

Abstract
G-protein coupled receptors (GPCRs) play an essential role in many biological processes. Despite an increase in the number of solved X-ray crystal structures of GPCRs, capturing a GPCR in its activated state for structural analysis has proven to be difficult. An unexplored paradigm is stabilization of one or more conformational states of a GPCR via binding a small molecule to the intracellular loops. A short tetrazole peptidomimetic based on the R*-bound structure of Gtα(340-350) was previously designed and shown to stabilize the photoactivated state of rhodopsin (R*), the GPCR involved in vision. A pharmacophore model derived from the designed tetrazole tetrapeptide was used for ligand-based virtual screening to enhance the possible discovery of novel scaffolds. Maybridge Hitfinder and National Cancer Institute (NCI) diversity libraries were screened for compounds containing the pharmacophore. Forty-seven compounds resulted from virtually screening the Maybridge library, whereas no hits resulted with the NCI library. Three of the 47 Maybridge compounds were found to stabilize the MII state. As these compounds did not inhibit binding of transducin to R*, they were assumed to be allosteric ligands. These compounds are potentially useful for crystallographic studies where complexes with these compounds might capture rhodopsin in its activated conformational state.
Keywords: ligand-based virtual screening, GPCRs, rhodopsin, transducin, MII stabilization
INTRODUCTION
G-protein coupled receptors (GPCRs) are the largest family of transmembrane (TM) receptor proteins and involved in many physiological processes in humans. GPCRs are involved in transducing extracellular (EC) signals such as light, hormones, neurotransmitters, amino acids, peptides, and odorants to intracellular (IC) effectors (1, 2). The binding of the agonist, or other signal mediator (e.g. light), causes a conformational change in the GPCR. This physical change activates the GPCR and allows its G-protein to bind to the IC interface of the GPCR and catalyze the exchange of GDP to GTP on the α-subunit of the G-protein. The α-subunit then dissociates from the βγ complex of the G protein and activates an effector protein that modulates IC second messengers (3).
GPCRs are extremely important pharmaceutical targets (1, 4). Out of approximately 35,000 genes in the human genome, about 720 genes encode GPCRs of which roughly 400 are thought to be potential drug targets (3). Currently, 50% of marketed drugs (e.g. Inderal, an adrenoreceptor antagonist, and Zantac, a histamine-receptor antagonist) target the GPCR family (1). However, the full potential of GPCR therapeutics has not been realized due to many uncharacterized orphan GPCRs and difficulties in obtaining X-ray crystallographic data for structural determination of these receptors (1). Based on a recent review, only six GPCRs in the PDB have solved crystal structures (5). Despite the lack of structural data, ab initio (6, 7) and homology models (7-13) have been shown to be of use for structure-based virtual screening of EC agonist and antagonists. Furthermore, there have been other cases of ligand-based virtual screening that have been successful in finding EC agonists and antagonists (4, 11, 14).
Over the past decade, it has become clear that GPCRs can adopt multiple biologically active states that modify their coupling to intracellular effectors. Such states can be stabilized by ligands that bind to topographically different (allosteric) sites than their normal biological ligands (15-18). These observations in multiple GPCR systems require a multistate model of receptor activation in which ligand-specific conformations are capable of differentially activating distinct IC signaling partners. Such orthosteric and allosteric binding sites offer new opportunities for modulating GPCR signal transduction.
The focus of this research was stabilization of a GPCR-activated state via a small molecule binding intracellularly to the GPCR IC activated loops, rather than targeting the EC or TM portions of a GPCR (19, 20). This potentially provides a method for stabilizing GPCR structures in their activated states for crystallization studies and a starting point for developing novel compounds that can modulate the interaction between a GPCR and its G-protein. Although the number of X-ray crystal structures of GPCRs has grown over the past few years (5), capturing the GPCR in an active conformation in which it interacts with its G-protein has proven to be difficult (21). Previously, we have used computational modeling and structure-based virtual screening to find small molecules that stabilize the activated state of rhodopsin and also modulate the interaction between a GPCR and its G-protein on the IC side (20). In this study, we use a ligand-based virtual screening method to find other compounds that stabilize the activated state of a GPCR. Ligand-based virtual screening is a fast and powerful tool and, most important, applicable even when the structure of the receptor is unknown (4, 11, 14). In particular, the data presented herein supports modulation of GPCR conformational states at multiple binding sites by intracellular ligands.
