

Abstract
The cellular source(s) and the clinical significance of persistent low-level viremia, below 50 HIV-RNA copies per ml of plasma, achieved in many patients with high adherence to highly active antiretroviral therapy (HAART) remain unclear. Also, it is not clear if residual plasma HIVs during HAART can become predominant populations in the rebounding plasma viral loads after therapy interruption. Since, different HIV quasispecies tend to compartmentalize in various cell types and tissue locations in patients during chronic infection, the phylogenetic relationships between HIV sequences amplified from residual plasma viruses and CD4 T cells of five patients on long-term suppressive therapy were examined. Three of these patients stopped therapy voluntarily for three weeks, but only one of them demonstrated viral load rebound in plasma. In phylogenetic analyses, the residual plasma viruses were found to be distinct genetically from the majority of CD4 T cell-associated virus populations in 4 of 5 patients. The compartmental analyses revealed that in all patients, plasma- and CD4 T cell-derived viral sequences were compartmentalized separately. Interestingly, the plasma sequences obtained before and after HAART-off in two patients were produced apparently from the same compartment, which was different from the circulating CD4 T cell-compartment. These results suggest the possibility that residual plasma viruses in patients on long-term suppressive HAART may be produced persistently from a cellular source yet to be identified, and are capable of spreading quickly in vivo, accounting for the rapid rebound of viral loads in plasma after therapy interruption.
Keywords: HIV+ patients, suppressive therapy, residual plasma viral loads, HAART-interruption, virus compartmentalization
INTRODUCTION
HIV-1 is member of the Lentivirus genus of the family Retroviridae and gradually depletes CD4 T cells in infected humans, leading to immunodeficiency and ultimately death. HAART consisting of viral protease and reverse-transcriptase inhibitors reduces the viral loads in plasma in many patients to below the detection limit (~50 copies of HIV-RNA per ml) of currently available HIV RNA load assays [Gulick et al., 1997; Hammer et al., 1997; Luzuriaga et al., 1997]; however, it cannot completely eliminate HIV from patients [Blankson et al., 2002]. It is clear that virus production continues at levels below 50 copies/ml of plasma during suppressive HAART [Dornadula et al., 1999; Gunthard et al., 1999; Martinez-Picado et al., 2000; Hermankova et al., 2001; Havlir et al., 2003; Palmer et al., 2003; Kieffer et al., 2004; Nettles et al., 2004; Nettles et al., 2005; Tobin et al., 2005] and plasma viral loads return to detectable levels in most cases when therapy is stopped [Davey et al., 1999; Garcia et al., 1999; Mata et al., 2005]. Although a very small percentage of highly stable, resting memory CD4+ T lymphocytes (~1–10 cells per 106 CD4 cells) are known to carry latent, but replication-competent proviral DNA, even after long-term drug treatment [Chun et al., 1997; Wong et al., 1997; Hermankova et al., 2003], the cellular source(s) of persistent low-level viremia below 50 copies/ml of plasma during suppressive therapy remain unclear.
In the presence of protease and reverse transcriptase inhibitors used in HAART, the residual viruses circulating in blood likely remain immature and therefore, non-infectious, but after therapy withdrawal, they should be produced as mature and infectious particles. However, it is unclear if these residual plasma viruses can spread rapidly in the body after HAART-withdrawal, resulting in the quick return of detectable plasma viral loads in patients. Because of error-prone HIV replication, a high-degree of genetic heterogeneity in viral populations is observed, even within an infected individual [Saag et al., 1988; Shankarappa et al., 1999]. The viral quasispecies also tend to compartmentalize in the various tissues and cell-types in patients during chronic infections [refs in Zarate et al., 2007]. It is not clear if the viruses present in plasma during suppressive therapy are compartmentalized in their cellular source(s). This study attempted to investigate the phylogenetic relationships between HIVs present in CD4 T cells versus residual plasma HIVs in five patients on suppressive HAART, as well as plasma HIVs isolated 3 weeks after HAART-withdrawal from three of them.
