (DCFH-DA) and dihydrorhodamine 123 (Rho123; Invitrogen, Carlsbad, CA; LeBel et al., 1992; Wang and Joseph, 1999). DCFH-DA and Rho123 were prepared as 100 mM and 10 mM stocks, respectively, in DMSO and stored in the dark at −20°C. After cells had been treated with arachidonic acid for 15–18 hr in the presence or absence of vehicle, K1 or MK-4, DCFH-DA (100 μM), or dihydrorhodamine (10 μM) was added directly to the cells and incubated with the cells for 20 min at 37°C. The extracellular DCFH-DA or dihydrorhodamine was then removed by washing the cells twice with EBSS. The fluorescence of the cells loaded with DCFH-DA was measured by using a multiwell fluorescence plate reader (FluoroCount; Packard Instrument Co.) with excitation λ = 485 nm; emission λ = 530 nm. For fluorescent imaging of oxidized Rho123, cells were immediately visualized with a fluorescence microscope (Olympus IX71) equipped with an Olympus DP70 digital camera. For images, the microscope settings, such as brightness, contrast, and exposure time, were held constant to compare the relative intensity of oxidized Rho123 across all treatment conditions.
12-LOX Activity Assay
Cellular assay
12-LOX activation in pre-OLs was evaluated by quantifying the major metabolic product of 12-LOX, 12-HETE, using a correlated ELISA kit (Assay Designs Inc., Ann Harbor, MI) as described previously (Wang et al., 2004). Cells in 60-mm dishes were treated as indicated for 15–20 hr. Culture medium and cells were collected to evaluate the arachidonic acid metabolite 12-HETE. Cell suspension was sonicated and an aliquot was saved for protein concentration determination. Lipids were extracted and analyzed for 12-HETE according to the protocol of the manufacturer. The concentration of 12-HETE in each sample was calculated based on standard curves and normalized by the level of proteins in the sample.
In vitro cell-free assay
The direct effect of vitamin K on 12-LOX enzymatic activity was evaluated in a cell-free system using purified 12-LOX. Briefly, purified porcine leukocyte 12-LOX (10 units) was preincubated with vitamin K or 12-LOX inhibitors as specified in the figure legend for 15 min at room temperature in a Tris buffer (50 mM Tris-HCl, pH 7.2) in a volume of 200 μl. Enzymatic reaction was initiated by adding arachidonic acid (final concentration was 100 μM) and allowed to proceed at 37°C for 20 min. The reaction was terminated by adding an equal volume of cold extraction solution [diethyl ether:methanol:1 M citric acid 30:4:1 (v/v); Khanna et al., 2003], followed by vortexing and centrifugation. The aqueous phase was extracted again, and the combined organic phase containing the lipids was concentrated by nitrogen air and spotted on a Whatman silica gel thin-layer chromatograph plate. The plate was developed with diethyl ether, petroleum ether, and acetic acid at a ratio of 85:15:0.1 (v/v) at −20°C. Authentic 12-HETE and arachidonic acid were also spotted onto the plate and used as standards to identify their positions. Spots were visualized with iodine vapor, scanned, and analyzed.
Statistical Analysis
All cell culture treatments were performed in triplicate. Results were analyzed by one-way ANOVA followed by Bonferroni’s post hoc multiple-comparisons test to determine statistical significance. Comparison between two experimental groups was based on two-tailed t-test. P < 0.05 was considered statistically significant.
