

Abstract
Inadequate oxygen availability or hypoxia induces a complex and still incompletely understood set of adaptations that influence cellular survival and function. Many of these adaptations are directly controlled by a master transcription factor, hypoxia inducible factor-alpha (HIF-α). In response to hypoxia, HIF-α levels increase and directly induce the transcription of >100 genes, influencing functions ranging from metabolism, survival, proliferation, migration, to angiogenesis, among others. Recently, it has been demonstrated that a specific set of microRNA molecules are upregulated by hypoxia, which we denote here as “hypoxamirs.” In particular, the HIF-responsive hypoxamir microRNA-210 (miR-210) is a unique microRNA that is evolutionarily conserved and ubiquitously expressed in hypoxic cell and tissue types. A number of direct targets of miR-210 have been identified by in silico, transcriptional and biochemical methods, a subset of which have been extensively validated. As a result, miR-210 has been mechanistically linked to the control of a wide range of cellular responses known to influence normal developmental physiology as well as a number of hypoxia-dependent disease states, including tissue ischemia, inflammation and tumorigenesis. Thus, reflecting the pleiotropic actions of HIF-α, miR-210 appears to function as a “master microRNA” relevant for the control of diverse functions in the hypoxic state.
Keywords: microRNA, hypoxia, miR-210, hypoxia inducible factor, metabolism, proliferation, angiogenesis, apoptosis, cell cycle, Pasteur effect
Introduction
Low oxygen exposure induces a complex set of adaptations in the mammalian cell that remain incompletely understood. In the short term, hypoxic adaptations are important for improving survival and function, and can be essential for optimal function during this common transient stress. These adaptations and their phenotypic consequences have been most strikingly demonstrated in cell autonomous development pathways, including hematopoietic, endothelial, myocyte, adipocyte, chondrocyte, trophoblast and neuronal differentiation programs.1 Chronic exposure to inadequate oxygen levels can, however, lead to cellular dysfunction for typically aerobic mammalian cells and cause adverse pathological consequences. In turn, hypoxia is a common causative feature in pathological conditions such as tissue ischemia, inflammation and solid tumorigenesis.
In recent years, our understanding of the molecular changes that dictate hypoxic adaptation has been greatly augmented. In large part, this has been aided by the study of the hypoxia-inducible factors (HIFs), master transcription factors that are upregulated under low oxygen tension and, consequently, induce a complex set of transcriptional responses that are conserved across evolution.
HIF is a heterodimer of bHLH-PAS proteins and consists of an unstable α subunit and a stable β subunit (also called ARNT1).1 Under normal oxygen tension (i.e., normoxia), HIF-α is transcribed and translated but is rapidly hydroxylated at specific proline amino acid sites for recognition by the von Hippel Lindau protein (pVHL) and immediate proteasomal degradation. However, with hypoxia, HIF-α protein is not hydroxylated and is, thus, stabilized for heterodimerization with HIF-β. Once formed, this heterodimer can bind specific sites in gene promoters termed hypoxia response elements (HREs), thereby inducing downstream transcription. More than 100 different genes have been identified as carrying functional HREs in their promoters. Consequently, HIF-dependent transcriptional changes induce a wide range of cellular adaptations involving metabolism/respiration, cell cycle, proliferation, apoptosis, autophagy, erythropoiesis, immune reaction, cytokine production and angiogenesis, among many others.2,3 Currently, three HIF-α proteins have been described in higher metazoans: HIF-1α and HIF-2α regulate the expression of overlapping and unique transcriptional target genes,4 while some splice variants of HIF-3α have been identified as dominant inhibitors of HIF-dependent gene transcription.5 Although non-HIF-dependent pathways are activated during hypoxia, it is clear that a large component of the hypoxic program in the metazoan cell is controlled by HIF activity.
MicroRNA are Non-Canonical Regulatory Factors of the Hypoxia Response
Although the regulation1 of and the global transcriptional responses6 to HIF-α upregulation have been extensively characterized, the downstream and more complex phenotypic changes associated with hypoxia are not as well understood. Recently, endogenous microRNA (miRNA) molecules have been identified as essential, non-canonical factors that control numerous cellular processes and are particularly relevant in the hypoxic response. MiRNA are small, non-coding RNA molecules approximately 19–23 nucleotides in length that are largely conserved through evolution. The human genome is predicted to encode for greater than 1,000 miRNA genes.7 It is estimated that greater than 30% of all mRNA transcripts are regulated by miRNA in some context,7 but the functions of specific miRNA molecules are still being determined. Transcription of miRNA genes leads to the creation of hairpin-looped, primary miRNA molecules (pri-miRNA). Pri-miRNA molecules are then processed in the nucleus into smaller precursors, named pre-miRNA.8–10 Following export to the cytoplasm, the pre-miRNA hairpin loop is removed by the RNA endonuclease Dicer, yielding a double-stranded miRNA duplex. The miRNA duplex then congregates with the RNA-induced silencing complex (RISC). At this point, the active strand of the miRNA duplex can anneal to its mRNA target, while the antisense miRNA strand (miRNA*) is either degraded or recognizes a different set of target transcripts for regulation of gene expression. As part of the RISC, miRNA molecules can negatively regulate gene expression typically by interacting with the 3′ untranslated regions (3′ UTR) of specific mRNA targets to allow for repression of translation and/or mRNA degradation (reviewed in ref. 11).
Over the past three years, a number of independent studies have demonstrated that expression of a specific set of miRNA is upregulated by hypoxia. We have termed these uniquely induced miRNA “hypoxamirs,” and they appear to control a network of multiple processes during the hypoxic response. Screening to identify hypoxamirs has initially focused on culturing a variety of transformed cell types and primary tumors12–19 as well as primary cell types20,21 under hypoxic conditions. The reported miRNA sets from each study are not completely identical, which likely reflects a combination of technical differences in sensitivities of miRNA detection, the range of miRNA screened, the duration and severity of oxygen deprivation, and cellular context. Additionally, some miRNA had previously been linked to indirect, hypoxia-associated stimuli such as inflammation (i.e., miR-146a/b22), presumably suggesting that their regulation does not result from direct induction by low oxygen tension itself. Nonetheless, more than 90 miRNA have been described as induced by hypoxic exposure in a variety of cellular contexts (Supplementary Table 1). Notably, most of these studies have focused on upregulated miRNA. Downregulated miRNA have also been identified in screens after hypoxic exposure;15–17,19 however, it is still unclear if these changes simply result from cell cycle arrest or reflect more specific regulatory mechanisms. Furthermore, given that most mature miRNA are quite stable, an extended duration of hypoxia may be necessary to identify a more complete set of downregulated miRNA, in contrast to the relatively short duration of hypoxia (6–72 hours) in previous cell culture studies. In turn, the currently identified upregulated hypoxamirs likely represent only a partial list of all “hypoxia-associated” miRNA. For instance, in this review, we have elected not to list a number of additional miRNA that have been identified in pathophysiological conditions of ischemia (as reviewed by Fasanaro and colleagues23), which are subject to a complex set of hypoxia-dependent and hypoxia-independent influences. It is likely that at least some of these “ischemia-regulated” miRNA are directly linked to hypoxia, but delineation of that subset is still pending. As a result, we expect that this list will continue to expand as screening technologies become more accurate and less expensive.
Although a number of hypoxamirs have been identified, most demonstrate modest induction, and many are upregulated only in certain cellular contexts. In contrast, all of these studies have identified microRNA-210 (miR-210) as a unique hypoxamir that is robustly and ubiquitously induced in both transformed and primary cell types.12–21,24–26 Notably, when compared directly, the absolute levels of miR-210 induced by hypoxia are largely similar across most cells;21 however, certain cell types, such as primary vascular endothelial cells, carry particularly low levels of miR-210 in normoxia, perhaps suggestive of an augmented action of this miRNA when induced by hypoxia. Nevertheless, while some heterogeneity exists in the baseline level of expression and relative degree of upregulation, its ubiquitous expression during hypoxia likely reflects fundamental and highly conserved actions for this miRNA.
