

Abstract
Increasing evidence suggests that glucagon-like peptide-1 (GLP-1), an incretin hormone of current interest in type 2 diabetes, is neuroprotective in both cell culture and animal models. To characterize the neuroprotective properties of GLP-1 and associated underlying mechanisms, we over-expressed the GLP-1 receptor (R) on human neuroblastoma SH-SY5Y cells to generate a neuronal culture system featuring enhanced GLP-1R signaling. In GLP-1R over-expressing SH-SY5Y (SH-hGLP-1R#9) cells, GLP-1 and the long-acting agonist exendin-4 (Ex-4) stimulated cell proliferation and increased cell viability by 2-fold at 24 h at physiologically relevant concentrations. This GLP-1R-dependent action was mediated via the protein kinase A (PKA) and phosphoinositide 3-kinase (PI3K) signaling pathways, with the mitogen-activated protein kinase (MAPK) pathway playing a minor role. GLP-1 and Ex-4 pretreatment dose-dependently protected SH-hGLP-1R#9 cells from hydrogen peroxide (H2O2)- and 6-hydroxydopamine (6-OHDA)-induced cell death. This involved amelioration of elevated caspase-3 activity, down-regulation of pro-apoptotic Bax and up-regulation of anti-apoptotic Bcl-2 protein. In the presence of 6-OHDA, GLP-1's ability to lower caspse-3 activity was abolished with the PI3K inhibitor, LY2940002, and partly reduced with the PKA inhibitor, H89. Hence, GLP-1R mediated neurotrophic and anti-apoptotic actions co-contribute to the neuroprotective property of GLP-1 in neuronal cell cultures, and reinforce the potential therapeutic value of GLP-1R agonists in neurodegenerative disorders involving oxidative stress.
Keywords: GLP-1, Exendin-4, oxidative stress, 6-hydroxydopamine, neuroprotection, Parkinson's disease
Introduction
Substantial evidence supports the hypothesis that increased oxidative stress plays an important role in the pathogenesis of numerous diseases prevalent in aging (Valko et al., 2007). Indeed, oxidative stress, often causing neuronal cell dysfunction and death, is one of the major factors contributing to the development of age-related neurodegenerative diseases, epitomized by Alzheimer's disease (AD) and Parkinson's disease (PD) (Haliwell 2006; Reddy et al., 2009; Sultana & Butterfield, 2009). In general, oxidative stress arises from an imbalance between the oxidant and antioxidant systems consequent to physiological stressors, environmental factors or disease. Oxidants, such as reactive oxygen species (ROS: e.g., superoxide radical anion (O2•), hydrogen peroxide (H2O2), and hydroxyl radical (•OH)) and reactive nitrogen species (RNS: e.g., nitric oxide (•NO) and peroxynitrite (ONOO-)), are routinely generated in small amounts under normal physiological conditions, and some (H2O2 and •NO) possess a physiological role (Sultana & Butterfield, 2009). Whereas, ROS and RNS are transient and efficiently neutralized by endogenous antioxidant enzyme systems in the healthy brain, either a reduction in endogenous antioxidants, an increased brain vulnerability or elevated oxidative stress during aging can promote disease.
H2O2 and 6-hydroxydopamine (6-OHDA) have been widely used in neuronal cell culture systems to induce oxidative stress and provide a model to evaluate neuroprotective agents (Albani et al., 2009). As part of the natural defense system, several peptide hormones have been shown to possess neuroprotective potential in numerous experimental cellular and animal models. A strategy to prevent or treat a neurodegenerative disease would be to enhance this endogenous mechanism by exogenous administration of such natural peptides as therapeutic agents. In particular, members of the secretin/glucagon/vasoactive intestinal peptide (VIP) superfamily have demonstrated neurotrophic and neuroprotective properties (Banks et al., 1998, 2002; Dejda et al. 2005). Peptides in this superfamily include pituitary adenylate cyclase-activating polypeptide (PACAP), VIP, activity-dependent neurotrophic factor (ADNF), activity-dependent neuroprotective protein (ADNP) and peptide histidine-isoleucine (PHI), as well as glucagon-like peptide-1 (GLP-1) (Banks et al., 1996; Ulrich et al., 1998).