METHODS
Study subjects
Five HIV-positive individuals (designated as P1, P2, P3, P4 and P5), who were treated with HAART for a long time in the Infectious disease clinic at UTMB and had viral loads of less than or around 50 copies per ml in plasma for the last two years or more (except any occasional blips in plasma viral loads), were studied. The study subjects signed the consent forms approved by the Internal Review Board (IRB), UTMB. About 50 ml of blood was collected from each by venipuncture at the enrollment. HAART was withdrawn from three of these patients (i.e., P3, P4 and P5) for a period of three weeks only and every week, blood samples were collected and CD4 counts and viral loads in plasma were monitored. The blood samples were collected in 2002 and later analyzed.
Blood processing and isolation of viral nucleic acids from plasma and CD4 T cells
Peripheral blood mononuclear cells (PBMCs) and plasma were separated using the ficoll-hypaque method. Then CD4 T-cells were isolated from PBMCs by a negative selection method (StemCell Technologies Inc, Canada) described elsewhere [Sahu et al., 2006]. Total DNA was isolated from CD4+ T cells by using the Wizard genomic DNA purification kit (Promega, USA) and stored at −20°C. Plasma was centrifuged at 850 × g for 20 min at 4°C in order to remove any contaminating blood cells or cell-debris. About 20 ml of plasma was then ultracentrifuged at 120,000 × g for 2h at 4°C to pellet the virus particles. Viral RNAs were isolated using a commercial kit (High Pure Viral RNA kit, Roche, USA).
Amplification, cloning and sequencing of HIV-tat regions
HIV-tat regions were amplified by nested reverse transcription (RT)-polymerase chain reaction (PCR) and nested PCR methods from about one-third volume of isolated plasma viral RNA (in 50 μl) and ~3 μg of CD4 T cells’ DNA, respectively. The superscript-III one-step RT-PCR system with platinum Taq DNA polymerase (Invitrogen) and the Expand High Fidelity PCR system (Roche) were used to amplify tat from plasma RNA and CD4 T-cell’s DNA, respectively. Five microliters of PCR product was taken for second round PCR in a 50μl volume using the Expand High Fidelity PCR system (Roche). The appropriate primers and programs were used. PCR-products were cloned and sequenced.
Phylogenetic and compartmental analyses
The nucleotide sequences were aligned using CLUSTAL-W algorithms and phylogenetic trees were constructed by using neighbor joining, maximum parsimony and maximum likelihood methods implemented in the PAUP 4.0 software package. The HKY85 formula was used for distance calculations [Swofford, 1998].
Compartmental analysis utilized the Slatkin-Maddison (SM) test implemented in the HyPhy package developed by Kosakovsky Pond, Frost and Muse [Pond et al., 2005]. The SM test [Slatkin and Maddison, 1989] determines the minimum number of migrations (M) between the separated sequence groups (compartments) in the constructed phylogenetic tree and the p-values are obtained using number of migration events (M) that can be expected in a randomly structured populations, derived by permuting sequences between groups. The p-values of <0.01 were considered as the evidence of compartmentalization.
RESULTS
Blood samples were collected from each patient at enrollment and viral loads were measured. All patients’ plasma viral loads remained below 50 copies per ml of plasma, except in one patient, P3, who had viral load of 66 copies/ml (Table I). The mean duration of undetectable viral loads prior to enrollment was ~2.5 yrs, except that each of patients P1 and P4 encountered only one blip in plasma viral loads of 476 and 793 copies/ml, respectively (Table I).
Table I.