As proof of concept, rhodopsin, the GPCR of the human eye that transduces signals in the vision cascade, was utilized. The absorbance of light causes the chromophore bound to rhodopsin, 11-cis-retinal, to isomerize to all-trans retinal in a series of seven spectroscopically detectable changes (22). This transformation changes the conformation of the IC loops of rhodopsin that complex and activate its G-protein, transducin (5). The metarhodopsin I state (MI) and metarhodopsin II state (MII) are in equilibrium following photoactivation, and stabilization of the MII state (R*) occurs when transducin binds (5). The 340-350 region of the α-subunit of transducin, the 11-membered peptide IKQNLKDCGLF (Gt〈(340-350)), was shown to stabilize the photoactivated state of rhodopsin (22). The three-dimensional structure of this peptide in complex with rhodopsin was determined by TrNOE and was found to contain a reverse-turn region (23), see Fig. 1a. A tetrazole-containing peptidomimetic designed to mimic the presumed surface of the reverse turn interacting with R* was designed, synthesized and found to stabilize the MII state of rhodopsin (24). We have used the designed conformation of this tetrazole tetrapeptide to generate a pharmacophore model to search the Maybridge HitFinder and NCI diversity libraries to expand the diversity of possible scaffolds. The search for this pharmacophore in the Maybridge HitFinder library yielded compounds that stabilized the MII state of rhodopsin. This study provides the first example of developing a unique pharmacophore from a TrNOE structure and using the pharmacophore for ligand-based virtual screening to find small drug-like molecules that allosterically stabilize the activated state of a GPCR.
Figure 1.
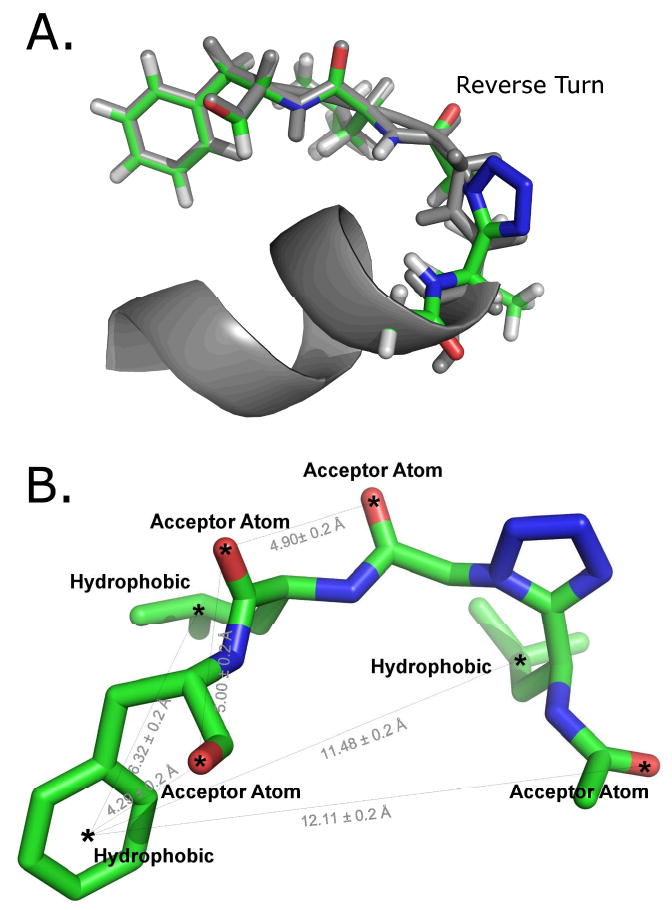
a. Tetrazole peptidomimetic aligned with the first model from the TrNOE NMR structure of Gtα(340-350) (PDB ID: 1AQG). The TrNOE structure is shown in gray, and the tetrazole peptidomimetic is shown in green. b. Pharmacophore model with spatial features and distance constraints. Rendered using Pymol (42).
MATERIALS AND METHODS
Database Creation
Two databases, one with compounds from the National Cancer Institute Diversity Library (1,992 compounds, accessed July 6, 2008) and the other with compounds from the Maybridge Hitfinder Library version 5 (14,400 compounds, accessed July 6, 2008), were imported into UNITY, a cheminformatic program within the Sybyl 7.3 software package (25). The molecular libraries were read into Sybyl as an sdf file and created with the default import options with a few exceptions recommended by the Tripos Bookshelf tutorial for Sybyl 7.3 (25). Chirality was determined when importing the files at carbon, nitrogen, and phosphorus atoms, as well as at double bonds. CONCORD, a program in Sybyl, was used to generate 3D coordinates.