GLP-1 is an incretin hormone, secreted from the enteroendocrine L cells in response to food intake, whose biological activities include (i) the stimulation of glucose-dependent insulin secretion and insulin biosynthesis, (ii) inhibition of glucagon secretion and gastric emptying, and (iii) inhibition of food intake (Baggio and Drucker 2007). GLP-1 is a tissue-specific posttranslational proteolytic product of the proglucagon gene and, once secreted, is rapidly inactivated by dipeptidyl peptidase-4 (DPP-4)-mediated cleavage at its position 2 alanine. In contrast, exendin-4 (Ex-4), a 53% homologous peptide originally isolated from the saliva of the Gila monster (Heloderma suspectum), is DPP-4 resistant and a long-acting, potent, selective GLP-1 receptor (GLP-1R) agonist. Ex-4, widely used in the arena of GLP-1 research, represents the first of a new drug class (incretin mimetics) for the treatment of type 2 diabetes (Exenatide or Byetta). Its target, the GLP-1R, belongs to the class B family of 7-transmembrane-spanning, heterotrimeric G-protein-coupled receptors. It is distributed in many tissues, including pancreas, lung, heart, kidney, stomach and intestine, and additionally is detected in many regions of the brain as well as in some peripheral neurons, as assessed by receptor autoradiography or in situ hybridization.
GLP-1 and Ex-4 have trophic properties in pancreas, stimulating pancreatic β-cell proliferation and inhibiting its apoptosis (Li et al. 2003). Both peptides can readily enter brain, and GLP-1R activation is neurotrophic, inducing neurite outgrowth in PC12 pheochromocytoma cells (Perry et al. 2002), and neuroprotective, rescuing cultured neurons from various toxic insults, including glutamate-, Aβ- and iron- induced apoptotic cell death. Such actions have translated to neuroprotective and neuroregenerative activity in a variety of animal models of acute and chronic neurological conditions (Perry et al. 2003; Perry and Greig 2004; Perry et al., 2007; Gault and Holscher 2008, Li et al., 2010), including 6-OHDA- and MPTP-induced PD (Bertilsson et al., 2008; Harkavyi et al., 2008; Li et al., 2009). Recent work has confirmed that this protective effect is GLP-1R-mediated by using GLP-1R knock-out mice (Li et al., 2009). Together, these studies provide strong evidence that GLP-1R signaling may play an important role in neural cells and brain function, and provide a treatment target for neurodegenerative diseases.
In the present study, the GLP-1R was stably over-expressed in human neuroblastoma SH-SY5Y cells to amplify GLP-1R signaling to aid characterize the neurotrophic actions of GLP-1 on neuronal cells and define the signaling pathways and mechanisms involved. Both GLP-1 and Ex-4 promoted cell proliferation in a protein kinase A (PKA) and phosphoinositide 3-kinase (PI3K) dependent manner. They inhibited both H2O2- and 6-OHDA-induced apoptosis by down-regulating the pro-apoptotic protein Bax, and up-regulating the anti-apoptotic protein, Bcl-2, in addition to ameliorating elevated caspase-3 activity.
Materials and Methods
Materials
The peptides, GLP-1, Ex-4 and Ex-9-39 were obtained from Bachem (Torrance, CA). The kinase inhibitors, H89, LY294002, PD98059 and U0126 derived from Calbiochem (Gibbstown, NJ). All other reagents were from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture
SH-SY5Y cells, obtained from American Type Culture Collection (ATCC, Manassa, VA), were grown in a 1:1 mixture of Eagle's Minimum Essential Medium and Ham's F12 Medium supplemented with 10% heat-inactivated fetal bovine serum (FCS) and 100 U/mL penicillin/streptomycin (Invitrogen, Carlsbad, CA). The cells were maintained at 37°C in a humidified incubator with 5% CO2 and 95% air. Medium was changed every other day and the cells were split in a 1:3 ratio every 5 days (0.25% trypsin, 0.53 mM EDTA solution) or when they reached approximately 80% confluence.