Independent pieces of data now exist that link the dynamic regulation of miR-210 directly to low oxygen levels as opposed to other mechanisms that are secondarily associated to hypoxia (i.e., oxidative stress, inflammation and others). Unlike inflammatory-driven, hypoxia-associated miRNA such as miR-146a and miR-146b (Chan and Loscalzo, unpublished data), there exists a dose dependent upregulation of miR-210 as oxygen levels decline,20,21 suggesting a direct dependence on hypoxia itself for expression. Reinforcing this principle, exposure of cultured cells to low pH, oxidant stress, growth factor deprivation, and osmotic stress to mimic hypoxia-associated processes does not induce miR-210.20 The genomic location of the miR-210 gene is also consistent with its regulation by low oxygen concentrations—it is embedded in the intronic sequence of a mRNA transcript AK123483 that itself is hypoxia-inducible,14 and miR-210 is flanked by two other genes, HRAS and RASSF7, with expression that correlates both with miR-210 levels and hypoxic exposure in vivo.26 Finally, independent biochemical and genetic data12,20,26 have indeed confirmed that the promoter of miR-210 carries a functioning HRE that is recognized by HIF-1α for induction of miRNA transcription upon hypoxic exposure. Multiple groups have found that miR-210 is specifically induced by HIF-1α;12,14,20,25,26 however some,25 but not all,14 investigators have found similar regulation by HIF-2α. Notably, a few other miRNA have also been verified as HIF-dependent,12,24 such as miR-373, but expression of these miRNA appears more restricted and less robust in response to hypoxia. Therefore, miR-210 is unique in its wide distribution, HIF dependence and robust upregulation in response to hypoxia.
Furthermore, observational evidence suggests that levels of miR-210 are specifically regulated in a number of human disease states, indicating an important role for this miRNA in modulating hypoxia-induced pathogenesis. For example, miR-210 is induced in certain pathophysiological conditions associated with hypoxia, such as placental tissue derived from patients with preeclampsia,27,28 macronodular adrenocortical disease,29 and tissue derived from murine models of experimentally induced hind-limb ischemia.18 MiR-210 is also upregulated in a significant percentage of human samples derived from primary solid tumors, including head and neck cancer,26 melanoma,30 glioma,31 small cell and lung cancer,31 lung adenocarcinoma32 and adrenocortical carcinoma.33 Correspondingly, secreted miR-210 has been proposed as a novel biomarker of diffuse large B-cell lymphoma34 and pancreatic cancer.35 Genomic deletions of miR-210 have also been reported in human epithelial ovarian cancer samples, and these deletions are suggested as a possible trigger to tumorigenesis.13 Correspondingly, Huang and colleagues have demonstrated that forced expression of miR-210 in implanted tumor tissue can repress tumor growth in immunodeficient mice.26 However, upregulated levels of miR-210 in human breast cancer samples display an inverse correlation with disease-free and overall patient survival,14 and, in a separate study, miR-210 levels correlate with breast cancer aggressiveness and metastatic potential.36 While somewhat paradoxical, these findings taken together may indicate that miR-210 can play either adaptive or maladaptive roles in repressing or disease conditions, respectively, depending upon the specific in vivo context. Notwithstanding the unresolved questions regarding its mechanisms of action, miR-210 certainly appears relevant not only during adaptation to hypoxic stress in normal physiology but also in a variety of hypoxia-induced diseases.
Numerous Direct Targets of miR-210
The recognition of target mRNA by miRNA in vivo requires variable degrees of Watson-Crick complementarity. Conserved base-pairing at the 5′ region of the miRNA (at positions 2–8), termed the seed sequence, is especially important for target binding;37 however, “seed matches” are not always sufficient nor required for target repression, and other general features of site context exist that appear to enhance site efficacy.38 Based on these features, multiple algorithms have been developed to predict mRNA targets of specific miRNA molecules and most carry roughly 50%–60% validation rates. As a result, experimental confirmation of relevant targets is necessary and can be technically challenging. Recently, a number of independent studies have identified a diverse array of both verified and putative targets of miR-210, consistent with the pleiotropic actions of this miRNA during the complex hypoxic response.
Most early studies of miR-210 have utilized primarily an in silico bioinformatic approach to screen for predicted targets with the highest likelihood for biological and physiological relevance. Because no particular published algorithm has been uniformly accepted as most accurate in predicting target transcripts, these studies have concentrated on studying the targets predicted by at least one,13,24,25 and often multiple, independent algorithms18,20,21 for further verification.
A more sophisticated protocol was employed by Zhang and colleagues,25 based on the observation that downregulated transcripts identified shortly after introduction of miR-210 are more likely to be direct miR-210 targets. In doing so, 31 such transcripts were identified as downregulated by miR-210 that also carry in silico predicted target sequences of this miRNA. Notably, one of these had previously been validated as a direct target (ephrin A3 or EFNA3).20 Furthermore, the c-MYC antagonist, MNT, was further validated by this group as a direct target, and mainly by transcriptional array analyses, downregulation of MNT was also demonstrated as functionally important in controlling the cell cycle through a reciprocal upregulation of c-MYC activity (see below for details). Validation of the other 29 transcripts as direct targets of miR-210 is, however, pending.
More recently, integrated approaches utilizing bioinformatics together with array-based technology have been successful in identifying a more complete list of biologically relevant, direct targets of miR-210. An advent in the screening tools available for miRNA targets includes the use of biochemical immunoprecipitation of Argonaute 2 (AGO2), one of the major protein components of the miRNA-containing RISC (as detailed above), followed by expression array analysis of AGO2-bound transcripts.39 Huang and colleagues adopted such an approach by comparing transformed cells exposed to either normoxia or hypoxia.26 In doing so, 246 transcripts were identified as enriched in hypoxia-exposed, AGO2-immunoprecipitates, 50 of which correspond with in silico predicted targets of miR-210. Some of these 3′UTR target sequences were then verified by trans-repression luciferase reporter assays, thereby confirming these sequences as recognized by miR-210 for gene repression. Verification of the remaining genes identified in this screen will be necessary to distinguish additional direct targets of miR-210 from other hypoxia-dependent, yet miR-210-independent, immunoprecipitated transcripts.
Another recent study by Fasanaro and colleagues40 utilized proteomic profiling of miR-210-expressing endothelial cells (human umbilical vein endothelial cells, HUVEC) in combination with transcriptional profiling of HUVECs after forced expression of miR-210 during normoxia and after antisense inhibition of miR-210 during hypoxia. After proteomic analysis, only 11 proteins were found reproducibly downregulated in miR-210-positive HUVECs. In comparison, after transcriptional array analysis, 51 transcripts were identified as both downregulated by forced expression of miR-210 during normoxia and upregulated after inhibition of miR-210 during hypoxia. Of these 62 gene products, very few are predicted as targets of miR-210 by published in silico algorithms—7 in total and only 1 of which (KLF12) is predicted by multiple algorithms. In contrast, when a low stringency search for miR-210 seed sequence matches was performed, 42 of the 62 potential targets were found to carry one or more potential target sites for miR-210. Notably, however, 29 of these targets carry these sites in the transcript coding region and 5 display such sequences in the 5′ UTR. Recent reports indicating that such locations of miRNA target sequences can still function to repress gene expression41–46 add credence to the notion that these mRNAs may represent functioning targets of miR-210 in vivo.
These results were then coupled with transcriptional profiling after AGO-2/RISC immunoprecipitation derived from miR-210-expressing transformed cells (human embryonic kidney HEK-293 cells). Of the 42 previously identified transcripts by transcriptional/proteomic analysis above, 16 were enriched for by AGO-2/RISC immunoprecipitation. Additionally, another 15 immunoprecipitated transcripts are predicted targets of miR-210 through in silico algorithm analysis. Of these 31 final putative target transcripts, all were found to be downregulated by hypoxia. After further verification through trans-repression luciferase reporter assays and/or modulation of hypoxic expression with miR-210 antisense inhibition, 25 of the 31 candidates were definitively identified as direct targets of miR-210. Notably, these targets identified by Fasanaro and colleagues demonstrate little overlap with those reported by Huang and colleagues, again potentially highlighting the differences of cellular context and/or experimental techniques. Nevertheless, taken together, these data certainly offer a more complete set of verified targets (Fig. 1) from which to base further validation studies of the actions of miR-210 in vivo.
Figure 1.