Generating the Pharmacophore
The TrNOE structure of the Gtα(340-350) peptide was used to model a hypothesized bound conformation of Ac-Leu-Ψ[CN4]-Gly-Leu-Phe-OH (Figure 1a). In the TrNOE structure, the α-helix on the Gtα(340-350) peptide was deleted, part of the turn was replaced with a tetrazole ring to impose a reverse turn, the cysteine was replaced with leucine, and the changed areas were minimized using default settings in Sybyl 7.3 (25). The designed peptidomimetics that showed MII stabilization were basically 4-residue peptides, LGLF and L-DAla-LF. The tetrazole ring mimics the cis-amide bond between Leu and Gly to force a cis-amide-like conformation (26-29) that placed the side chain of the first Leu close to the phenyl ring of the terminal phenylalanine. Despite the limited structural similarity to Gtα(340-350)-peptide in composition, both compounds stabilized the MII state. The compounds suffered from solubility problems, precluding an accurate EC50 measurement and promoting the development of an alternative scaffold that may be more soluble. Hydrophobic, hydrogen-bond acceptor, and donor features were placed on regions of the tetrazole compound assumed to be important, and distance constraints were added, each with a 0.20 Å tolerance (Figure 1b). This pharmacophore model was used to search both the NCI Diversity Library and the Maybridge Hitfinder Library using UNITY flex search in Sybyl 7.3 (25).
Rod Outer-Segment Preparation
W.L. Lawson Co., Lincoln, NE supplied the frozen, dark-adapted bovine retinas. The method of Papermaster and Dreyer (30) was used to prepare the ROS membranes, and the urea-washed ROS membranes were prepared as described previously (31, 32). A silver-stained SDS-PAGE gel determined the purity of the purified rhodopsin as >99%. Rhodopsin concentration was determined by using a molar extinction coefficient value ε500 of 40,600 m−1 cm−1, and a molecular weight of 40,000 Da. The ROS disk membranes were resuspended in the following buffer, then stored at −70 °C: 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 0.1 mM PMSF.
MII Stabilization Assay
Compounds obtained from Maybridge, with purity greater than 90%, were combined with rhodopsin in urea-washed ROS membranes (UM), and the absorbance spectra of rhodopsin was measured using UV/Vis spectroscopy. An analog of Gtα(340-350) with high affinity for the MII state, VLEDLKSCGLF (33), was used as a positive control, whereas a solution with no compound was used as a negative control. Compounds obtained from Maybridge were essentially insoluble in water and were dissolved in DMSO, then diluted using buffer. The final concentration of VLEDLKSCGLF was 1.5 mM, and the final concentration of the Maybridge compounds in the initial screen was 3 mM. In many cases, the Maybridge compounds became insoluble upon adding the assay buffer to the compound dissolved in DMSO, so the mixtures were filtered. For these compounds, saturating amounts of compound were used in the initial screen and the pH of the compound solution was not titrated to 7.7. The final buffer in the assay consisted of 20 mM Tris/HCl, 130 mM NaCl, 3% v/v DMSO, pH 7.7 at 4 °C. The final concentration of rhodopsin in UM was 5 μM.
After determining the active compounds in the initial screen, compounds 3, 4, and 5 were solubilized in buffer containing 5% DMSO so that accurate dose-response measurements could be determined. The pH of the compound in a 1:1 solution of DMSO:buffer was adjusted such that the pH was between 7.5 and 8.0 and dilutions were made from this stock. The compounds, at a variety of concentrations, were added to rhodopsin immediately before light activation. The samples were kept on ice for the duration of the experiment, and all solutions containing rhodopsin were handled under dim red light to prevent rhodopsin from bleaching before light activation. The experiment was conducted essentially as described previously (23, 34). All curve fitting was done using Kaleidagraph 4.0. The EC50 values reported are the average of two experiments done on different stocks of Maybridge compound with their pH determined separately. The error bars shown in Figure 4 were derived from one experiment performed in duplicate or greater number.
Figure 4.
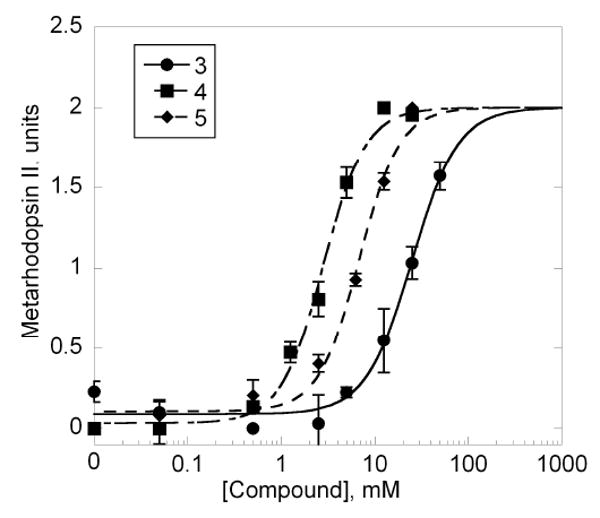
Dose-dependent stabilization of extra MII in the presence of specified compounds. The curves were fit using Kaleidagraph 4.0 with the maximal amount of extra MII being the saturating amount of extra MII for an individual compound and not the maximal MII from the peptide VLEDLKSCGLF. The EC50 values for compounds 1, 2, and 3 were 16.3±12.3 mM, 3.0±0.28 mM, and 11.8±6.7 mM, respectively.