HIV patients, antiretroviral regimens, duration of treatment, CD4 counts and plasma viral loads at enrollment in the study
| Patient ID | Antiretroviral drugs at enrollment | Duration of ART before enrollment (years) | Blood CD4 counts at enrollment (cells/μl) | PVL at enrollment (copies/ml) | Duration of undetectable PVL before sampling (years) | No. of viral blips in last 3 years before enrollment | Detection of HIV-RNA in plasma by RT-PCR at enrollment |
|---|---|---|---|---|---|---|---|
| P1 | AZT, IDV, 3TC | 7 | 986 | <50 | 2.5 | 1 (476 c/ml) | yes |
| P2 | AZT, IDV, 3TC | 6.9 | 923 | <50 | 3 | 0 | yes |
| P3 | AZT, 3TC, NFV | 5.8 | 342 | 66 | 2 | 0 | yes |
| P4 | ABC, AZT, 3TC | 3.8 | 602 | <50 | 2.2 | 1 (793 c/ml) | yes |
| P5 | ddI, d4T, NFV | 6.7 | 528 | <50 | 2.7 | 0 | no |
Abbreviations: ART, antiretroviral therapy; PVL, plasma viral loads; ABC, abacavir; AZT, zidovudine; NFV, nelfinavir; ddI, didanosine; d4T, stavudine; IDV, indinavir; 3TC, lamivudine. PVL were measured during HAART on every visit occurring at 3–4 month-intervals. However, these were measured every week after HAART-interruption. Viral loads of >400 copies/ml in plasma during HAART was included as viral blips
Although plasma viral loads remain undetectable by clinical tests in these patients during therapy, HIV-tat regions from purified plasma HIV RNAs in 4 of 5 patients still could be amplified (Table I) by following ultracentrifugation/RT-PCR-methods described by others [Kieffer et al., 2005]. Three weeks of HAART interruption in three patients (i.e., P3, P4 and P5) was done to examine the genetic relationship between rebounding virus populations in plasma versus CD4-associated viruses. Only in one patient (P3), viral loads in plasma increased marginally (up to 372 copies/ml), whereas the other two patients (P4 and P5) did not demonstrate any rises (i.e., above 50 copies/ml) in plasma viral loads during these three weeks of HAART-interruption (Fig. 1). However, viral sequences still could be amplified from plasma of patient P4, but not patient P5, after 3 weeks of HAART-off using ultracentrifugation/RT-PCR methods.
Figure 1.

Rebound of viral loads in plasma after HAART-interruption. Only patient P3 showed increasing viral loads up to 372 c/ml after therapy-off within three weeks. Despite failure to detect plasma viral loads in clinical tests (detection limit: 50 copies/ml) after therapy-off in patient P4 and P5, ultracentrifugation/PCR methods could only amplify viral sequences from P4’s plasma successfully, but not from P5’s plasma, at both time-points (before and 3wks after therapy-off). Based on the sensitivity of RT-PCR assays used, the level of viral RNA in P5’s plasma was calculated to be <1 copy/ml.
Various phylogenetic methods (such as neighbor joining, maximum parsimony and likelihood methods) showed that patient-specific tat sequences derived from plasma clustered separately, as expected, in all phylogenetic trees that were constructed (not shown). Using these methods, CD4 T cell-derived tat sequences were also found to cluster with plasma viral sequences in the patient-specific manner in the same clades of all phylogenetic trees, ruling out cross-contamination among samples or from laboratory strains (not shown). The tat region was chosen for PCR-amplification because, in addition to phylogenetic analyses, the functionality of isolated tat sequences in HIV LTR-trans activation assays could also be tested. Using routine nested PCR methods with high fidelity enzymes, heterogeneous viral populations from patients’ samples were amplified. For example, about 46–94% of amplified sequences were found to be unique (Table II). This suggests that intensive template-resampling bias in PCRs was not occurring as it ought to occur when the target copy number is low [Liu et al., 1996]. Although tat sequences isolated from each patient formed distinct groups in all phylogenetic trees expectedly (as mentioned above), compartment-specific (such as CD4 T cell- and plasma-derived) sequences in each patient appeared to cluster separately (Fig. 2). For example, in both patients P1 and P2, plasma viral populations formed a distinct cluster from the majority of the CD4 T cell-associated viruses in phylogenetic trees (Fig. 2). The plasma tat sequences derived from patients P3 and P4 also clustered relatively distantly from their CD4 T cell-associated tat sequences (either obtained from direct amplification in total CD4 T cells’ DNA or from replicating virus in culture)(Fig. 2). Interestingly, viral tat sequences isolated from patient P3’s CD4 T cell-depleted PBMCs grouped with the plasma-derived sequences obtained before and after HAART-withdrawal (Fig. 2).
Table II.