MiR-210 directly represses multiple transcripts associated with diverse cellular functions. To date, at least 35 unique transcripts (denoted by their official gene symbol) have been identified and further verified as direct targets of repression by miR-210. These transcripts are mechanistically linked to a wide array of fundamental cellular processes (denoted in the gray ovals) that are critical to hypoxic survival and adaptation of the mammalian cell. Here, verified targets of miR-210 are listed if previously found to be downregulated by miR-210, either through gain-of-function (forced expression of miR-210 during normoxia) or loss-of-function (inhibition of endogenous miR-210 during hypoxia) assays, coupled with either confirmation of specific recognition of the predicted target sequence by miR-210 (i.e., transactivation luciferase assays) or biochemical immunoprecipitation of the target transcript by miR-210-enriched AGO2/RISC. Notably, additional in silico-predicted transcripts have been further implicated as possible targets of miR-210 through either miR-210-dependent immunoprecipitation of AGO2/RISC or transcriptional/proteomic high-throughput analysis after manipulation of miR-210 expression. We expect that a significant percentage of these candidates will be confirmed as targets in the future, and as a result, the number of associated cellular processes controlled by this versatile hypoxamir will continue to expand.
Pleiotropic Actions of miR-210 Control the Hypoxic Response
The broad number of putative and verified gene targets of miR-210 identified to date further enforces the idea of this miRNA as a master regulator of the hypoxic response (Fig. 1). Furthermore, transcriptional profiling of miR-210-specific effects on overall hypoxic programming in HUVECs as detailed by Fasanaro and colleagues40 and in transformed cells as detailed by Zhang and colleagues25 indicate that miR-210-modulated genes are associated with RNA and DNA processing, binding and repair; cytoplasmic and nuclear trafficking; mitochondrial function; metabolism; cell cycle control and proliferation; apoptosis and cell survival; and differentiation and development. After performing an in silico analysis of gene interactions within these pathways, Fasanaro and colleagues have further highlighted processes associated with ATM (ataxia telangiectasia mutated), FAS, and TNFR1 apoptotic pathways; integrin and agrin pathways; and motility and cytoskeletal organization.40 Notably, the pathway, “hypoxia-inducible factor in the cardiovascular system,” was significantly enriched, correlating well with the separate observation of the robust hypoxic enrichment of miR-210 especially seen in the vascular endothelial compartment.21 Furthermore, a number of these gene ontology (GO) functions and additional pathways intersect with known hypoxia-induced gene programs (Fig. 2), further highlighting the pleiotropic functions of this miRNA in hypoxic gene reprogramming.
Figure 2.

HIF-dependent gene pathways coincide with miR-210-dependent gene pathways. Pathways of gene regulation in endothelial cells by miR-210 as assessed by transcriptional and proteomic analyses40 intersect with a majority of well-characterized gene expression changes induced by HIF-1α during hypoxic exposure.1 While some HIF-dependent processes have not been definitively implicated in the actions of miR-210 (i.e., direct erythropoiesis), it is still possible that a subset of these actions may be linked to miR-210 in separate cell-specific or environmental contexts. Furthermore, while HIF-1α appears to play a predominant role in the induction of miR-210, baseline, endogenous levels of miR-210 are higher in certain cell types21 and may carry some biological activity during normoxia.20 Thus, HIF-independent effects of miR-210 are possible and await further clarification.
Understanding the overall functions of this miRNA has been greatly advanced through interrogation of the mechanistic basis of how target gene expression affects downstream phenotypic responses. The following discussion of these actions is primarily based on specific studies of a few relevant target genes; however, these studies are far from complete, especially as more direct targets are clarified. Furthermore, caveats to the study of further downstream biology are warranted. Although miR-210 appears ubiquitously upregulated in nearly all hypoxic cell types, some of its functions may be significantly different depending upon cellular context. These functional differences may stem from varying levels of miR-210 upregulation in response to hypoxia21 and/or from differences in basal expression of potential targets. Such stoichiometric differences between miRNA and its targets could theoretically favor the importance of distinct subsets of targets, depending upon cell type or environmental milieu. For example, while HOXA9 may be a critical factor as a miR-210-dependent target in specific tumor cells,26 HOXA9 transcript is not detectable in pulmonary arterial endothelial cells (Chan SY and Loscalzo J, unpublished data) and, thus, presumably has a minimal role in control of miR-210-dependent actions in the latter cell type. Therefore, depending upon the regulation of the stoichiometric ratios of miR-210 and any given target, certain actions of miR-210 may be universally observed in all hypoxic cells, while other actions may be unique to specific cell types.
Metabolism
In the nineteenth century, Louis Pasteur first described a fundamental cellular response to hypoxia resulting from a metabolic shift from mitochondrial oxidative phosphorylation to glycolysis.47 This “Pasteur effect” profoundly influences cell survival and function, and is thought to optimize the efficiency by which energy is generated (ATP) while avoiding excessive generation of toxic reactive oxygen species (ROS).48 Its underlying molecular mechanisms, however, are incompletely understood with only a limited number of factors,48 including HIF-1α,49,50 that have been identified as regulating shifts in hypoxic mitochondrial respiration. Recently, we identified miR-210 as a key factor regulating HIF-dependent control of the Pasteur effect via its repression of mitochondrial metabolism in multiple cell types exposed to hypoxia21 (Fig. 3A).
Figure 3.

MiR-210 inhibits mitochondrial metabolism via repression of ISCU1/2 and iron-sulfur cluster-dependent function. (A) Louis Pasteur first described a fundamental cellular response to hypoxia, resulting in a metabolic shift from mitochondrial oxidative phosphorylation to glycolysis. The molecular mechanisms underlying this complex event have been incompletely characterized to date. (B) Iron-sulfur cluster assembly proteins ISCU1/2 are essential for iron-sulfur cluster biogenesis. These moieties are incorporated into a wide variety of proteins, many of which are involved in mitochondrial metabolism and aerobic energy production. While the function of iron-sulfur clusters has been more extensively studied in bacteria and yeast, less is known regarding their dynamic regulation and function in hypoxic mammalian cells. (C) MiR-210 regulates iron-sulfur cluster-dependent metabolic function during hypoxia via direct repression of ISCU1/2, leading to downregulate iron-sulfur cluster biogenesis and iron-sulfur-dependent metabolic enzyme activity. In doing so, miR-210 disrupts mitochondrial respiration and potentially other iron-sulfur cluster (ISC)-dependent functions such as iron metabolism, among others. As a result, miR-210 modulates a unique constellation of essential metabolic functions that predominates in the Pasteur effect, and influences cellular adaptation to hypoxia in the mammalian cell.
This unique metabolic function of miR-210 derives from its direct repression of the iron sulfur cluster assembly proteins ISCU1 and ISCU2. Initially, by analysis of multiple bioinformatic algorithms, we found that a highly conserved binding site for miR-210 is predicted in the 3′ UTR of the ISCU transcripts, which was then verified as a functioning target sequence through a combination of hypoxia-dependent and miR-210-dependent luciferase reporter gene assays and endogenous ISCU expression studies.21 In separate studies, ISCU1/2 has been confirmed as a hypoxia- and HIF-dependent gene,51 and has been verified as a direct target of miR-210, both by independent transcriptional screens25 and miR-210-enriched AGO2-immunoprecipitation of ISCU transcript.40 Notably, ISCU1/2 is expressed ubiquitously and has been identified as a relevant miR-210 target in a wide array of cell types, in culture and in vivo.21 Unlike target transcripts with a more limited range of expression, modulation of ISCU1/2 appears to reflect a more universal function for this otherwise pleiotropic miRNA.
In mammalian cells, two splice isoforms of ISCU exist. Both transcripts carry an identical 3′ UTR, but the translated proteins differ in their location: ISCU1 is located in the cytosol, whereas ISCU2 is located in mitochondria.52 In order to highlight their functional and structural similarities, these proteins are described here by a single term, ISCU1/2. ISCU1/2 are essential scaffolding proteins that collaborate with a large set of evolutionarily conserved assembly and transfer factors53 to produce [4Fe-4S] and [2Fe-2S] iron-sulfur clusters. These prosthetic groups promote electron transport and oxidation-reduction reactions integral to numerous cellular processes, ranging from ribosome biogenesis, purine catabolism, heme biosynthesis, DNA repair, to iron metabolism, among others.54 In particular, iron-sulfur clusters are incorporated into enzymes that are responsible for mitochondrial respiration and energy production. These include the enzymatic isoforms of aconitase, which are integral to the TCA cycle, and the mitochondrial respiratory complexes (Complexes I, II and III), which facilitate electron transport54 (Fig. 3B).