R*-Gt Binding and Release Assay
To test if the compounds directly inhibited the interaction between rhodopsin and transducin, a binding-and-release assay was performed as described before with some alterations (35). The assays were performed with 150 μg of washed native rod outer segment membranes (WM) dissolved in ROS-Hypo buffer (10 mM Tris-HCl pH 7.4, 0.5 mM MgCl2, 1 mM DTT, 0.1 mM PMSF). The same 1:1 DMSO:buffer stock solution used for the MII stabilization assay was used for this assay, and the final concentration of DMSO in the experiment was 5%. The concentration of compound where the MII stabilization was maximal was used to test for inhibition of transducin (3 – 25 mM, 4 – 12.5 mM, 5 – 12.5 mM). The reaction was initiated by exposure of the samples to bright ambient light, followed by a 3 min incubation at 4 °C. Following light activation, the remainder of the assay was done in the light. WM were centrifuged at 45,000 rpms in a Beckman TLA45 rotor using an Optima TL ultracentrifuge, at 4 °C, for 10 min. The pellet was washed twice with ROS-Hypo buffer. WM with Gt bound was resuspended in ROS-Hypo buffer containing 0.25 mM GTPγS, incubated on ice for 3 min and centrifuged as described above. Immunoblotting was used to analyze the supernatant for the presence of G-protein subunits.
RESULTS
Virtual Screening Results
A potential bound conformation of the tetrazole molecule was modeled from the TrNOE structure of Gtα(340-350). The TrNOE structure of Gtα(340-350) is nearly identical to the X-ray crystal structure of the mutant Gtα(340-350) with Lys341 changed to Leu when bound to opsin, as shown in the supplemental information by Scheerer et al. (36). It should be noted, however that the crystallographic resolution of the complex was only 3.2 Å, making interpretation of any perceived differences problematic. It is intriguing that the crystal structure of the complex which has been assumed to be representative of the photoactivated state is stabilized by the eleven-residue peptide and show the same movement of 6-7 Å for transmembrane helix-6 found for R* by Altenbach et al. using SDSL EPR studies (21). A pharmacophore model that assumed important features of the peptidomimetic was generated in Sybyl 7.3. The Maybridge HitFinder and the NCI Diversity Libraries were searched using UNITY within Sybyl 7.3 for compounds exhibiting the same pharmacophore signature. The flexible search option within UNITY has a stochastic component, so UNITY was run 9 times to insure adequate sampling. The following 47 compounds, listed by product code, were virtual hits from Maybridge: RH 01652, KM 06188, SCR 00616, PD 00556, SPB 06654, NRB 00350, AW 00932, HTS 01763, BTB 15187, CD 09278, KM 06189, RJC 01340, HTS 04162, HTS 00241, DSHS 00855, NRB 00264, SPB 08099, BLT 00208, KM 03270, AW 00987, HTS 00495, SPB 08081, GK 02096, KM 05703, HTS 00036, KM 04213, NRB 00351, HTS 03612, HTS 12751, SCR 00611, KM 10347, JFD 03000, HTS 00437, HTS 08293, HTS 05143, SPB 02443, RH 02224, SPB 08098, RJC 02196, HTS 08665, RDR 03021, KM 01884, BTB 13772, DSHS 00248, BTB 01696, HTS 09453, and BTB 08637. No additional compound hits were obtained by searching the NCI Diversity Library. As a control, we tested another 6 compounds chosen at random from the Maybridge library. None of these compounds stabilized the MII state of rhodopsin.
Experimental Testing
The compounds from Maybridge found by ligand-based virtual screening were assayed using the extra MII stabilization assay (20, 37). Many of the compounds were sparingly soluble in the buffer typically used in this assay. The compounds were first dissolved in DMSO and then diluted into the assay buffer. The solutions were centrifuged at maximal speed for 10 min in a benchtop microfuge, and the supernatant was removed and used in the assay. Previous experiments demonstrated the extra MII stabilization assay could tolerate up to 50% DMSO (20). With increasing concentrations of DMSO, the equilibrium between MI and MII was shifted toward MII, which increases the background levels of MII in the assay. The extra MII stabilization assay was used to determine if a compound was active when compared to the positive control peptide, VLEDLKSCGLF. After the initial screen, 1 (BTB 15187), 2 (RJC 02196), 3 (RJC 01340), 4 (KM 05703), and 5 (AW 00987) all exhibited significant levels of extra MII stabilization when compared to the negative control and were considered hits (see structures in Figure 2). However, after adjusting the compound stock solution for pH, only compounds 3, 4, and 5 retained activity as shown in Figure 3 and 4.
Figure 2.