RESULTS
Arachidonic Acid Induces Oxidative Injury to pre-OLs
Pre-OLs are intrinsically vulnerable to oxidative damage (Back et al., 1998, 2007). Exposure of pre-OLs to increasing concentrations of arachidonic acid resulted in gradual loss of cell viability within 24 hr (Fig. 1A). To examine whether this pre-OL death was due to oxidative stress, the known antioxidant vitamin E was added together with arachidonic acid. Cell death was completely abrogated (Fig. 1B), which is consistent with an oxidative cell death pathway. Coenzyme Q (ubiquinone) is an essential component of the electron transport chain, but it also acts as an efficient lipophilic antioxidant in its reduced form, ubiquinol (Ernster and Dallner, 1995). Treatment of pre-OLs with micromolar concentration of coenzyme Q4 (CoQ4) also prevented arachidonic acid-induced toxicity (Fig. 1B). Consistently with an oxidative cell death pathway, ROS were significantly elevated in pre-OLs treated with arachidonic acid (Fig. 1C). To investigate whether pre-OLs underwent apoptotic cell death, we tested the effect of the pan caspase inhibitor z-VAD-fmk and found that the drug had no protective effect (Fig. 1D). In addition, z-VAD-fmk also did not prevent oxidative pre-OL death caused by cystine depletion (not shown). Deprivation of cystine in the culture medium results in a decreased level of cysteine, the precursor for glutathione biosynthesis, depletion of glutathione, production of ROS, and oxidative cell death (Yonezawa et al., 1996).
Fig. 1.

Arachidonic acid induces oxidative death of pre-OLs. A: Arachidonic acid (AA) induced loss of pre-OL viability in a concentration-dependent manner. Data represent mean ± SEM of six independent experiments. B: Antioxidants vitamin E (0.1 μM) and coenzyme Q4 (1 μM) completely abolished the arachidonic acid-induced pre-OL death. Data are representative of four independent experiments. C: Reactive oxygen species were generated in pre-OLs challenged with arachidonic acid (100 μM). Data represent two separate experiments. D: The pan-caspase inhibitor z-VAD-fmk did not prevent arachidonic acid-induced cell death. Pre-OLs were treated with arachidonic acid (60 μM) in the presence or absence of z-VAD-fmk (50 μM) for 24 hr, and cell viability was analyzed. Data are representative of three separate experiments.
Vitamin K Potently Protects Against Oxidative Injury Induced by Arachidonic Acid
Vitamin K1 and MK-4, at subnanomolar concentrations, prevent oxidative injury to pre-OLs and developing neurons (Li et al., 2003). Because arachidonic acid also induces oxidative death of pre-OLs, we first examined whether K1 and MK-4 also protect pre-OLs against arachidonic acid-induced toxicity with a similar potency. Both K1 and MK-4 potently prevented arachidonic acid toxicity in a concentration-dependent manner (Fig. 2A; EC50 for MK-4 was <10 nM and for K1 ~25 nM). The effective doses were in a range similar to that required to prevent cystine deprivation-induced oxidative pre-OL death (Li et al., 2003). Under control conditions, K1 and MK-4 had no effect on pre-OL proliferation, insofar as the same number of pre-OLs per ×200 field was found 24 hr after K1 and MK-4 treatment (control 250 ± 55 pre-OLs/field vs. K1 256 ± 35 and MK-4 248 ± 42 pre-OLs/field; mean ± SD, n = 4–6). K1 and MK-2 also did not influence pre-OL morphology but completely blocked aracidonic acid toxicity (Fig. 2B). It should be mentioned that MK-4 and K1 did not reverse glutathione depletion induced by cystine deprivation (Li et al., 2003). As expected, K1 and MK-4 prevented arachidonic acid-induced accumulation of ROS (Fig. 3A,B). CoQ4, which blocked arachidonic acid toxicity (Fig. 1B), was also effective in preventing ROS generation in pre-OLs (Fig. 3B). The rationale for selecting CoQ4 instead of other ubiquinones is that CoQ4 has the same number of isoprenoid units (n = 4) in its side chain as MK-4, thus sharing some structural similarity with MK-4. In addition, both CoQ4 and MK-4 are not antioxidants by themselves but possess potent antioxidant capacities when reduced to QH2 (ubiquinol) and KH2 (dihydroquinone), respectively, in cells (Ernster and Dallner, 1995; Li et al., 2003).
Fig. 2.