A sophisticated molecular picture of iron-sulfur cluster biogenesis, trafficking and enzyme function has previously been established, derived mostly from studies in bacteria and yeast where these clusters are more easily quantified and manipulated.55 Genetic disruption of cluster biogenesis has profound consequences for cellular survival,54 indicating their fundamental biological importance in both prokaryotes and eukaryotes. Furthermore, even after biogenesis and enzyme incorporation, the integrity of iron-sulfur clusters is acutely sensitive to changes in oxidation-reduction states, leading to direct interactions with intracellular oxygen and oxygen radicals56 as well as intracellular nitric oxide (NO)57–61 and reactive nitrogen species, such as peroxynitrite (ONOO−).57,62 As a result, in single-cell organisms, iron-sulfur clusters have been proposed as intracellular sensors of oxygen56 and nitric oxide;63 however, due in large part to the difficulty of studying iron-sulfur clusters in multi-cellular organisms, the regulation of iron-sulfur cluster biogenesis and ISCU1/2, particularly in mammalian cells, has been less well characterized. Currently, our in vivo knowledge of deficient iron-sulfur cluster biogenesis in mammalian cells is indirect, based mostly on studies of frataxin, a protein which cooperates with ISCU2 and mitochondrial biogenesis machinery in mitochondrial iron-sulfur cluster production. Studies in frataxin-deficient mice64 have revealed its critical role in augmenting mitochondrial energy production through electron transport and oxidative phosphorylation, as well as in altering iron homeostasis. In correlation, a mutation in ISCU1/2 has recently been linked to a human genetic syndrome leading to myopathy and lactic acidosis;65 however, prior to our most recent studies of miR-210, the dynamic regulation and function of ISCU1/2 in the control of iron-sulfur cluster activity during hypoxia has been unappreciated.
Upon identifying ISCU1/2 as a miR-210 target, it has been shown that HIF-expressing tissues in vivo demonstrate increased levels of miR-210, decreased expression of ISCU1/2, and consequent disruption of the integrity of iron-sulfur clusters, as assessed by electron paramagnetic resonance spectroscopy. In turn, through repressing ISCU1/2 during hypoxia, miR-210 decreases the activity of prototypical iron-sulfur enzymes controlling mitochondrial metabolism, including Complex I and aconitase. Consequently, miR-210 represses mitochondrial respiration, which, in the presence of normal oxygen levels, leads to decreased ATP levels. In contrast, during hypoxia, when repression of electron transport corrects the mismatch with reduced oxygen tension, miR-210 appears to increase ATP levels21 and optimize energy production in the hypoxic cell via the Pasteur effect48 (Fig. 3C).
When interpreted together with our prior understanding of iron-sulfur cluster biology, such a miR-210/ISCU1/2 pathway highlights important mechanistic connections among microRNA, iron-sulfur cluster assembly, hypoxia and mitochondrial function, with broad implications for understanding cellular metabolism and cellular adaptation to metabolic stress. First, the characterization of this type of prosthetic group as a key determinant of this metabolic shift during hypoxia suggests that a combination of acute sensitivity of iron-sulfur cluster integrity to oxidative stress55 along with repression of iron-sulfur cluster biogenesis via downregulated ISCU1/2 may provide a real-time redox-sensitive “switch” that is triggered by hypoxia to induce downstream metabolic changes. Second, in addition to its effects on mitochondrial metabolism, the miR-210-ISCU1/2 axis may regulate additional hypoxia-dependent and iron-sulfur cluster-dependent processes. These may include iron metabolism, which is controlled by ISCU1/2 activity66 and hypoxia,67 but the mechanisms have only been partially delineated. Correlating with the signaling pathways highlighted by transcriptional array analysis,40 these may also include DNA/RNA and amino acid catabolism and cellular proliferation. Moreover, hypoxic repression of mitochondrial function by miR-210 and ISCU1/2 carries the theoretical potential to trigger mitophagy—a form of autophagy where defective mitochondria are selectively delivered to lysosomes for degradation. Mitophagy has been characterized as a HIF-dependent mechanism.68 It remains to be seen if miR-210 itself induces such mitophagy and if that mitophagy would lead to further adaptive or degenerative processes. Finally, future studies will be necessary to delineate the more complex effects of miR-210 and ISCU1/2 in vivo under physiological and hypoxia-dependent disease states. For example, in the short term, the miR-210-dependent Pasteur effect improves survival and allows for hypoxic cellular adaptation. This effect may be especially important in specialized developmental and differentiation processes that rely upon distinct hypoxic cues for initiation and maintenance. By contrast, chronic repression of mitochondrial function during hypoxia has been linked to numerous adverse pathologic consequences, especially in the vascular compartment in cases of hypoxia-ischemic injuries,69 stroke70 and diabetes mellitus.71,72 As a result, perhaps mirroring the varying effects of HIF-1α itself, we predict that the downstream phenotypic consequences of the metabolic actions of miR-210 in vivo will be significant and important in both normal adaptive physiology and hypoxia-dependent pathogenic conditions; however, these effects may be drastically different, depending upon a complex range of factors, including tissue type, duration of expression, and ambient oxygen concentration.
Cellular survival
The control of cellular survival during hypoxic stress also appears integral to the function of miR-210. Even prior to the verification of any miR-210 targets, miR-210 had been identified as protective to transformed cells,12 primary cells20 and mesenchymal stem cells73 in inhibiting apoptosis in response to hypoxic stress. Much like its control of the Pasteur effect, this overall cytoprotective action of miR-210 sheds light on an evolutionarily conserved adaptive process in response to cellular stress. Furthermore, this action carries significant implications in pathophysiological processes during hypoxia that may link to protective mechanisms of augmented cellular survival. Notably, these include solid tumor biology, where hypoxic cancer cells remain viable despite outgrowth of their blood supply; pulmonary vascular disease, where hypoxic vessels survive and remodel; and conditions in which inflammatory signals persist despite persistently low oxygen tension. As a result, improved understanding of the mechanism(s) by which miR-210 controls cell survival may offer a novel therapeutic target for these disease states.
Nonetheless, causative attribution of particular targets of miR-210 to this mechanism is a challenging process, as a complex set of diverse pathways likely influence apoptotic potential in the hypoxic state. One such relevant target includes ISCU1/2 and its control of mitochondrial electron transport and respiration. It has been proposed that hypoxia initially upregulates mitochondrial ROS flux and consequently induces HIF-1α due to a mismatch of electron transport (and ROS leakage) and decreased oxygen partial pressure. Upon induction of HIF-1α and HIF-dependent compensatory mechanisms, however, repression of electron transport corrects the mismatch with reduced oxygen tension, thereby decreasing ROS production, decreasing apoptosis, and optimizing energy production in the hypoxic cell.48 Based on this model, the repression of ISCU1/2 and iron-sulfur cluster biogenesis by miR-210 during normoxia would create a mismatch in electron transport and oxygen concentration that leads to an increase in toxic ROS production and increased apoptosis, as corroborated by our data in pulmonary vascular endothelial cells.21 It remains to be seen if this pro-apoptotic phenotype is also present in other primary and transformed cell types. By contrast, during hypoxia, when repression of electron transport corrects the mismatch with reduced oxygen tension, miR-210 appears to decrease ROS flux, to increase ATP levels,21 and to protect against apoptosis in both primary and transformed cells.12,20 These data support a model in which miR-210 carries a maladaptive role during normoxia at least in certain cell types, causing a mismatch of ambient oxygen tension and electron transport, but an adaptive role during hypoxia, due, at least in part, to the homeostatic correction of the imbalance of electron transport and oxygen tension (Fig. 4A).
Figure 4.