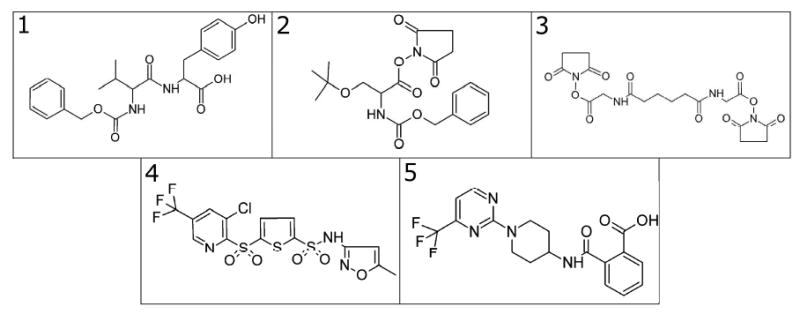
The hit compounds from the initial screen. Compounds 3, 4, and 5 stabilized the MII state.
Figure 3.
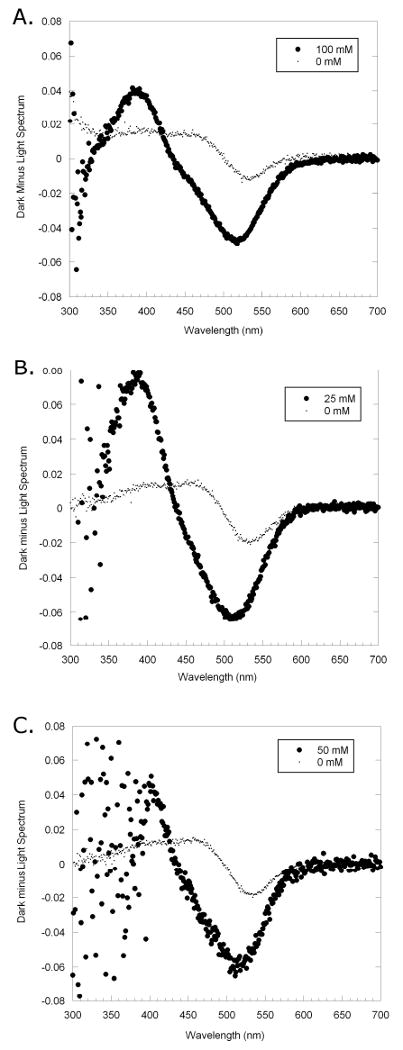
Experimental results of extra MII stabilization. The dark spectrum from the UV/Vis scan was subtracted from the spectrum taken after the rhodopsin was exposed to light. Compounds: A. 3 (100mM), B. 4 (25mM), C. 5 (50mM) are shown. The light gray trace shows the MII stabilization and MI depletion with no compound, and the dark trace shows MII stabilization and MI depletion when compound is present. In this test, there is a large difference between MII stabilization in the sample containing the compound versus the sample with no compound.
The dose response of the compounds was measured using various concentrations of compounds to derive the EC50. The EC50 values were calculated based on the maximal amount of MII stabilization for each compound and on separate experiments done in duplicate or greater number. The EC50 values of compounds 3, 4, and 5 were 16.3±12.3 mM, 3.0±0.28 mM, and 11.8±6.7 mM. The binding curves shown in Figure 4 show a representative experiment repeated at least two times. The assay was very sensitive to pH, leading to variation between separate experiments whose pH values were determined independently. For comparison, the EC50 value of Gtα(340-350) was reported as ∼100 μM (23), and the EC50 value for the high affinity peptide, VLEDLKSCGLF, was reported as ∼3.5 μM.(33).
Compounds 3, 4, and 5 were also screened using the R*-Gt Binding and Release Assay (35), which measured the ability of the compound to inhibit transducin from binding to the MII state (R*). As in the extra MII stabilization assay, the compounds were dissolved in DMSO and then diluted into assay buffer. The same 1:1 stock solution was used for this assay and the MII stabilization assay. Using 5% DMSO did not affect the results in this assay (data not shown). In the western blot shown in Figure 5, none of the compounds inhibited transducin binding to R* completely. The wells that contained the samples and the positive control (no compound added) look nearly identical. A slight decrease in band density might have been present at the highest concentrations tested, but the decrease in transducin binding was not more than 30%. If the compounds had inhibited transducin binding fully, the well containing the compound would have looked similar to the negative control.
Figure 5.