Vitamin K1 and menaquinone-4 completely abolish oxidative pre-OL death. A: Vitamin K1 and MK-4 prevent arachidonic acid-induced pre-OL death in a concentration-dependent manner. Pre-OLs were challenged with arachidonic acid (100 μM) in the presence of increasing concentrations of K1 or MK-4 for 24 hr, and cell survival was determined as described. B: Representative phase-contrast images of pre-OLs treated as indicated for 20–24 hr. Pre-OLs were treated with arachidonic acid (100 μM) in the absence or presence of K1 (1 μM) and MK-4 (0.1 μM) as indicated. Data are representative of at least three experiments. Scale bar = 50 μm.
Fig. 3.

Vitamin K1 and menaquinone-4 prevent arachidonic acid-induced ROS accumulation in pre-OLs. Pre-OLs were treated as indicated for 18–20 hr, and ROS accumulation was visualized with dihydrorhodamine 123 (A) and quantified with DCFH-DA (B). The concentrations of K1, MK-4, and CoQ4 were 1 μM. DCFH-DA and dihydrorhodamine are nonfluorescent but become fluorescent upon oxidation by peroxides generated in cells. Data are representative of two separate experiments. Scale bar = 50 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Essential Role of 12-LOX in Arachidonic Acid-Induced Pre-OL Cell Death
12-LOX has been implicated in oxidative death of primary neurons and developing OLs (Li et al., 1997; Canals et al., 2003; Wang et al., 2004; Zhang et al., 2004; Kwon et al., 2005). We first tested and confirmed that the LOX inhibitor AA861 (Yoshimoto et al., 1982; Nakadate et al., 1985; Li et al., 1997) effectively blocked arachidonic acid-induced cell death (Fig. 4A). Because we did not detect the presence of 5-LOX in pre-OLs previously, and other 5-LOX inhibitors did not protect the toxicity of arachidonic acid, the result is consistent with our previous findings indicating that only 12-LOX is required for arachidonic acid induced toxicity (Wang et al., 2004). Cyclooxygenase inhibitors did not have protective effects (data not shown). Blockade of 12-LOX with AA861 also efficiently prevented ROS accumulation (Fig. 4B), suggesting an essential role for 12-LOX in ROS generation and cell death. To evaluate 12-LOX activity directly under various treatment conditions, we then analyzed the level of 12-HETE, the stable reaction product of 12-LOX, in cultures challenged with arachidonic acid. We found that the level of 12-HETE was increased by three- to fivefold in cells treated with arachidonic acid compared with controls and that AA861 significantly prevented this elevation (Fig. 4C). Next we tested whether vitamin K1 and MK-4 prevented cell death by inhibiting 12-LOX activation. At concentrations effective for preventing ROS generation and cell death, K1 and MK-4 markedly blocked 12-HETE production and, thus, 12-LOX activation in pre-OLs exposed to arachidonic acid (Fig. 4D). These results indicate that vitamin K1 and MK-4 act at the level of or upstream of 12-LOX activation.
Fig. 4.

Vitamin K1 and menaquinone-4 inhibit arachidonic acid-induced 12-lipoxygenase activation. A,B: The 12-LOX inhibitor AA861 prevented arachidonic acid-induced pre-OL death and ROS accumulation. Pre-OLs were treated as indicated with or without arachidonic acid (200 μM) in the absence or presence of AA861 (10 μM). Cell viability and ROS generation were evaluated 24 and 20 hr later, respectively. C: The 12-LOX-specific inhibitor AA861 blocked arachidonic acid-induced activation of 12-LOX and accumulation of its product 12-HETE. **P < 0.01 compared with controls. D: Vitamin K1 and MK-4 prevented arachidonic acid-induced 12-HETE accumulation in pre-OLs. Data are representative of three experiments. *P < 0.05, **P < 0.001 vs. the controls.