PCR-amplification of heterogeneous sequences from patients
| Patient ID | Source of amplified HIV-tat sequences (% of unique sequences amplified by PCR) | ||
|---|---|---|---|
| CD4 T cells | Plasma before HAART-off | Plasma after HAART-off | |
| P1 | 76% (13/17) | 60% (6/10) | N/A |
| P2 | 94% (15/16) | 50% (5/10) | N/A |
| P3 | 46% (6/13) | 78% (7/9) | 80% (4/5) |
| P4 | 50% (2/4) | 50% (5/10) | 78% (7/9) |
N/A, not applicable.
Figure 2.
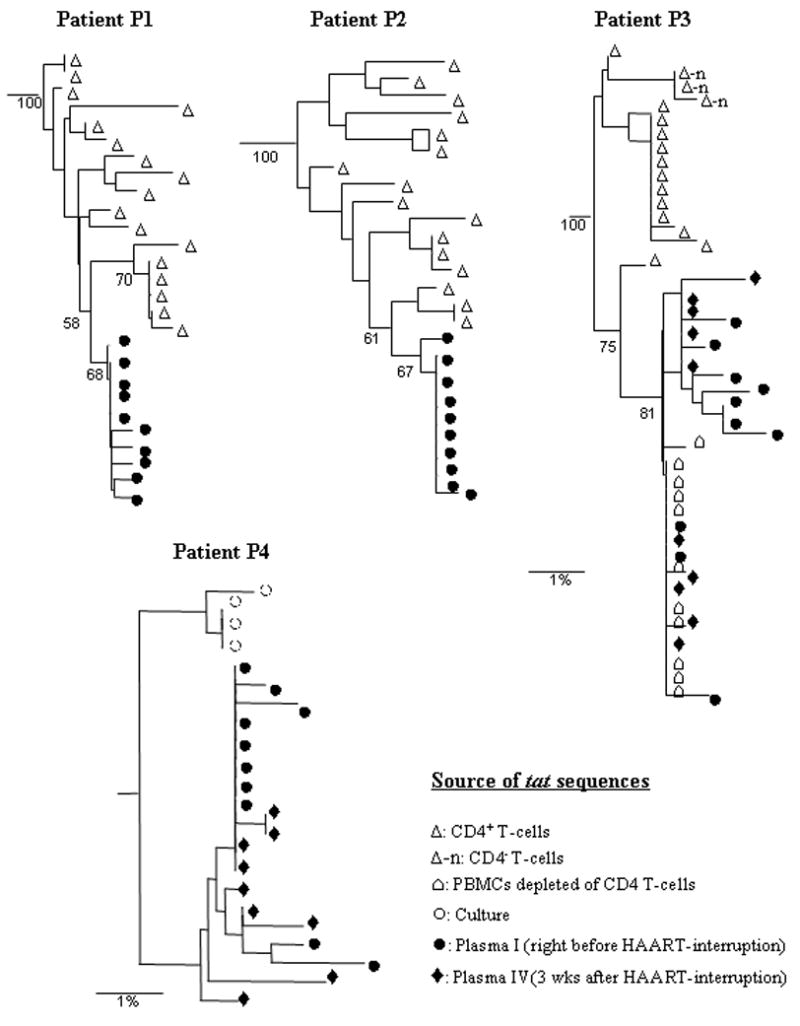
Phylogenetic analyses of tat sequences derived from patients’ CD4 T cells and plasma using neighbor joining methods. Numbers indicate bootstrap values for groups to the right, and the scale shows 1% nucleotide sequence divergence.