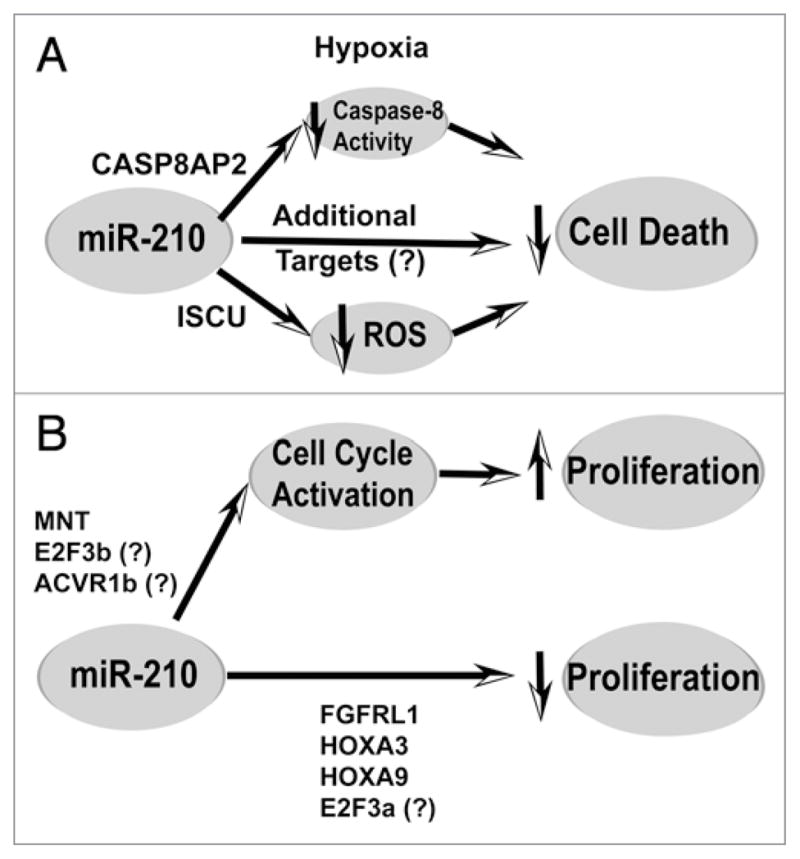
Complex control of cellular survival and proliferation by miR-210. (A) MiR-210 increases cell survival during hypoxia by modulating multiple direct targets involved in apoptosis and cell death. By repressing CASP8AP2 and directly modulating caspase-8 activity, miR-210 improves survival of mesenchymal stem cells after engraftment into infarcted cardiac tissue. Furthermore, by repressing ISCU1/2, miR-210 appears to decrease ROS levels and thereby improve survival of human pulmonary vascular endothelial cells exposed to hypoxia. There likely exist additional direct targets of miR-210 that further contribute to its pro-survival and adaptive functions during hypoxic stress. (B) MiR-210 may induce cell type-specific control of proliferation. In certain transformed cells, inhibition of direct target MNT and perhaps E2F3b and ACVR1b by miR-210 leads to an activation of G1/S cell cycle progression and increased cellular proliferation. However, in other transformed and tumor cells, inhibition of FGFRL1, HOXA3, and, perhaps, HOXA9 and E2F3a appear to reduce cellular proliferation. It is unclear how to reconcile these seemingly paradoxical findings. However, it is reasonable to hypothesize that a given cell type or basal milieu may carry different subsets of the total complement of targets of miR-210 and, thus, differentially respond to the control of cellular proliferation based on those distinct targets.
An additional direct target of miR-210, FLASH/caspase-8 associated protein-2 (CASP8AP2), can also influence cellular survival (Fig. 4A).73 Human CASP8AP2 is a FLICE-associated protein (FLASH) homolog and can regulate Fas-mediated apoptosis via interaction with the death-effector domain (DED) of caspase-8. Once activated, caspase-8 is released from the complex and participates in the activation of effector caspases, including caspases-3, 4, 6, 7, 9 and 10.74 Following multiple rounds of ischemic preconditioning, Kim and colleagues have reported that rat mesenchymal stem cells (MSCs) exhibit increased levels of HIF-1α and miR-210 along with a specific downregulation of CASP8AP2 that coincides with its translocation from the nucleus to the cytoplasm. These events correlate with improved MSC survival under subsequent anoxic conditions following engraftment into infarcted cardiac tissue; these improvements in cellular survival are also mimicked by RNA antisense inhibition of CASP8AP2, independent of miR-210 or ischemic preconditioning. At present, it remains to be seen if this miR-210-dependent action is conserved in other mammalian species, and if it is active in more differentiated primary and transformed cell types.
While both ISCU1/2 and CASP8AP2 have been directly linked to the control of cellular survival, other, as yet uncharacterized, direct targets of miR-210 likely influence these apoptotic pathways. Notably, the repression of ISCU1/2 only partially induces this effect, as constitutive expression of 3′UTR-deficient transcripts of ISCU1 and ISCU2 only partially rescues the miR-210-dependent apoptotic phenotype.21 Upon analysis of the recently identified miR-210 targets, at least 10 genes in addition to ISCU1/2 and CASP8AP2 (Fig. 1) have been linked to cellular survival. Unraveling the complexities of these combinations of targets will certainly be necessary prior to establishing an acceptable therapeutic avenue for hypoxia-dependent disease conditions.
Proliferation
Similarly, and possibly related to its cytoprotective effects, control of proliferation of hypoxic cells has also been linked to miR-210 (Fig. 4B). In cases of both squamous cell carcinoma of the head and neck as well as adenocarcinoma of the pancreas, in vivo tumor growth in immunodeficient mice is substantially repressed after forced expression of miR-210. This phenotype correlates with the known induction of cell cycle arrest at the G1/S transition during hypoxia.75–77 However, Zhang and colleagues have found that this miRNA appears to induce, rather than repress, the G1/S transition in other transformed cells, leading to a decrease in proliferation.25 Therefore, it is unclear if miR-210 acts primarily as a positive or negative regulator of proliferation, and if this action may vary depending upon contextual cues. A proliferative phenotype carries obvious therapeutic implications in hypoxic tumor growth; additionally, if present in primary cell types, such as in the vasculature, this phenotype may carry equally important ramifications in the progression of a variety of vascular diseases marked by dysregulated proliferation (e.g., atherosclerosis and pulmonary hypertension).
A careful delineation of the mechanisms at play in cell cycle control and proliferation is required for exploitation of this miRNA for therapeutic gain. As in the case of cellular survival, the control of proliferation by miR-210 is likely induced by repression of a number of direct target transcripts. A few of these targets have been interrogated in sufficient detail to establish plausible mechanisms by which proliferation is affected during hypoxia. First, Huang and colleagues have identified the novel targets FGFRL1 and HOXA3 as factors downregulated by miR-210, which, in turn, appear to decrease tumor initiation and growth.26 FGFRL1 is the fifth and most recently discovered fibroblast growth factor receptor (FGFR), a family of receptors important in a wide range of complex biological actions involving growth, differentiation and organogenesis.78,79 Recent work suggests that FGFRL1 may act as a negative regulator of FGF signaling, and, potentially, is a decoy receptor for FGF ligands.80 HOXA3 belongs to the HOX family of transcription factors with well-described roles in control of morphogenesis during embryonic development. Interesting, HOXA3 has been implicated in both angiogenesis and wound repair via promotion of endothelial and epithelial migration.81 As a result, the independent identification of these genes as direct targets of miR-210 correlates well with a function of this miRNA as a critical regulator of growth and differentiation. Alternatively, Zhang and colleagues have verified MNT as a target of miR-210 with profound effects on gene expression relevant to cell cycle and proliferation.25 MNT is a known antagonist to the proto-oncogene c-MYC, which itself is a positive regulator of the cell cycle and cellular proliferation. Through transcriptional array analysis in transformed cells, a significant overlap exists between the gene expression signatures of miR-210 and c-MYC, as would be predicted by miR-210-dependent downregulation of MNT. Notably in this setting, induction of miR-210 and subsequent downregulation on MNT positively activates the G1/S transition, leading to cell cycle progression and tumor proliferation. Furthermore, because c-MYC has also been described as a negative regulator of HIF-dependent signaling, a model has been proposed by which miR-210 constitutes a negative feedback mechanism for the hypoxic response by reducing MNT and thereby increasing c-MYC activity. It remains unclear how this model of miR-210- and MNT-dependent initiation of cell cycling and growth may be integrated into the findings of miR-210- and HOXA9/FGFRL1-dependent repression of tumor growth (Fig. 4B).26 Again, it is possible that some of these targets are not ubiquitously expressed in all cell types. For example, if activation of c-MYC by miR-210 inhibits direct HIF-dependent gene transcription in transformed cell types, this may not be the case in all cell types, as the effects of miR-210 on such direct HIF-dependent gene expression appear minimal, at least in primary vascular endothelial cells.20 Thus, in the case of proliferation, specific cellular context may again dictate the negative or positive regulatory effects of miR-210.