Vitamin K itself Does Not Directly Inhibit 12-LOX Enzymatic Activity In Vitro
To test the possibility that the observed inhibition of 12-LOX by MK-4 in pre-OLs may due to a direct interaction between 12-LOX and MK-4, we used a cell-free analysis system. In this assay system, the 12-LOX enzymatic reaction was initiated by adding the 12-LOX substrate arachidonic acid to purified 12-LOX that was preincubated with MK-4 or known 12-LOX inhibitors, and formation of the product, 12-HETE, was assayed by thin-layer chromatography. MK-4, even when used at 10 μM, did not directly block 12-LOX enzymatic reaction in the cell-free system (Fig. 5A). The known LOX inhibitor AA861, which blocked arachidonic acid toxicity and 12-HETE formation in cultured pre-OLs, only partially inhibited 12-HETE formation even when used at concentrations 10–50 μM in the cell-free assay system (Fig. 5A; and data not shown). It is interesting to note that 30 μM AA861 inhibited only approximately 20% of porcine leukocyte and rat lung 12-LOX activity in another cell-free assay system (Yoshimoto et al., 1982). The failure of MK-4 and the partial inhibition of 12-LOX activity by AA861 are not due to the insensitivity of the 12-LOX to its inhibitors in the cell-free system. In fact, a specific 12-LOX inhibitor, BHPP (Honn et al., 1994; Nie et al., 2000), which effectively prevented glutathione-depletion as well as arachidonic acid-induced 12-LOX activation and pre-OL death (Wang et al., 2004), potently inhibited 12-HETE formation in the cell-free system in a concentration-dependent manner (Fig. 5B). These results suggest that BHPP inhibits 12-LOX by directly interacting with the enzyme, whereas MK-4 and AA681 may act upstream of 12-LOX activation or require metabolism to inhibit 12-LOX activity.
Fig. 5.
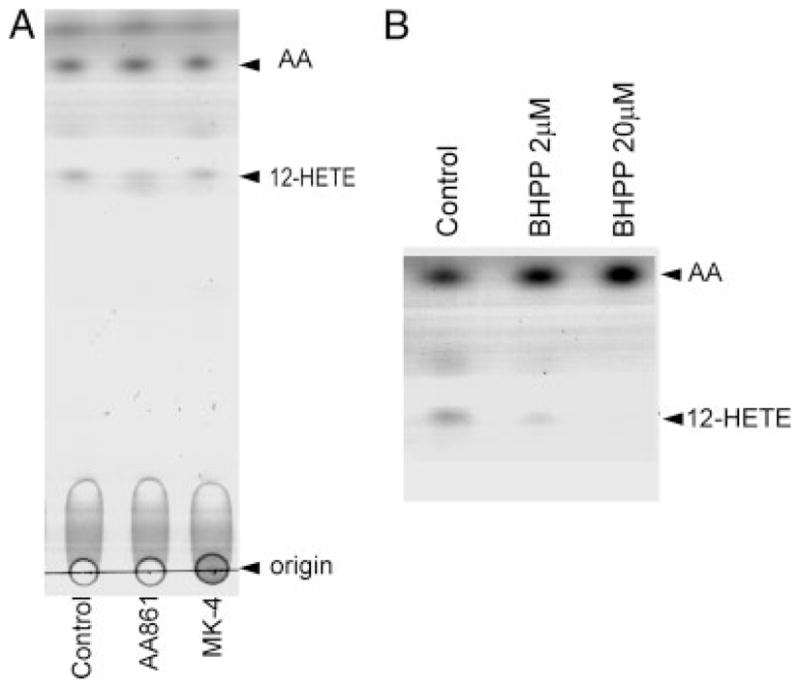
Vitamin K1 and menaquinone-4 do not directly inhibit 12-lipoxygenase enzymatic activity in a cell-free system. A: Purified 12-LOX (20 U) was incubated with AA861 (10 μM) or MK-4 (10 μM) for 30 min prior to addition of the 12-LOX substrate arachidonic acid (100 μM). The reaction mix was incubated for 20 min at 37°C. 12-HETE was extracted and analyzed by thin-layer chromatography as described. Positions of origins of sample application, 12-HETE, and free arachidonic acid are indicated by arrows. B: The 12-LOX-specific inhibitor BHPP (2–20 μM) dose dependently blocked 12-HETE production. Data are from one experiment representative of three that were performed.