To check if plasma and CD4 T cell-associated sequences are actually compartmentalized, compartmental analysis was done using the Slatkin-Maddison (SM) test (see materials and methods). Essentially, the p-values and mean number of migration events (M) between two separated sequence-groups were calculated (Fig. 3). The p-values of <0.01 were considered evidence of compartmentalization. The lower M values indicate less traffic between two separated groups, providing greater evidence for close phylogenetic relatedness between sequences within each group. Patient P1 and P2: Using CD4 T cell- and plasma-derived sequences, SM test yielded p- and M-values of <0.001 and 1, respectively, in both these patients (Fig. 3, panel P1 and P2), suggesting that these sequences are compartmentalized in their corresponding sources. Patient P3: This patient showed moderate rebound in plasma viral loads after 3 weeks of HAART-interruption. When the CD4 sequence group (shown as a box) was compared with any other sequence-groups (see Fig. 3, panel P3), p values always remained less than 0.001 and M values remained at 1, suggesting that CD4 sequences form a distinct compartment. When two different plasma sequence groups (lower two boxes in Fig 3, panel P3) and PBMC-minus CD4 T cell-derived sequence-group (top right box) were compared in various combinations (broken arrows), p-values always remained higher than 0.01, demonstrating that these three sequence-groups actually represent a single compartment. These data suggest that the viruses circulating in plasma at low-levels during HAART were capable of spreading infection in the body after HAART-interruption and emerged as the predominant population in rebounding viral loads. Furthermore, it is also suggestive that non-CD4 T cells could be the cellular source for residual plasma viruses during suppressive therapy. Patient P4: Although this patient did not show rebound in plasma viral loads (i.e. >50 copies/ml) after 3 weeks of HAART-off, the viral sequences in plasma were amplified and compared them with the sequences obtained from plasma prior to therapy-interruption. The data obtained from the SM test (Fig. 3, panel P4) suggest that, similar to patient P3, this patient also had plasma and CD4-derived sequences compartmentalized. The residual plasma viral sequences amplified before or after HAART-off were genetically similar, suggesting that they were chronically produced from the same compartment (p = 0.038, M= 4).
Figure 3.

Compartmental analysis of viral sequences. This was done by Slatkin-Maddison test implemented in the HyPhy package (see materials and methods). p<0.01 is considered evidence of compartmentalization. M represents mean number of migration events between two separated populations. The lower M values indicate the less traffic between separated groups and therefore, the greater evidence of close phylogenetic relatedness between the sequences within each group. In all patients, CD4 T cell- and plasma-derived sequences were compartmentalized.
DISCUSSION
The successful amplifications of HIV sequences from plasma having viral loads of <50 copies/ml showed that viral production still continues at very low-levels in patients receiving long-term effective HAART, which is in agreement with previous reports by others [Dornadula et al., 1999; Gunthard et al., 1999; Martinez-Picado et al., 2000; Hermankova et al., 2001; Havlir et al., 2003; Palmer et al., 2003; Kieffer et al., 2004; Nettles et al., 2004; Nettles et al., 2005; Tobin et al., 2005]. This study has shown that low-level viruses in the plasma of 4 of 5 patients receiving long-term suppressive HAART were phylogenetically distinct from the majority of viral populations present in CD4 T cells, a major cellular reservoir for HIV (Fig 2 and 3). Using the SM test for sequence compartmentalization, it was found that sequences derived from CD4 T cells or plasma of these patients were, in fact, compartmentalized in their sources. Many studies have observed this compartmentalization phenomenon in various tissue locations, providing evidence for cell type-specific viral adaptation during chronic HIV infections [refs in Zarate et al., 2007]. Moreover, because of suppressive therapy, the virus spreading between various tissue sites in the body are impeded severely, but viral replication still can be active locally due to sub-optimal concentrations of drugs, resulting in micro-evolution and therefore, increase in genetic distances between viral populations residing in such sites [Si-Mohamed et al., 2000; Smit et al., 2004; Delobel et al., 2005]. The low-level plasma viruses in these patients on suppressive HAART could be produced from such a cell type in which viral populations might have evolved independently, but have been restricted to this reservoir in the presence of HAART. An alternative explanation is that the amplified plasma viral sequences may not be representative of all the viral populations residing in CD4 T cells and therefore, the observation can be misinterpreted as sequence compartmentalization. In all 4 patients, heterogeneous viral sequences from both CD4 T cells and plasma were amplified by PCR (Table II), and none of these patients showed any exception to sequence compartmentalization. Furthermore, sequences amplified from plasma that were collected at two different time-points in patient P3 and P4 showed a high degree of genetic similarity among each other in phylogenetic and SM tests, relative to the sequences amplified from CD4 T cells. This demonstrated that the observed compartmentalization of HIV sequences was not an artifact. These findings suggested that the production of low-level plasma viruses during effective HAART was not because of time-to-time reactivation of CD4 T-cell-associated latent viral quasispecies. If this would occur, then the plasma and CD4 T cell-derived viruses would not be compartmentalized separately.