The known functions of other confirmed targets of miR-210 may also be predicted to influence the effects of miR-210 on cellular proliferation. E2F3 is such a target of miR-210 that belongs to the E2F family of transcriptional activators that control cell cycle progression via influencing genes required for DNA synthesis at the G1/S phases. Alternative promoters of the E2F3 gene generate two forms (a and b) that are identical, apart from their first exon, and both carry the same 3′UTR target sequence recognized by miR-210.82 E2F3a is an “activating” factor that can drive G0 cells to cycle and is induced when quiescent cells are stimulated to divide. E2F3b is a “repressive” factor that maintains G0 cells in quiescence. How miR-210 regulates E2F3a and E2F3b to modulate the cell cycle and proliferation under hypoxia is still under investigation. ACVR1b is yet another confirmed miR-210 target in primary osteoblasts that may influence proliferation and differentiation.83 ACVR1b is a member of the transforming growth factor-β (TGFβ) receptor superfamily, a large and multi-functional family of receptors that can influence both cellular proliferation and differentiation. In osteoblasts, miR-210 appears to induce differentiation by inhibiting AVCR1b and, thus, downstream TGF/activin signaling. Because proliferation and differentiation can be inversely correlated, proliferation may also be affected through this mechanism. Again, more detailed and sophisticated studies will be required to determine how these targets and their downstream signals interact and influence with one another to converge ultimately upon the net proliferative capacity of the hypoxic cell.
DNA repair
Recent work suggests that miR-210 may also control the DNA repair capacity of tumor cells during hypoxia.24 Hypoxia can promote genetic instability in tumor cells exposed to hypoxia.84,85 RAD52 is a DNA repair factor that has been demonstrated to be a direct target of miR-210, and is thought to aid in forming nucleoprotein filaments for repair of double-stranded breaks in chromosomal DNA. Correspondingly, another HIF-dependent miRNA, miR-373, can downregulate RAD23b, a key component of the XPC/RAD23b complex which mediates DNA damage recognition. Direct data linking these miRNA to assays of DNA damage is limited. Furthermore, it is unclear if decreasing the DNA reparative capacity would act as an adaptive or maladaptive mechanism during hypoxia. Some theories suggest a beneficial impact of repressing DNA repair on ATP levels and/or autophagy, which could be important for survival in acute hypoxia; however, these putative mechanisms await more definitive corroboration.
Endothelial angiogenesis and chemotaxis
While most of the above functions are fundamental to the cellular hypoxic response in general, certain actions of miR-210 are specialized to certain cell or tissue types. Relevant for tumor biology as well as peripheral and pulmonary vascular diseases, hypoxic exposure significantly increases angiogenic potential, thereby offsetting limited oxygen and nutrient access and increasing cellular survival and growth. Notably, Fasanaro and colleagues have demonstrated that forced expression of miR-210 in cultured HUVECs stimulates chemotaxis in response to vascular endothelial growth factor (VEGF) as well as the formation of capillary-like tubular structures in Matrigel.20 At least in part, these processes are controlled by the downregulation of the receptor tyrosine kinase ligand ephrin-A3 (EFNA3), a direct target of miR-210. Ephrin ligands and their receptors have prominent roles in the development of the cardiovascular system, vascular remodeling and angiogenesis86,87 with known interactions of ephrin-A3 with such pathways.88 However, the specific actions of ephrin-A3 in angiogenesis and chemotaxis are currently unclear.
Furthermore, as capillary tube formation and chemotaxis in cell culture are surrogate measures of true angiogenic potential, corroboration that miR-210 can direct angiogenesis in vivo will be important in settings of tumor growth as well as primary vascular disease. It is also possible that such downstream, specialized phenotypes of cellular migration and angiogenesis are subject to control by multiple targets and signaling pathways of miR-210. For example, thrombospondin, a c-MYC regulated gene involved in angiogenesis,89 is also downregulated by miR-21025 and theoretically may also affect an angiogenic phenotype via control by MNT (a target mentioned previously). Finally, given the robust upregulation of miR-210 in endothelium and the dependence of endothelial function on fundamental miR-210-dependent processes, such as mitochondrial metabolism (see above), it is tempting to speculate that multiple specialized endothelial processes beyond angiogenesis (such as thrombotic potential, solute permeability, and vascular smooth muscle phenotype modulation, among others) may also be sensitive to the effects of this miRNA.
Erythroid fetal γ-globin production
In addition to specialized endothelial functions, developing erythroid cells may also depend upon miR-210 to direct specific globin expression. Through miRNA profiling assays, Bianchi and colleagues have reported that miR-210 is preferentially upregulated in human erythroid precursor cells displaying a hereditary persistence of fetal hemoglobin (HPFH).90 This upregulation is replicated by treatment of erythroid precursors in cell culture with mithramycin, which correlates with induction of fetal γ-globin expression. Hypoxia has previously been reported to alter progression of the erythroid program91 and is certainly a driving stimulus in erythropoeisis (reviewed in ref. 92). However, it is unclear if this upregulation of miR-210 in HPFH erythroid cells is dependent upon HIF-1α or if miR-210 and its direct targets are causatively responsible for this regulation.
Conclusions and Future Directions
In just three years since the first reports of the upregulation of miR-210 by hypoxic exposure, a wealth of findings has been generated from multiple independent research groups regarding the regulation, target transcripts, and downstream functions of this unique and pleiotropic miRNA. Presently, miR-210 represents one of the few, and likely the predominant, hypoxamir that is robustly upregulated by HIF-1α. By virtue of its apparent diverse range of targets, this miRNA influences a wide range of hypoxia-induced gene programs and cellular activities.
Although this discussion has focused mainly on the more detailed characterizations of miR-210 in relation to a few of its fully validated targets, there now exists a substantial list of predicted targets that have been confirmed biochemically by AGO2/miR-210-immunoprecipitation or have been confirmed functionally as either directly or indirectly regulated by miR-210 expression. As these studies are by no means exhaustive, it should be expected that even a larger number of targets will be reported in the coming years, as these screening techniques are applied to a more diverse set of cell and tissue types. When referenced to their known functions, these additional targets suggest an even broader scope of action of miR-210. While these findings add to an ever-increasing appreciation of the importance of this miRNA in the complex events induced by hypoxia, detailed interrogation of these functions remains a challenge. First, many of the putative targets and associated pathways of gene regulation carry multiple intersecting functions themselves and at times lead to opposite downstream phenotypic consequences. Thus, predicting a clear downstream function based on a single validated target of this miRNA is challenging. Second, owing to varying levels of a basal target’s expression, it is likely that miR-210 may preferentially regulate differing subsets of genes depending upon cellular or tissue context. It is conceivable that seemingly opposite actions of miR-210 can result, depending upon the specific cellular and environmental context leading to a complex network of interactions that may be shifted due to differences in one or a few upstream targets.
In light of these complexities, we propose that further studies of the functions of miR-210 may be aided by the in silico utilization of a systems biological approach to predict appropriately physiological and pathophysiological consequences of perturbing these complex networks. In its simplest form, a biological “interaction” model can be constructed based on the in silico predicted targets of miR-210 and the known interactions of downstream signaling factors (previously validated by focused biochemical studies and/or yeast two-hybrid screening). By correlating that map with the targets present in a given physiological setting or cell type, accurate predictions of downstream consequences can be made. Furthermore, this interaction model can be expanded to connect to the known molecular pathways regulated in hypoxia and dysregulated in hypoxia-dependent disease states, such as cancer, stroke and pulmonary hypertension, among many others. These interactions and downstream functional consequences can then be more easily ascertained for further mechanistic verification. Such strategies of discovery have been successful in highlighting the most promising molecular mechanisms that control disease phenotypes in other complex biological settings,93–99 and we predict that a similar approach may be useful in future studies of this miRNA.