DISCUSSION
Here we have investigated the mechanism by which vitamin K1 and MK-4 protect developing oligodendrocytes against oxidative stress-induced cell death. We demonstrated that activation of 12-LOX is essential to arachidonic acid-induced ROS generation and subsequent pre-OL death and that vitamin K efficiently blocked 12-LOX activation as determined by 1) diminished production of the 12-LOX stable product 12-HETE, 2) inhibition of ROS generation, and 3) abrogation of pre-OL death in cultures. Our results suggest that vitamin K1 and MK-4, or their metabolites, act upstream of 12-LOX to inhibit 12-LOX and prevent oxidative injury to pre-OLs.
Using an in vitro assay system consisting of purified porcine leukocyte 12-LOX, we investigated whether MK-4 interacts directly with 12-LOX to block 12-LOX enzymatic activity and production of 12-HETE. We did not observe any inhibitory effect of MK-4 on 12-LOX in the cell-free system, suggesting that vitamin K by itself does not interact directly with 12-LOX. Interestingly, the reported LOX inhibitor AA861 also had limited inhibitory effect on 12-LOX activity. In contrast, another 12-LOX-specific inhibitor, BHPP, blocked 12-LOX activity in the same assay system. It should be noted that both AA861 and BHPP were effective for preventing oxidative OL death in which 12-LOX plays a causal role in cell death (Wang et al., 2004; Zhang et al., 2006). A previous study showed that AA861 was a potent inhibitor for 5-LOX, with an ID50 approximately two orders of magnitude lower than that for leukocyte 12-LOX, and that AA681 did not block platelet 12-LOX when analyzed in vitro (Yoshimoto et al., 1982). As mentioned previously, 12-LOX is the predominant LOX in the brain (Hambrecht et al., 1987; Watanabe et al., 1993; Bendani et al., 1995) and in neurons and OLs (Nishiyama et al., 1993; Wang et al., 2004). Activation of 12-LOX rather than 5-LOX is responsible for oxidative pre-OL death (Wang et al., 2004).
There are a few possible explanations for the failure of AA861 and vitamin K to block 12-LOX activity in vitro directly while they are capable of inhibiting 12-LOX in cultures of intact cells. First, the 12-LOX in pre-OLs from rat forebrain may possess properties different from those of the porcine leukocyte 12-LOX used in the assay mixture, insofar as 12-LOX in the brain exhibits 71% identity and 84% similarity to porcine leukocyte 12-LOX (Watanabe et al., 1993). Second, metabolism of AA861 and vitamin K1 and MK-4 in cells may be required for converting them into metabolites that are effective for preventing 12-LOX activation in cells. Vitamin K is known to undergo redox cycling through the vitamin K cycle and is a cofactor for a single known enzyme, γ-glutamylcarboxylase, that catalyzes the post-translational conversion of glutamic acid to γ-carboxyglutamic acid in vitamin K-dependent proteins involved in blood coagulation and bone metabolism (Suttie, 1991). Besides this canonical pathway, vitamin K has been shown to be metabolized into unidentified derivatives in fibroblasts (Canfield et al., 1987; Ross et al., 1991), and, more recently, accumulating evidence suggests that vitamin K acts as a signaling molecule (Ichikawa et al., 2007; Igarashi et al., 2007) and modulates cellular signaling transduction pathways (Saxena et al., 1997; Osada and Carr, 2001; Wang et al., 2001; Tsujioka et al., 2006). We previously demonstrated that the protective function of vitamin K against oxidative pre-OL injury is independent of the vitamin K cycle (Li et al., 2003). Here we present evidence that vitamin K prevents 12-LOX activation in pre-OLs without direct inhibition in vitro. The exact molecular mechanism by which vitamin K inhibits 12-LOX remains to be defined but may involve metabolites of vitamin K or alternatively interference with signaling pathways that lead to 12-LOX activation. Specific cellular pathways may be involved in redox activation of 12-LOX (Shornick and Holtzman, 1993; Li et al., 1997; Canals et al., 2003; Khanna et al., 2003). For example, in mature OLs, intracellular zinc release and activation of ERK1/2 have been found to result in 12-LOX activation and ultimately cell death in OLs exposed to the potent oxidant peroxynitrite (Zhang et al., 2006).