RT-PCR
Total RNA was extracted from both SH-SY5Y and SH-hGLP-1R#9 cells cultured separately in 60-mm plates by utilizing Trizol reagent (Invitrogen). RNA quality and quantity were assessed by spectrophotometer at 260 and 280 nM λ, and 1 ug total RNA was used for RT-PCR. SuperScript III First-strand Synthesis Supermix (Invitrogen) was used for the reverse transcription step. Primers utilized for PCR were: human GLP-1R, forward: 5′ TCAAGGTCAACGGCTTATTAG 3′ and reverse: 5′ TAACGTGTCCCTAGATGAACC 3′, expected PCR product is 480bp; GAPDH, forward: 5′ TCCACCACCCTGTTGCTGTAG 3′ and reverse: 5′ GACCACAGTCCATGACATCACT 3′, expected PCR product is 452bp.
Stable transfection
SH-SY5Y cells were grown in complete media on 100-mm plates until reaching 60-70% confluence, and then cells were serum starved for 24 h before transfection. They were transfected with 10 μg DNA of pcDNA3.1 vector control or pcDNA-hGLP-1R (pcDNA3.1 that expresses full-length human GLP-1R cDNA) (a kind gift from Dr. Daniel Drucker, University of Toronto) by using FuGENE HD Transfection Reagent (Roche, Indianapolis, IN) at a ratio of 4:1 of transfection reagent to DNA, as per the manufacturer's protocol. Stably transfected clones were selected in complete culture media containing 500 μg/ml G418 (Roche), individual clones were picked and those that over-expressed hGLP-1R were identified by RT-PCR and Western blotting analysis.
cAMP assay
Cells grown in 6-well plates were serum-starved overnight and treated with various concentrations of GLP-1/Ex-4 (10-9,10-8,10-7,10-6,10-5 M), or 10 μM Forskolin in serum-free media for 30 min at 37°C. Cells were then lysed with 0.1 M HCl containing 0.5% Triton x-100 for 10 min at room temperature. Cell lysates were collected and centrifuged at 600 × g at room temperature to remove cell debris. Supernatants were directly analysed by cAMP assay. To measure the time course of cAMP production induced by 100 nM GLP-1 or Ex-4, samples were collected at 0, 10, 30, 45 and 60 min after treatment and processed as described above for the preparation of samples. Intracellular cAMP content was then determined using the cAMP EIA kit (Assay Designs, Inc, Ann Arbor, MI), as per the manufacture's protocol for the acetylated version, and results were normalized by total protein content.
Cell viability/proliferation assays
Cell viability was assessed by either lactate dehydrogenase (LDH) or 3-(4, 5-dimethylthiazol-2-yl) -5-(3-carboxymethoxyphenyl) -2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Cell proliferation was determined by measuring 5-bromo-2-deoxyuridine (BrdU) incorporation.
For the LDH assay, experiments were performed in 24-well plates, and SH-SY5Y cells were serum-starved for 24 h before pretreatment with various concentrations of Ex-4 for 2 h. Cells were then exposed to different concentrations of 6-OHDA or H2O2 for 24 h. Samples of conditioned media were collected for the measurement of secreted LDH levels (LDH assay kit, Sigma), an accepted indicator of membrane integrity and cell viability.
For the MTS assay, SH-SY5Y cells in 96-well plates were serum-starved for 24 h before pretreatment with various concentrations of GLP-1, Ex-4 or Ex-4 + Ex9-39 (a selective GLP-1R antagonist (Li et al., 2003; Baggio and Drucker, 2007)) for 2 h. Thereafter, cells were exposed to different concentrations of 6-OHDA or H2O2 for 24 h. For MTS assay, a CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) was utilized to measure a formazan product, which is directly proportional to the cell viability.