In patient P3 who encountered viral load rebound after therapy withdrawal (Fig. 1), the data showed greater evidence of close phylogenetic relatedness between plasma viral sequences obtained before and after therapy withdrawal, than between plasma sequences from any time points versus CD4-derived sequences (Fig 2 and 3). These data demonstrate, for the first time, that the residual viruses are capable of rekindling infection and spreading in the body when drug-mediated inhibition is lifted. This rekindling causes quick rebound of viral loads in plasma and much of those viruses do not arise from CD4 T cell-associated viral populations, as similarly suggested by others [Chun et al., 2000; Ramratnam et al., 2000]. However, the resumption of baseline viral loads after HAART-off likely requires virus production from CD4 T-cells [Riva et al., 2003; Tierney et al., 2003]. Although another patient P4 did not show any rises in plasma viral loads (i.e., >50 copies/ml) after therapy-off, the closer phylogenetic relatedness between plasma sequences isolated before and after therapy cessation, relative to CD4 T cell-associated replicating viruses, led us to speculate that a stable non-CD4 cellular site might be making HIVs persistently at low-levels, even during therapy.
Although ~11% of tat clones isolated from patient P3’s plasma were defective in transcriptional trans-activation function (not shown), the corresponding viruses were being produced still from its’ cellular reservoir, which was either activated persistently, or did not require active Tat for low-level HIV-production. Moreover, protease region analyses revealed that none of the clones isolated from plasma possessed mutations known to confer resistance to nelfinavir, a protease inhibitor, which was used in the HAART regimen to treat this patient (not shown). These further indicate that a stable cellular source producing drug-sensitive HIVs persistently may be responsible for low-level viruses in plasma during therapy.
It is not clear which cell-type(s) could be the source for low-level plasma viruses in patients on suppressive HAART. However, intriguingly, in patient P3 the residual viruses were found to be very close in terms of phylogenetic relatedness to the sequences obtained from CD4 T cell-depleted PBMCs, which mainly consist of monocytes and NK cells as additional cellular reservoirs for HIV. Since these cell types are known to produce HIV RNA in patients during HAART [Sonza et al., 2001; Zhu et al., 2002], it can be speculated that they might be candidates for low-level viruses in plasma during therapy. Additionally, HIV-infected CD4 T cells residing in the gut may also be such a source [Poles et al., 2006]. Although the sample size (n=5) in this study is small, the provocative data obtained in the study may inspire studies on a larger number of patients to identify any specific cellular source(s) responsible for residual plasma viruses during therapy. If successful, its identification would help in developing strategies for complete suppression of residual viremia in patients during pronged effective HAART, which could delay the emergence of drug-resistant mutants during low-adherence to HAART and also prevent rapid spreading of infection during HAART interruptions.
Acknowledgments
This work was partially funded by General Clinical Research Center, UTMB, and partially by National Institutes of Health (grant: AI 062453). SDWF was supported by the National Institutes of Health (grants AI47745, AI57167).