Because of its essential role in regulating coagulation and bone metabolism, vitamin K has been widely used in the United States to prevent hemorrhage in newborns and in Japan to treat patients with hypoprothrombinemia and osteoporosis. Administration of MK-4 has been recently shown to ameliorate experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis (Moriya et al., 2005). Because the release of arachidonic acid and resulting oxidative stress are associated with ischemic injury and various neurological diseases, the observed effectiveness of the natural forms of vitamin K in preventing oxidative injury to OLs suggests the potential of vitamin K as a safe therapeutic intervention for oxidative cell injury.
Acknowledgments
Contract grant sponsor: NIH; Contract grant number: P01NS38475 (to J.J.V., P.A.R.); Contract grant number: HD18655; Contract grant sponsor: Hearst Foundation (to J.L.); Contract grant sponsor: United Cerebral Palsy Foundation (to J.L.).
We thank Ling Dong and Ping Xu for technical assistance.
References
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Khan R, Gan X, Rosenberg PA, Volpe JJ. A new alamar blue viability assay to rapidly quantify oligodendrocyte death. J Neurosci Methods. 1999;91:47–54. doi: 10.1016/s0165-0270(99)00062-x. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Mallinson RA, O’Malley JP, Wallen LD, Frei B, Morrow JD, Petito CK, Roberts CT, Jr, Murdoch GH, Montine TJ. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58:108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- Back SA, Riddle A, McClure MM. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke. 2007;38:724–730. doi: 10.1161/01.STR.0000254729.27386.05. [DOI] [PubMed] [Google Scholar]
- Bendani MK, Palluy O, Cook-Moreau J, Beneytout JL, Rigaud M, Vallat JM. Localization of 12-lipoxygenase mRNA in cultured oligodendrocytes and astrocytes by in situ reverse transcriptase and polymerase chain reaction. Neurosci Lett. 1995;189:159–162. doi: 10.1016/0304-3940(95)11482-c. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med. 2006;40:388–397. doi: 10.1016/j.freeradbiomed.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- Canfield LM, Johnson TM, Martin GS, Gunn JM. Absorption and metabolism of vitamin K in Swiss 3T3 mouse fibroblasts—a model system for study of vitamin K absorption and metabolism. Biochem Biophys Res Commun. 1987;147:731–739. doi: 10.1016/0006-291x(87)90991-0. [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid Redox Signal. 2004;6:277–287. doi: 10.1089/152308604322899341. [DOI] [PubMed] [Google Scholar]
- Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Giri S, Khan M, Rattan R, Singh I, Singh AK. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res. 2006;47:1478–1492. doi: 10.1194/jlr.M600084-JLR200. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford: Oxford University Press; 2006. [Google Scholar]
- Hambrecht GS, Adesuyi SA, Holt S, Ellis EF. Brain 12-HETE formation in different species, brain regions, and in brain microvessels. Neurochem Res. 1987;12:1029–1033. doi: 10.1007/BF00970932. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi Y, Tanii H, Koriyama Y, Mizukami Y, Yoshimoto T. Arachidonic acid promotes glutamate-induced cell death associated with necrosis by 12-lipoxygenase activation in glioma cells. Life Sci. 2007;80:1856–1864. doi: 10.1016/j.lfs.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Honn KV, Tang DG, Grossi IM, Renaud C, Duniec ZM, Johnson CR, Diglio CA. Enhanced endothelial cell retraction mediated by 12(S)-HETE: a proposed mechanism for the role of platelets in tumor cell metastasis. Exp Cell Res. 1994;210:1–9. doi: 10.1006/excr.1994.1001. [DOI] [PubMed] [Google Scholar]