To study the mechanism underpinning GLP-1/Ex-4 actions on cell proliferation, we pretreated cells with various kinase inhibitors for 20 min prior to GLP-1/Ex-4 addition to cells. The concentrations of inhibitors used were: PKA inhibitor H89 (10 μM), PI3K inhibitor LY294002 (10 μM) or MEK inhibitors U0126 (5 μM) (MEK1/2) and PD98059 (20 μM) (MEK1). The MTS assay was utilized to assess cell number 24 h after GLP-1/Ex-4 treatment.
Cell proliferation was assessed by measuring BrdU incorporation into newly synthesized DNA strands of actively proliferating cells using a BrdU cell proliferation assay kit from CHEMICON (Millipore, Billerica, MA). Specifically, cells were plated at a density of 5 × 104 cells/well in a 96-well plate using low-serum media that contained 0.5% FCS. Cells were then treated with varies concentrations of GLP-1 or Ex-4 for 24 h, BrdU was added 2 h after the treatment with a total incubation time of 22 h. BrdU incorporation was measured immunochemically by mouse monoclonal anti-BrdU antibody followed by peroxidase conjugated secondary antibody and colorimetric substrate detection. The plate was read at 450/550 nm λ using a spectrophotometer microplate reader, with increasing OD being proportional to higher BrdU concentration in the sample.
Western Blotting
Cells grown in 100-mm plates and approximately 2 × 106 cells were used to extract total protein. First, cells were washed twice with cold PBS buffer, and then lysed in RIPA buffer (Sigma) for 5 min on ice. Total protein was extracted according to the manufacture's protocol. For all sample, about 40 μg protein extracts were resolved by Tris-glycine 10-20% precast gel (Invitrogen, Carlsbad, CA) and transferred onto 0.2 μm PVDF membrane (Invitrogen). The blots were first blocked in 5% milk in TBST at room temperature for 1 h, and then incubated in the same blocking solution containing primary antibodies overnight at 4°C (GLP-1R antibody from Abcam (Cambridge, MA) used at a dilution of 1:500; Bax, Bcl-2 and ATF-4 antibodies from Cell Signaling (Danvers, MA) used at a dilution of 1:1000; β-actin antibody from Sigma used at a dilution of 1: 2000). After sufficient washes with TBST, blots were incubated with appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. Blots were again washed in TBST and, thereafter, signals were detected by using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). Finally, blots were exposed to high-performance chemiluminescence film (GE Healthcare, Piscataway, NJ) for an appropriate period of time, and densitometric quantification of the protein bands was performed by using a PC version of NIH IMAGE (ImageJ software).
Caspase-3 assay
Caspase-3 activity was measured using a colorimetric caspase-3 assay kit (Sigma), as per the manufacturer's protocol for a 96-well plate microassay. Briefly, cell lysates from ∼1 × 106 cells were centrifuged at 20,000 × g for 10 min at 4°C and the supernatant was used for caspase-3 reaction in a 96-well plate. Specifically, the supernatant was incubated with caspase 3 substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) in the presence or absence of caspase-3 inhibitor, Ac-DEVD-CHO, overnight at 37 °C; the release of p-nitroaniline (pNA) from caspase-3 hydrolysis was measured by absorbance at 405 nm wavelength. The remaining cell lysate was used to measure total cellular protein concentrations by BCA™ protein assay kit (Pierce) with bovine serum albumin (BSA) as standards. The final results of caspase-3 activity were normalized by protein content.
Statistical Analysis
Data are presented as mean ± S.E.M. As detailed in the Figure legends, unpaired student's t-test and one-way ANOVA are used for statistical evaluation. A Dunnett's t-test was utilized for comparison of multiple samples.