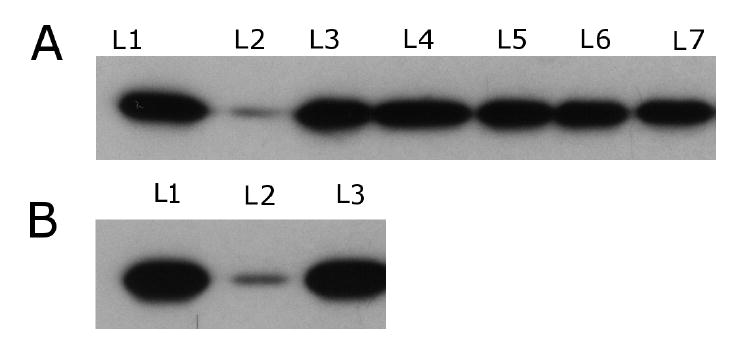
Binding and Release Assay Results. A. Results of compounds found via ligand-based virtual screening L1. Positive Control; L2. Negative Control; L3. 1 (12.5 mM), L4. 2 (50 mM); L5. 3 (25 mM); L6. 4 (12.5 mM); L7. 5 (12.5 mM). B. Results from the high affinity peptide L1. Positive Control; L2. Negative Control; L3. 1 mM. All of the bands with compound added look similar to the positive control. If the compound inhibited transducin from binding to rhodopsin, the bands would have looked similar to the negative control. At these concentrations, the compounds do not inhibit transducin from binding to rhodopsin.
Acid-trapping experiments were done on the three hit compounds to determine if they stabilized just the MII state, or multiple states of rhodopsin. Unfortunately, all three compounds precipitated when the acid was added. Therefore, the necessary spectra were not obtained.
DISCUSSION
Through the use of ligand-based virtual screening, three novel compounds that stabilized the activated state of a GPCR were found. In this study, we tested ligand-based virtual screening using the GPCR, rhodopsin, and its G-protein, transducin, which are found in the eye and involved in vision. Ligand-based virtual screening utilized a pharmacophore pattern based on important regions on the designed peptidomimetic (Figure 1b) previously found to bind and stabilize the MII state of rhodopsin (24). Because the precise bound structure of the peptidomimetic was not determined experimentally, its bound conformation was modeled by superimposing the peptidomimetics onto the reverse-turn region of the TrNOE structure of Gtα(340-350) (Figure 1a). The pharmacophoric pattern derived from the peptidomimetic was used to search the Maybridge HitFinder Library of 14,400 compounds, as well as the smaller NCI diversity library.
The search of the Maybridge HitFinder Library yielded 47 compounds that were tested experimentally for their ability to stabilize the MII state of rhodopsin and inhibit transducin binding. Of the 47 compounds, three stabilized the MII state and none inhibited the interaction between rhodopsin and transducin. Another 6 compounds were randomly chosen from the Maybridge HitFinder Library and showed no activity. Obviously, the pharmacophore model derived from the peptidomimetic compound encoded some aspects of the recognition motif of Gtα(340-350) that also does not completely block transducin binding. Gtα(340-350) and VLEDLKSCGLF do not completely block transducin binding (38), and concentrations 20- to 30-fold higher than their respective EC50 values in the extra MII stabilization assay are needed to see some inhibition (39). Because the EC50 values of the compounds found in this ligand-based virtual screen are so high, the compound concentration necessary to see significant inhibition of transducin has not been reached. It has been hypothesized that there may be a sequential mechanism of transducin binding to rhodopsin (39) and multiple binding poses for Gtα(340-350) are thought to exist (40). One of the multiple-binding sites for Gtα(340-350) and compounds 3, 4, and 5 must be an allosteric site that allows stabilization of photoactivated rhodopsin without inhibiting transducin binding. Allosteric modulaters of GPCRs have shown great promise in recent years due to their novel mechanism of control and may lead to therapies that treat multiple psychiatric and neurological disorder (17).
Compounds found in this study were very different from compounds that modulated the interaction between rhodopsin and transducin found in our previous structure-based screening (20). The structures of the current hits are much more “peptide-like” compared to the previous study where compounds contained conjugated rings. As a result, the peptide-like compounds are much more soluble and higher concentrations can be used. There is the possibility that all of these compounds may not be stable in aqueous solution, depending on time and experimental conditions; however, their results for extra MII stabilization were highly reproducible.
This study complements our previous study, which used a structure-based approach to find inhibitors of the rhodopsin/transducin interaction (20). As the MII-bound state of the C-terminal recognition motif of the α-subunit was known (it is much easier to determine the bound conformation of a ligand by TrNOE NMR than to obtain the X-ray crystal structure of the complex with the entire receptor), a pharmacophore model was generated and used to virtually search molecule libraries for leads. There is much sequence homology between Gtα(340-350) and most subclasses of C-terminal regions of G-protein α-subunits (24). In fact, the TrNOE structure of Gtα(340-350) (23) is very similar in structure to that seen in the crystal structure of Giα a rhodopsin-family G-protein (41) with backbones differing by only 0.5 Å RMSD. Potentially, a homology model of the desired C-terminal region of Gtα could be generated and used to generate similar pharmacophoric models. Given that GPCRs are important pharmaceutical targets, this alternative method for generating hits that stabilize the activated state of a GPCR may prove to be effective for other GPCR proteins. The method could be used to design small molecules that stabilize the activated state of a GPCR for crystallographic purposes as well.