- Ichikawa T, Horie-Inoue K, Ikeda K, Blumberg B, Inoue S. Vitamin K2 induces phosphorylation of protein kinase A and expression of novel target genes in osteoblastic cells. J Mol Endocrinol. 2007;39:239–247. doi: 10.1677/JME-07-0048. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Yogiashi Y, Mihara M, Takada I, Kitagawa H, Kato S. Vitamin K induces osteoblast differentiation through pregnane X receptor-mediated transcriptional control of the Msx2 gene. Mol Cell Biol. 2007;27:7947–7954. doi: 10.1128/MCB.00813-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Inder T, Mocatta T, Darlow B, Spencer C, Volpe JJ, Winterbourn C. Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatr Res. 2002;52:213–218. doi: 10.1203/00006450-200208000-00013. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Slivka A, Craft TKS, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL, Sen CK. Neuroprotective properties of the natural vitamin E alphatocotrienol. Stroke. 2005;36:e144–e152. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer BC, Yabut JA, Cheong J, Jnobaptiste R, Robakis T, Olanow CW, Mytilineou C. Toxicity of glutathione depletion in mesencephalic cultures: a role for arachidonic acid and its lipoxygenase metabolites. Eur J Neurosci. 2004;19:280–286. doi: 10.1111/j.1460-9568.2004.03111.x. [DOI] [PubMed] [Google Scholar]
- Kwon K, Jung Y, Lee S, Moon C, Baik E. Arachidonic acid induces neuronal death through lipoxygenase and cytochrome P450 rather than cyclooxygenase. J Neurosci Res. 2005;81:73–84. doi: 10.1002/jnr.20520. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin K in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–5826. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci USA. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya M, Nakatsuji Y, Okuno T, Hamasaki T, Sawada M, Sakoda S. Vitamin K2 ameliorates experimental autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 2005;170:11–20. doi: 10.1016/j.jneuroim.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Nakadate T, Yamamoto S, Aizu E, Kato R. Inhibition of mouse epidermal 12-lipoxygenase by 2,3,4-trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-1,4-benzoquinone (AA861) J Pharm Pharmacol. 1985;37:71–73. doi: 10.1111/j.2042-7158.1985.tb04938.x. [DOI] [PubMed] [Google Scholar]
- Nie D, Tang K, Diglio C, Honn KV. Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood. 2000;95:2304–2311. [PubMed] [Google Scholar]
- Nishiyama M, Watanabe T, Ueda N, Tsukamoto H, Watanabe K. Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. J Histochem Cytochem. 1993;41:111–117. doi: 10.1177/41.1.8417106. [DOI] [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada S, Carr BI. Mechanism of novel vitamin K analog induced growth inhibition in human hepatoma cell line. J Hepatol. 2001;34:676–682. doi: 10.1016/s0168-8278(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- Paller MS, Jacob HS. Cytochrome P-450 mediates tissue-damaging hydroxyl radical formation during reoxygenation of the kidney. Proc Natl Acad Sci USA. 1994;91:7002–7006. doi: 10.1073/pnas.91.15.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Piomelli D. Arachidonic acid in cell signaling. Curr Opin Cell Biol. 1993;5:274–280. doi: 10.1016/0955-0674(93)90116-8. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Dean A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology. 2002;22:106–132. doi: 10.1046/j.1440-1789.2002.00438.x. [DOI] [PubMed] [Google Scholar]