References
- Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–93. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- Chun TW, Davey RT, Jr, Ostrowski M, Shawn Justement J, Engel D, Mullins JI, Fauci AS. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med. 2000;6:757–61. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]
- Davey RT, Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:15109–14. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delobel P, Sandres-Saune K, Cazabat M, L’Faqihi FE, Aquilina C, Obadia M, Pasquier C, Marchou B, Massip P, Izopet J. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. Aids. 2005;19:1739–50. doi: 10.1097/01.aids.0000183125.93958.26. [DOI] [PubMed] [Google Scholar]
- Dornadula G, Zhang H, VanUitert B, Stern J, Livornese L, Jr, Ingerman MJ, Witek J, Kedanis RJ, Natkin J, DeSimone J, Pomerantz RJ. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. 1999;282:1627–32. doi: 10.1001/jama.282.17.1627. [see comment] [DOI] [PubMed] [Google Scholar]
- Garcia F, Plana M, Vidal C, Cruceta A, O’Brien WA, Pantaleo G, Pumarola T, Gallart T, Miro JM, Gatell JM. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS. 1999;13:F79–86. doi: 10.1097/00002030-199907300-00002. [DOI] [PubMed] [Google Scholar]
- Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New England Journal of Medicine. 1997;337:734–9. doi: 10.1056/NEJM199709113371102. [see comment] [DOI] [PubMed] [Google Scholar]
- Gunthard HF, Frost SD, Leigh-Brown AJ, Ignacio CC, Kee K, Perelson AS, Spina CA, Havlir DV, Hezareh M, Looney DJ, Richman DD, Wong JK. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. Journal of Virology. 1999;73:9404–12. doi: 10.1128/jvi.73.11.9404-9412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, Chodakewitz JA, Fischl MA. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. New England Journal of Medicine. 1997;337:725–33. doi: 10.1056/NEJM199709113371101. [see comment] [DOI] [PubMed] [Google Scholar]
- Havlir DV, Strain MC, Clerici M, Ignacio C, Trabattoni D, Ferrante P, Wong JK. Productive infection maintains a dynamic steady state of residual viremia in human immunodeficiency virus type 1-infected persons treated with suppressive antiretroviral therapy for five years. J Virol. 2003;77:11212–9. doi: 10.1128/JVI.77.20.11212-11219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermankova M, Ray SC, Ruff C, Powell-Davis M, Ingersoll R, D’Aquila RT, Quinn TC, Siliciano JD, Siliciano RF, Persaud D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. Jama. 2001;286:196–207. doi: 10.1001/jama.286.2.196. [DOI] [PubMed] [Google Scholar]
- Hermankova M, Siliciano JD, Zhou Y, Monie D, Chadwick K, Margolick JB, Quinn TC, Siliciano RF. Analysis of human immunodeficiency virus type 1 gene expression in latently infected resting CD4+ T lymphocytes in vivo. Journal of Virology. 2003;77:7383–92. doi: 10.1128/JVI.77.13.7383-7392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, Persaud D, Siliciano RF. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J Infect Dis. 2004;189:1452–65. doi: 10.1086/382488. [DOI] [PubMed] [Google Scholar]
- Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. G-->A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol. 2005;79:1975–80. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Rodrigo AG, Shankarappa R, Learn GH, Hsu L, Davidov O, Zhao LP, Mullins JI. HIV quasispecies and resampling. Science. 1996;273:415–6. doi: 10.1126/science.273.5274.415. [DOI] [PubMed] [Google Scholar]
- Luzuriaga K, Bryson Y, Krogstad P, Robinson J, Stechenberg B, Lamson M, Cort S, Sullivan JL. Combination treatment with zidovudine, didanosine, and nevirapine in infants with human immunodeficiency virus type 1 infection. New England Journal of Medicine. 1997;336:1343–9. doi: 10.1056/NEJM199705083361902. [DOI] [PubMed] [Google Scholar]
- Martinez-Picado J, DePasquale MP, Kartsonis N, Hanna GJ, Wong J, Finzi D, Rosenberg E, Gunthard HF, Sutton L, Savara A, Petropoulos CJ, Hellmann N, Walker BD, Richman DD, Siliciano R, D’Aquila RT. Antiretroviral resistance during successful therapy of HIV type 1 infection. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10948–53. doi: 10.1073/pnas.97.20.10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata RC, Viciana P, de Alarcon A, Lopez-Cortes LF, Gomez-Vera J, Trastoy M, Cisneros JM. Discontinuation of antiretroviral therapy in patients with chronic HIV infection: clinical, virologic, and immunologic consequences. AIDS Patient Care & Stds. 2005;19:550–62. doi: 10.1089/apc.2005.19.550. [DOI] [PubMed] [Google Scholar]