CONCLUSIONS AND FUTURE DIRECTIONS
In this study, computational and experimental techniques were used to successfully find small molecules that allosterically stabilized R*. Using receptor-bound ligand-based virtual screening, three compounds that stabilized the MII-photoactivated state of rhodopsin were found. This receptor-bound ligand-based approach for finding lead compounds could readily be adapted for other GPCRs given the large sequence homology of all C-terminal regions of the G-protein α-subunits. Obviously, this approach can be used as an adjunct that complements structure-based virtual screening of GPCR homology models to find different lead motifs, especially when structural information on the protein target is limited.
Acknowledgments
This work was partially supported by National Institutes of Health National Research Service Awards (NRSA) Postdoctoral Fellowship F32GM082200 (C.M.T.), National Institutes of Health grants GM68460, GM53630, and EY1211301 (G.R.M.), Research to Prevent Blindness and NIH GM63203 grants (O.K.G.), and the University of Missouri-St. Louis Students and Teachers as Research Scientists (STARS) program (C.L.). We thank Gregory Nikiforovich for advice and critical reading of the paper. We are also indebted to our long-term collaborators, Prof. Janusz Zabrocki and Dr. Krystof Kaczamarek of the Technical University of Lodz, Poland, for synthesis and characterization of the two tetrazole peptidomimetics referred to in this paper, and to Dr. Reiko Arimoto who did the preliminary MII-stabilization bioassays during her thesis research.
Abbreviations Footnote
- GPCR
G-protein coupled receptor
- NCI
National Cancer Institute
- R*
MII-photoactivated rhodopsin
- TrNOE
transferred nuclear Overhauser effect
- IC
intracellular
- EC
extracellular
- UM
urea-washed rod outer-segment membranes
- ROS
rod-outer segment
- Gt
transducin
- Gtα(340-350)
transducin α-subunit C-terminal region
References
- 1.Howard AD, McAllister G, Feighner SD, Liu Q, Nargund RP, Van der Ploeg LH, et al. Orphan G-protein-coupled receptors and natural ligand discovery. Trends Pharmacol Sci. 2001;22(3):132–40. doi: 10.1016/s0165-6147(00)01636-9. [DOI] [PubMed] [Google Scholar]
- 2.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21(1):90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 3.Kroeze WK, Sheffler DJ, Roth BL. G-protein-coupled receptors at a glance. J Cell Sci. 2003;116(Pt 24):4867–9. doi: 10.1242/jcs.00902. [DOI] [PubMed] [Google Scholar]
- 4.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3(10):928–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Mustafi D, Palczewski K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Mol Pharmacol. 2009;75(1):1–12. doi: 10.1124/mol.108.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, et al. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci U S A. 2004;101(31):11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ananthan S, Zhang W, Hobrath JV. Recent advances in structure-based virtual screening of G-protein coupled receptors. AAPS J. 2009;11(1):178–85. doi: 10.1208/s12248-009-9094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bissantz C, Schalon C, Guba W, Stahl M. Focused library design in GPCR projects on the example of 5-HT(2c) agonists: comparison of structure-based virtual screening with ligand-based search methods. Proteins. 2005;61(4):938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 9.Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets? Proteins. 2003;50(1):5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 10.Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J Med Chem. 2004;47(22):5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 11.Evers A, Hessler G, Matter H, Klabunde T. Virtual screening of biogenic amine-binding G-protein coupled receptors: comparative evaluation of protein- and ligand-based virtual screening protocols. J Med Chem. 2005;48(17):5448–5465. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 12.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. J Med Chem. 2005;48(4):1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 13.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, et al. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J Med Chem. 2003;46(21):4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 14.Klabunde T, Giegerich C, Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J Med Chem. 2009;52(9):2923–32. doi: 10.1021/jm9001346. [DOI] [PubMed] [Google Scholar]
- 15.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100(20):11406–11. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valant C, Sexton PM, Christopoulos A. Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol Interv. 2009;9(3):125–35. doi: 10.1124/mi.9.3.6. [DOI] [PubMed] [Google Scholar]
- 17.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehlert FJ. On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(4-6):549–77. doi: 10.1007/s00210-008-0260-4. [DOI] [PubMed] [Google Scholar]
- 19.Prevost GP, Lonchampt MO, Holbeck S, Attoub S, Zaharevitz D, Alley M, et al. Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Galpha/Gbetagamma protein complex. Cancer Res. 2006;66(18):9227–9234. doi: 10.1158/0008-5472.CAN-05-4205. [DOI] [PubMed] [Google Scholar]
- 20.Taylor CM, Barda Y, Kisselev OG, Marshall GR. Modulating G-protein coupled receptor/G-protein signal transduction by small molecules suggested by virtual screening. J Med Chem. 2008;51(17):5297–303. doi: 10.1021/jm800326q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci U S A. 2008;105(21):7439–7944. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez DM, Karnik SS. Multiple signaling states of G-protein-coupled receptors. Pharmacol Rev. 2005;57(2):147–61. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- 23.Kisselev OG, Kao J, Ponder JW, Fann YC, Gautam N, Marshall GR. Light-activated rhodopsin induces structural binding motif in G protein alpha subunit. Proc Natl Acad Sci U S A. 1998;95(8):4270–4275. doi: 10.1073/pnas.95.8.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arimoto R. Thesis in Biomedical Computing. St. Louis: Washington University D.Sc.; 2002. The structural basis of the G-protein activation mechanism: rhodopsin-transducin interface. [Google Scholar]
- 25.SYBYL7.3. In: Tripos International, 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA.