Finally, although these recent years have resulted in much information regarding the mechanism of action of this miRNA, future studies will be essential to verify these functions in vivo. While many of the already validated actions affect fundamental processes of cell biology and are best interrogated in cell culture (i.e., mitochondrial respiratory complex function), it is also likely that these actions profoundly affect physiological and developmental processes, as well as pathophysiological disease states. Recently, some progress has been made in describing the effects of miR-210 in transplanted tumor tissue growth,26 but the diverse in vivo functions of this miRNA in other cellular and tissue types remain unclear. We expect these studies will be both revealing and necessary for a more complete molecular understanding of the hypoxic response. Additionally, it is conceivable that such studies may further identify miR-210 and/or its downstream targets as attractive therapeutic candidates for pharmacological manipulation in a variety of ischemia-related human diseases. However, because of its pleiotropic nature and because its actions in vivo are largely unexplored and may be unpredictable from a reductionist perspective, we would advocate that attempts at therapeutic manipulation of this miRNA should proceed with caution until our understanding of this hypoxamir is more firmly solidified.
In conclusion, recent studies have rapidly and convincingly demonstrated the unique, robust and complex nature of miR-210 in the control of the hypoxic response. Further investigation of its regulation, its targets, and its physiological and/or pathogenic effects in hypoxia are eagerly anticipated in the coming years.
Supplementary Material
Acknowledgments
This work was supported by AHA grant 0825906D (S.Y.C.), and NIH grants HL 61795, NO1 HV 28178, PO1 81587, U54 HL 70819 (J.L.). No conflicts of interest are reported. We apologize to those authors whose work is not cited due to space restrictions. We thank the Loscalzo laboratory for fruitful discussions; T. Lis for graphic design; S.K. Chan and J.W. Snow for critical reading of the manuscript; and S. Tribuna for expert administrative assistance.
Abbreviations
- ATP
adenosine triphosphate
- AGO2
argonaute 2
- miRNA*
antisense miRNA strand
- ATM
ataxia telangiectasia mutated
- DED
death-effector domain
- EFNA3
ephrin A3
- FGFR
fibroblast growth factor receptor
- CASP8AP2
FLASH/caspase-8 associated protein-2
- FLASH
FLICE-associated protein homolog
- GO
gene ontology
- HPFH
hereditary persistence of fetal hemoglobin
- HEK-293 cells
human embryonic kidney 293 cells
- HUVEC
human umbilical vein endothelial cells
- HIF-α
hypoxia inducible factor-alpha
- HREs
hypoxia response elements
- ISCU1/2
iron sulfur cluster assembly proteins 1 and 2
- miRNA
microRNA
- MSCs
mesenchymal stem cells
- miR-210
microRNA-210
- NO
nitric oxide
- ONOO−
peroxynitrite
- pri-miRNA
primary miRNA molecules
- ROS
reactive oxygen species
- RISC
RNA-induced silencing complex
- TGFβ
transforming growth factor-β
- VEGF
vascular endothelial growth factor
- pVHL
Von Hippel Lindau protein
- 3′ UTR
3′ untranslated regions
References
- 1.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621–7. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 3.Adams JM, Difazio LT, Rolandelli RH, Lujan JJ, Hasko G, Csoka B, et al. HIF-1: a key mediator in hypoxia. Acta Physiol Hung. 2009;96:19–28. doi: 10.1556/APhysiol.96.2009.1.2. [DOI] [PubMed] [Google Scholar]
- 4.Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene upregulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65:3299–306. doi: 10.1158/0008-5472.CAN-04-4130. [DOI] [PubMed] [Google Scholar]
- 5.Maynard M, Evans A, Hosomi T, Hara S, Jewett M, Ohh M. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is downregulated in renal cell carcinoma. FASEB J. 2005;19:1396–406. doi: 10.1096/fj.05-3788com. [DOI] [PubMed] [Google Scholar]
- 6.Manalo D, Rowan A, Lavoie T, Natarajan L, Kelly B, Ye S, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–69. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 7.Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk R, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–4. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–9. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 9.Gregory R, Yan K, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–40. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 10.Denli A, Tops B, Plasterk R, Ketting R, Hannon G. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–5. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 11.van Rooij E, Olson E. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–76. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulshreshtha R, Ferracin M, Wojcik S, Garzon R, Alder H, Agosto-Perez F, et al. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–67. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giannakakis A, Sandaltzopoulos R, Greshock J, Liang S, Huang J, Hasegawa K, et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther. 2008;7:255–64. doi: 10.4161/cbt.7.2.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–8. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- 15.Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PloS one. 2006;1:116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donker RB, Mouillet JF, Nelson DM, Sadovsky Y. The expression of Argonaute2 and related microRNA biogenesis proteins in normal and hypoxic trophoblasts. Mol Hum Reprod. 2007;13:273–9. doi: 10.1093/molehr/gam006. [DOI] [PubMed] [Google Scholar]
- 17.Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol Cancer. 2007;6:5. doi: 10.1186/1476-4598-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulkkinen K, Malm T, Turunen M, Koistinaho J, Yla-Herttuala S. Hypoxia induces microRNA miR-210 in vitro and in vivo ephrin-A3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett. 2008;582:2397–401. doi: 10.1016/j.febslet.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 19.Guimbellot JS, Erickson SW, Mehta T, Wen H, Page GP, Sorscher EJ, et al. Correlation of microRNA levels during hypoxia with predicted target mRNAs through genome-wide microarray analysis. BMC Med Genomics. 2009;2:15. doi: 10.1186/1755-8794-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283:15878–83. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–84. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taganov K, Boldin M, Chang K, Baltimore D. NFkappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F. microRNA: Emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther. 2009 doi: 10.1016/j.pharmthera.2009.10.003. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 24.Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009;69:1221–9. doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle. 2009;8:2756–68. doi: 10.4161/cc.8.17.9387. [DOI] [PubMed] [Google Scholar]
- 26.Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, et al. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell. 2009;35:856–67. doi: 10.1016/j.molcel.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pineles B, Romero R, Montenegro D, Tarca A, Han Y, Kim Y, et al. Distinct subsets of microRNAs are expressed differentially in the human placentas of patients with preeclampsia. Am J Obstet Gynecol. 2007;196:261–6. doi: 10.1016/j.ajog.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Zhu XM, Han T, Sargent IL, Yin GW, Yao YQ. Differential expression profile of microRNAs in human placentas from preeclamptic pregnancies vs. normal pregnancies. Am J Obstet Gynecol. 2009;200:661–7. doi: 10.1016/j.ajog.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 29.Bimpaki EI, Iliopoulos D, Moraitis A, Stratakis CA. MicroRNA signature in massive macronodular adrenocortical disease and implications for adrenocortical tumorigenesis. Clin Endocrinol (Oxf) 2009 doi: 10.1111/j.1365-2265.2009.03725.x. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satzger I, Mattern A, Kuettler U, Weinspach D, Voelker B, Kapp A, et al. MicroRNA-15b represents an independent prognostic parameter and is correlated with tumor cell proliferation and apoptosis in malignant melanoma. Int J Cancer. 2009 doi: 10.1002/ijc.24960. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 31.Malzkorn B, Wolter M, Liesenberg F, Grzendowski M, Stuhler K, Meyer HE, et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol. 2009 doi: 10.1111/j.1750-3639.2009.00328.x. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho WC, Chow AS, Au JS. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer. 2009;45:2197–206. doi: 10.1016/j.ejca.2009.04.039. [DOI] [PubMed] [Google Scholar]
- 33.Tombol Z, Szabo PM, Molnar V, Wiener Z, Tolgyesi G, Horanyi J, et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer. 2009;16:895–906. doi: 10.1677/ERC-09-0096. [DOI] [PubMed] [Google Scholar]
- 34.Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141:672–5. doi: 10.1111/j.1365-2141.2008.07077.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Chen J, Chang P, LeBlanc A, Li D, Abbruzzesse JL, et al. MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev Res. 2009;2:807–13. doi: 10.1158/1940-6207.CAPR-09-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105:13021–6. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis B, Burge C, Bartel D. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 38.Grimson A, Farh K, Johnston W, Garrett-Engele P, Lim L, Bartel D. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karginov F, Conaco C, Xuan Z, Schmidt B, Parker J, Mandel G, Hannon G. A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci USA. 2007;104:19291–6. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fasanaro P, Greco S, Lorenzi M, Pescatori M, Brioschi M, Kulshreshtha R, et al. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem. 2009;284:35134–43. doi: 10.1074/jbc.M109.052779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–71. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Tsai NP, Lin YL, Wei LN. Micro-RNA mir346 targets the 5′UTR of RIP140 mRNA and upregulates its protein expression. Biochem J. 2009;424:411–8. doi: 10.1042/BJ20090915. [DOI] [PubMed] [Google Scholar]