- Ross PJ, Shearer MJ, Diplock AT, Schey SA. A fibroblast cell culture model to study vitamin K metabolism and the inhibition of vitamin K epoxide reductase by known and suspected antagonists. Br J Haematol. 1991;77:195–200. doi: 10.1111/j.1365-2141.1991.tb07977.x. [DOI] [PubMed] [Google Scholar]
- Saxena SP, Fan T, Li M, Israels ED, Israels LG. A novel role for vitamin K1 in a tyrosine phosphorylation cascade during chick embryogenesis. J Clin Invest. 1997;99:602–607. doi: 10.1172/JCI119202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Wolfe LS. Arachidonic acid cascade and signal transduction. J Neurochem. 1990;55:1–15. doi: 10.1111/j.1471-4159.1990.tb08813.x. [DOI] [PubMed] [Google Scholar]
- Shornick LP, Holtzman MJ. A cryptic, microsomal-type arachidonate 12-lipoxygenase is tonically inactivated by oxidation-reduction conditions in cultured epithelial cells. J Biol Chem. 1993;268:371–376. [PubMed] [Google Scholar]
- Siesjo BK, Katsura K. Ischemic brain damage: focus on lipids and lipid mediators. Adv Exp Med Biol. 1992;318:41–56. doi: 10.1007/978-1-4615-3426-6_5. [DOI] [PubMed] [Google Scholar]
- Soliven B, Takeda M, Shandy T, Nelson DJ. Arachidonic acid and its metabolites increase Cai in cultured rat oligodendrocytes. Am J Physiol Cell Physiol. 1993;264:C632–C640. doi: 10.1152/ajpcell.1993.264.3.C632. [DOI] [PubMed] [Google Scholar]
- Suttie J. Handbook of vitamins. Basel: Marcel Dekker, Inc; 1991. Vitamin K; pp. 145–194. [Google Scholar]
- Takeda M, Soliven B. Arachidonic acid inhibits myelin basic protein phosphorylation in cultured oligodendrocytes. Glia. 1997;21:277–284. [PubMed] [Google Scholar]
- Tsujioka T, Miura Y, Otsuki T, Nishimura Y, Hyodoh F, Wada H, Sugihara T. The mechanisms of vitamin K2-induced apoptosis of myeloma cells. Haematologica. 2006;91:613–619. [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol. 2004;207:3149–3154. doi: 10.1242/jeb.01064. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Cerebral white matter injury of the premature infant-more common than you think. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.176. [DOI] [PubMed] [Google Scholar]
- Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20:2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Southwick EC, Wang M, Kar S, Rosi KS, Wilcox CS, Lazo JS, Carr BI. Involvement of Cdc25A phosphatase in Hep3B hepatoma cell growth inhibition induced by novel K vitamin analogs. Cancer Res. 2001;61:7211–7216. [PubMed] [Google Scholar]
- Watanabe T, Medina JF, Haeggstrom JZ, Radmark O, Samuelsson B. Molecular cloning of a 12-lipoxygenase cDNA from rat brain. Eur J Biochem. 1993;212:605–612. doi: 10.1111/j.1432-1033.1993.tb17699.x. [DOI] [PubMed] [Google Scholar]
- Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. 1996;67:566–573. doi: 10.1046/j.1471-4159.1996.67020566.x. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Yokoyama C, Kenkichi O, Yamamoto S, Maki Y, Ashida Y, Terao S, Shiraishi M. 2,3,5-Trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-l,4-benzoquinone (AA861), a selective inhibitor of the 5-lipoxygenase reaction and the biosynthesis of slow-reacting substance of anaphylaxis. Biochim Biophys Acta Lipids Lipid Metab. 1982;713:470–473. [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Dong L, Xu P, Chen W, Neve RL, Volpe JJ, Rosenberg PA. Intracellular zinc release and ERK phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J Biol Chem. 2006;281:9460–9470. doi: 10.1074/jbc.M510650200. [DOI] [PubMed] [Google Scholar]