- Nettles RE, Kieffer TL, Kwon P, Monie D, Han Y, Parsons T, Cofrancesco J, Jr, Gallant JE, Quinn TC, Jackson B, Flexner C, Carson K, Ray S, Persaud D, Siliciano RF. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA. 2005;293:817–29. doi: 10.1001/jama.293.7.817. [DOI] [PubMed] [Google Scholar]
- Nettles RE, Kieffer TL, Simmons RP, Cofrancesco J, Jr, Moore RD, Gallant JE, Persaud D, Siliciano RF. Genotypic resistance in HIV-1-infected patients with persistently detectable low-level viremia while receiving highly active antiretroviral therapy. Clin Infect Dis. 2004;39:1030–7. doi: 10.1086/423388. [DOI] [PubMed] [Google Scholar]
- Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–6. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poles MA, Boscardin WJ, Elliott J, Taing P, Fuerst MM, McGowan I, Brown S, Anton PA. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. J Acquir Immune Defic Syndr. 2006;43:65–8. doi: 10.1097/01.qai.0000230524.71717.14. [DOI] [PubMed] [Google Scholar]
- Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Ramratnam B, Mittler JE, Zhang L, Boden D, Hurley A, Fang F, Macken CA, Perelson AS, Markowitz M, Ho DD. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat Med. 2000;6:82–5. doi: 10.1038/71577. [DOI] [PubMed] [Google Scholar]
- Riva E, Antonelli G, Scagnolari C, Pistello M, Capobianchi MR, Monforte A, Pezzotti P, Dianzani F. Human immunodeficiency virus (HIV) DNA load and level of immunosuppression in treatment-naive HIV-1-infected patients. J Infect Dis. 2003;187:1826–8. doi: 10.1086/375251. [DOI] [PubMed] [Google Scholar]
- Saag MS, Hahn BH, Gibbons J, Li Y, Parks ES, Parks WP, Shaw GM. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature. 1988;334:440–4. doi: 10.1038/334440a0. [DOI] [PubMed] [Google Scholar]
- Sahu GK, Lee K, Ji J, Braciale V, Baron S, Cloyd MW. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology. 2006;355:127–37. doi: 10.1016/j.virol.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Mohamed A, Kazatchkine MD, Heard I, Goujon C, Prazuck T, Aymard G, Cessot G, Kuo YH, Bernard MC, Diquet B, Malkin JE, Gutmann L, Belec L. Selection of drug-resistant variants in the female genital tract of human immunodeficiency virus type 1-infected women receiving antiretroviral therapy. J Infect Dis. 2000;182:112–22. doi: 10.1086/315679. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Maddison WP. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics. 1989;123:603–13. doi: 10.1093/genetics/123.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, Saksena NK. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol. 2004;78:10133–48. doi: 10.1128/JVI.78.18.10133-10148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonza S, Mutimer HP, Oelrichs R, Jardine D, Harvey K, Dunne A, Purcell DF, Birch C, Crowe SM. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. Aids. 2001;15:17–22. doi: 10.1097/00002030-200101050-00005. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates; Sunderland, Massachusetts: 1998. [Google Scholar]
- Tierney C, Lathey JL, Christopherson C, Bettendorf DM, D’Aquila RT, Hammer SM, Katzenstein DA. Prognostic value of baseline human immunodeficiency virus type 1 DNA measurement for disease progression in patients receiving nucleoside therapy. J Infect Dis. 2003;187:144–8. doi: 10.1086/345870. [DOI] [PubMed] [Google Scholar]
- Tobin NH, Learn GH, Holte SE, Wang Y, Melvin AJ, McKernan JL, Pawluk DM, Mohan KM, Lewis PF, Mullins JI, Frenkel LM. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. Journal of Virology. 2005;79:9625–34. doi: 10.1128/JVI.79.15.9625-9634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–5. doi: 10.1126/science.278.5341.1291. [see comment] [DOI] [PubMed] [Google Scholar]
- Zarate S, Pond SL, Shapshak P, Frost SD. Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. J Virol. 2007;81:6643–51. doi: 10.1128/JVI.02268-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins JI, Corey L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. Journal of Virology. 2002;76:707–16. doi: 10.1128/JVI.76.2.707-716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]