- 43.Rigoutsos I. New tricks for animal microRNAS: targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res. 2009;69:3245–8. doi: 10.1158/0008-5472.CAN-09-0352. [DOI] [PubMed] [Google Scholar]
- 44.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou X, Duan X, Qian J, Li F. Abundant conserved microRNA target sites in the 5′-untranslated region and coding sequence. Genetica. 2009;137:159–64. doi: 10.1007/s10709-009-9378-7. [DOI] [PubMed] [Google Scholar]
- 46.Lee I, Ajay SS, Yook JI, Kim HS, Hong SH, Kim NH, et al. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009;19:1175–83. doi: 10.1101/gr.089367.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aisenberg AC, Potter VR. Studies on the Pasteur effect II. Specific mechanisms. J Biol Chem. 1957;224:1115–27. [PubMed] [Google Scholar]
- 48.Semenza G. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 49.Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, et al. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–44. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chavez A, Miranda LF, Pichiule P, Chavez JC. Mitochondria and hypoxia-induced gene expression mediated by hypoxia-inducible factors. Ann N Y Acad Sci. 2008;1147:312–20. doi: 10.1196/annals.1427.021. [DOI] [PubMed] [Google Scholar]
- 51.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587–602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tong WH, Rouault T. Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 2000;19:5692–700. doi: 10.1093/emboj/19.21.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–8. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 54.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beinert H, Holm RH, Munck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science. 1997;277:653–9. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 56.Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J Biol Chem. 1996;271:2762–8. doi: 10.1074/jbc.271.5.2762. [DOI] [PubMed] [Google Scholar]
- 57.Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem. 1994;269:29409–15. [PubMed] [Google Scholar]
- 58.Hidalgo E, Demple B. An iron-sulfur center essential for transcriptional activation by the redox-sensing SoxR protein. EMBO J. 1994;13:138–46. doi: 10.1002/j.1460-2075.1994.tb06243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welter R, Yu L, Yu CA. The effects of nitric oxide on electron transport complexes. Arch Biochem Biophys. 1996;331:9–14. doi: 10.1006/abbi.1996.0276. [DOI] [PubMed] [Google Scholar]
- 60.Kennedy MC, Antholine WE, Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem. 1997;272:20340–7. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- 61.Cruz-Ramos H, Crack J, Wu G, Hughes MN, Scott C, Thomson AJ, et al. NO sensing by FNR: regulation of the Escherichia coli NO-detoxifying flavohaemoglobin, Hmp. EMBO J. 2002;21:3235–44. doi: 10.1093/emboj/cdf339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–8. [PubMed] [Google Scholar]
- 63.Chiang CY, Darensbourg MY. Iron nitrosyl complexes as models for biological nitric oxide transfer reagents. J Biol Inorg Chem. 2006;11:359–70. doi: 10.1007/s00775-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 64.Thierbach R, Schulz T, Isken F, Voigt A, Mietzner B, Drewes G, et al. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum Mol Genet. 2005;14:3857–64. doi: 10.1093/hmg/ddi410. [DOI] [PubMed] [Google Scholar]
- 65.Mochel F, Knight M, Tong W, Hernandez D, Ayyad K, Taivassalo T, et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–60. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 67.Meyron-Holtz E, Ghosh M, Rouault T. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004;306:2087–90. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun. 2003;304:551–9. doi: 10.1016/s0006-291x(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 70.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40:511–26. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 71.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duchen MR. Roles of mitochondria in health and disease. Diabetes. 2004;53:96–102. doi: 10.2337/diabetes.53.2007.s96. [DOI] [PubMed] [Google Scholar]
- 73.Won Kim H, Haider HK, Jiang S, Ashraf M. Ischemic preconditioning augments survival of stem cells via MIR-210 expression by targeting caspase-8 associated protein 2. J Biol Chem. 2009;284:33161–8. doi: 10.1074/jbc.M109.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, Yonehara S. The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature. 1999;398:777–85. doi: 10.1038/19709. [DOI] [PubMed] [Google Scholar]
- 75.Hammer S, To KK, Yoo YG, Koshiji M, Huang LE. Hypoxic suppression of the cell cycle gene CDC25A in tumor cells. Cell Cycle. 2007;6:1919–26. doi: 10.4161/cc.6.15.4515. [DOI] [PubMed] [Google Scholar]
- 76.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–69. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics. 2000;69:275–9. doi: 10.1006/geno.2000.6332. [DOI] [PubMed] [Google Scholar]
- 79.Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P, et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271:171–82. doi: 10.1016/s0378-1119(01)00518-2. [DOI] [PubMed] [Google Scholar]
- 80.Steinberg F, Zhuang L, Beyeler M, Kalin RE, Mullis PE, Brandli AW, et al. The FGFRL1 receptor is shed from cell membranes, binds FGFs and antagonizes FGF signaling in Xenopus embryos. J Biol Chem. 2009 doi: 10.1074/jbc.M109.058248. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mace KA, Hansen SL, Myers C, Young DM, Boudreau N. HOXA3 induces cell migration in endothelial and epithelial cells promoting angiogenesis and wound repair. J Cell Sci. 2005;118:2567–77. doi: 10.1242/jcs.02399. [DOI] [PubMed] [Google Scholar]
- 82.Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, et al. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol. 2000;20:3626–32. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mizuno Y, Tokuzawa Y, Ninomiya Y, Yagi K, Yatsuka-Kanesaki Y, Suda T, et al. miR-210 promotes osteoblastic differentiation through inhibition of AcvR1b. FEBS Lett. 2009;583:2263–8. doi: 10.1016/j.febslet.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Bindra RS, Crosby ME, Glazer PM. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007;26:249–60. doi: 10.1007/s10555-007-9061-3. [DOI] [PubMed] [Google Scholar]
- 85.Bindra RS, Glazer PM. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: role of the Myc/Max network. Cancer Lett. 2007;252:93–103. doi: 10.1016/j.canlet.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 86.Kuijper S, Turner CJ, Adams RH. Regulation of angiogenesis by Ephephrin interactions. Trends Cardiovasc Med. 2007;17:145–51. doi: 10.1016/j.tcm.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 87.Pandey A, Shao H, Marks RM, Polverini PJ, Dixit VM. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNFalpha-induced angiogenesis. Science. 1995;268:567–9. doi: 10.1126/science.7536959. [DOI] [PubMed] [Google Scholar]
- 88.Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, et al. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816–9. doi: 10.1126/science.7973638. [DOI] [PubMed] [Google Scholar]
- 89.Watnick RS, Cheng YN, Rangarajan A, Ince TA, Weinberg RA. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell. 2003;3:219–31. doi: 10.1016/s1535-6108(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 90.Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Expression of miR-210 during erythroid differentiation and induction of gamma-globin gene expression. BMB Rep. 2009;42:493–9. doi: 10.5483/bmbrep.2009.42.8.493. [DOI] [PubMed] [Google Scholar]
- 91.Rogers HM, Yu X, Wen J, Smith R, Fibach E, Noguchi CT. Hypoxia alters progression of the erythroid program. Exp Hematol. 2008;36:17–27. doi: 10.1016/j.exphem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114:2015–9. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- 93.Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–14. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 94.Grimbs S, Selbig J, Bulik S, Holzhutter HG, Steuer R. The stability and robustness of metabolic states: identifying stabilizing sites in metabolic networks. Mol Syst Biol. 2007;3:146. doi: 10.1038/msb4100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu X, Jain VV, Finn PW, Perkins DL. Hubs in biological interaction networks exhibit low changes in expression in experimental asthma. Mol Syst Biol. 2007;3:98. doi: 10.1038/msb4100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qi Z, Miller GW, Voit EO. Computational systems analysis of dopamine metabolism. PloS One. 2008;3:2444. doi: 10.1371/journal.pone.0002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qi Z, Miller GW, Voit EO. Computational analysis of determinants of dopamine (DA) dysfunction in DA nerve terminals. Synapse. 2009;63:1133–42. doi: 10.1002/syn.20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xie L, Li J, Xie L, Bourne PE. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5:1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loscalzo J, Kohane I, Barabasi A. Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.