- 26.Beusen DD, Zabrocki J, Slomczynska U, Head RD, Kao JL, Marshall GR. Conformational mimicry: synthesis and solution conformation of a cyclic somatostatin hexapeptide containing a tetrazole cis amide bond surrogate. Biopolymers. 1995;36(2):181–200. doi: 10.1002/bip.360360207. [DOI] [PubMed] [Google Scholar]
- 27.Smith GD, Zabrocki J, Flak TA, Marshall GR. Conformational mimicry. II. An obligatory cis amide bond in a small linear peptide. Int J Pept Protein Res. 1991;37(3):191–7. doi: 10.1111/j.1399-3011.1991.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 28.Marshall GR, Hodgkin EE, Langs DA, Smith GD, Zabrocki J, Leplawy MT. Factors governing helical preference of peptides containing multiple alpha, alpha-dialkyl amino acids. Proc Natl AcadSci USA. 1990;87(1):487–91. doi: 10.1073/pnas.87.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zabrocki J, Smith GD, Dunbar JB, Jr, Iijima H, Marshall GR. Conformational mimicry. 1. 1,5-Disubstituted tetrazole ring as a surrogate for the cis amide bond. J Am Chem Soc. 1988;110(17):5875–80. [Google Scholar]
- 30.Papermaster DS, Dreyer WJ. Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry. 1974;13(11):2438–2444. doi: 10.1021/bi00708a031. [DOI] [PubMed] [Google Scholar]
- 31.Willardson BM, Pou B, Yoshida T, Bitensky MW. Cooperative binding of the retinal rod G-protein, transducin, to light-activated rhodopsin. Journal of Biological Chemistry. 1993;268:6371–6382. [PubMed] [Google Scholar]
- 32.Yamazaki A, Bartucca F, Ting A, Bitensky MW. Reciprocal effects of an inhibitory factor on catalytic activity and noncatalytic cGMP binding sites of rod phosphodiesterase. Proc Natl Acad Sci U S A. 1982;79(12):3702–3706. doi: 10.1073/pnas.79.12.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J Biol Chem. 1996;271(1):361–6. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- 34.Anderson MA, Ogbay B, Arimoto R, Sha W, Kisselev OG, Cistola DP, et al. Relative strength of cation-pi vs salt-bridge interactions: the Gtalpha(340-350) peptide/rhodopsin system. J Am Chem Soc. 2006;128(23):7531–7541. doi: 10.1021/ja058513z. [DOI] [PubMed] [Google Scholar]
- 35.Kisselev OG, Downs MA. Rhodopsin-interacting surface of the transducin gamma subunit. Biochemistry. 2006;45(31):9386–9392. doi: 10.1021/bi060806x. [DOI] [PubMed] [Google Scholar]
- 36.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455(7212):497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 37.Kisselev O, Ermolaeva M, Gautam N. Efficient interaction with a receptor requires a specific type of prenyl group on the G protein gamma subunit. J Biol Chem. 1995;270(43):25356–25358. doi: 10.1074/jbc.270.43.25356. [DOI] [PubMed] [Google Scholar]
- 38.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241(4867):832–5. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 39.Kisselev OG, Meyer CK, Heck M, Ernst OP, Hofmann KP. Signal transfer from rhodopsin to the G-protein: evidence for a two-site sequential fit mechanism. Proc Natl Acad Sci U S A. 1999;96(9):4898–903. doi: 10.1073/pnas.96.9.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikiforovich GV, Taylor CM, Marshall GR. Modeling of the complex between transducin and photoactivated rhodopsin, a prototypical G-protein-coupled receptor. Biochemistry. 2007;46(16):4734–44. doi: 10.1021/bi700185p. [DOI] [PubMed] [Google Scholar]
- 41.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89(2):251–61. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA, USA: DeLano Scientific; 2002. [Google Scholar]