Results
Characterization of hGLP-1R over-expressing cell line SH- hGLP-1R#9
The SH-SY5Y human neuroblastoma cell line is widely used in the study of neurotrophic and neuroprotective mechanisms. Normal SH-SY5Y cells express only low levels of GLP-1R mRNA and protein (Figure 1A-C). In order to enhance GLP-1R signaling in SH-SY5Y cells, over-expression of human GLP-1R (hGLP-1R) was undertaken by stable transfection. The hGLP-1R expressing plasmid, pcDNA-hGLP-1R, was used to transfect SH-SY5Y cells, several individual clones were isolated and their hGLP-1R expression was verified by RT-PCR and Western Blotting. From such studies, a clone expressing stable high levels of hGLP-1R, designated SH-hGLP-1R#9 was chosen for further analysis. RT-PCR was performed to determine hGLP-1R expression at the mRNA level, and hGLP-1R mRNA levels were significantly increased in SH-hGLP-1R#9 versus SH-SY5Y parent cells (Figure 1A). Furthermore, as illustrated by Western blotting in Figure 1B and C (normalized to β-actin), hGLP-1R protein levels were elevated 2-fold in SH-hGLP-1R#9 versus parent (original) cells.
Figure 1.
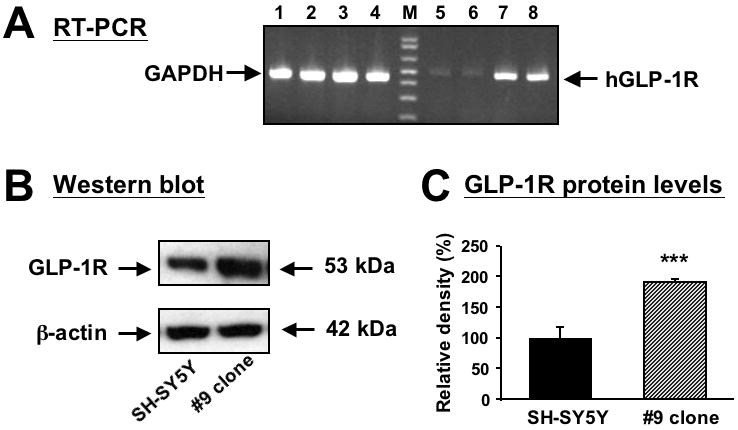
Over-expression of human GLP-1R RNA and protein in stably transfected SH-SY5Y cells. (A) RT-PCR showing human GLP- 1R mRNA expression (lanes 5 to 8) in original and human GLP-1 stably transfected SH-SY5Y (SH-hGLP-1R#9) cells. The expected RT-PCR product size is 480 bp. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (lanes 1-4) was utilized as an external control and showed equal expression across lanes. Lanes 1, 2, 5 and 6 are non-transfected (original) cells and lanes 3,4,7 are 8 are SH-hGLP-1R#9 cells; M is a molecular ladder. (B) A representative Western blot analysis of human GLP-1R protein levels (∼53 kD) shows that hGLP-1R protein is over-expressed in SH-hGLP-1R#9 clones when compared with original SH-SY5Y cells; (C) Quantitative analysis of Western blots shows that protein levels of hGLP-1R are increased 2 fold in #9 clone as compared to original SH-SY5Y cells (normalized by β-actin levels), which was significantly different (***p<0.001, Student's t-test).
To establish whether or not the GLP-1R was functional in the SH- hGLP-1R#9 clone, cells were probed with 100 nM of Ex-4 to activate adenylate cyclase and induce intracellular cAMP production. As illustrated in Figure 2A, Ex-4 induced a rapid and transient elevation of intracellular cAMP levels that peaked at 15 min and then returned towards baseline by 60 min, demonstrating the presence of a functional GLP-1R. Peak cAMP induction proved to be Ex-4 dose-dependent (Fig. 2B), achieving a maximal level at 1 × 10-7 M, with higher concentrations providing no further affect. A similar dose response curve was obtained with GLP-1 incubation (data not shown). By contrast, in parent SH-SY5Y cells, the GLP-1/Ex-4-induced cAMP response proved to be weaker: peak induction of cAMP by 100 nM Ex-4 similarly occurred at 15 min and was 44% of hGLP-1 over-expressing cells (Fig. 2A single bar). Together, these results demonstrate the generation of an hGLP-1R over-expressing SH-SY5Y clone in which GLP-1R signaling was functionally enhanced, providing a tool to permit the study of GLP-1R signaling pathways involved in neural cells.
Figure 2.
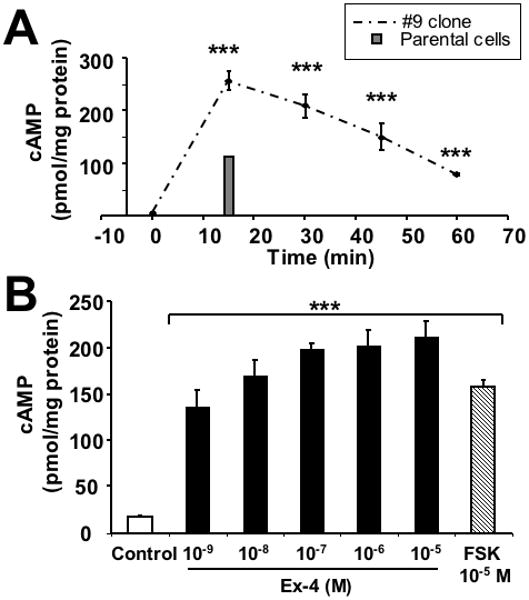
Human GLP-1R over-expression cell line is functional (A) Time course of cAMP production in SH-hGLP-1R#9 cells. Following treatment with 100 nM of Ex-4 at time zero, cells were collected every 15 min at 0, 15, 30, 45 and 60 min to measure intracellular cAMP. For comparison, the peak cAMP response of the parental line to 100 nM Ex-4 is shown by the grey bar at 15 min. Data shown are means from three independent experiments; (B) Intracellular cAMP levels in SH-hGLP-1R#9 cells after treatment with increasing concentrations of Ex-4 (10-9, 10-8,10-7,10-6,10-5 M) or 10 μM of forskolin for 30 min. Controls received PBS vehicle for 30 min. Results are normalized by total protein levels and presented in pmol/mg protein. Statistical analysis: (A,B) each treatment was significantly different from its respective control value at ***p<0.001 (Dunnett's t-test).
Neurotrophic effect of GLP-1/Ex-4 and its downstream signaling pathways
To ascertain whether GLP-1/Ex-4 have neurotrophic effects in SH-hGLP-1R#9 cultures, cells were incubated with increasing concentrations of GLP-1/Ex-4 and cell proliferation and viability were assessed by BrdU and MTS assays. A dose-dependent increase in cell proliferation was observed after GLP-1 or Ex-4 treatment (Figure 3A), as reflected by elevated BrdU concentrations that rose by up to 20% of control values. This increase was matched by a significant rise in cell viability, assessed by MTS in Figure 3B. Both GLP-1 and Ex-4 proved to be potent GLP-1R agonists by stimulating proliferation and cell survival at 0.1 to 1.0 nM concentrations. The action of both peptides on cell survival proved to be saturable at a concentration of 1 nM, with greater amounts providing no greater elevation beyond increased cell viabilities of 180% and 210% of vehicle treated control cells, respectively. The greater induction of cell viability by Ex-4, versus equimolar GLP-1, may relate to the enhanced resistance of the former to DPP-4 cleavage. The specific GLP-1R antagonist, Ex-9-39 (1 μM), abolished the neurotrophic actions of Ex-4 (Fig. 3C), demonstrating that this effect is mediated through the GLP-1R. A similar challenge of parental SH-SY5Y cells to Ex-4 resulted in no change in BrdU activity or cell viability, versus control cells, except at a high Ex-4 (1×10-6 M) concentration (Figure 3A&B right-hand side).
Figure 3.
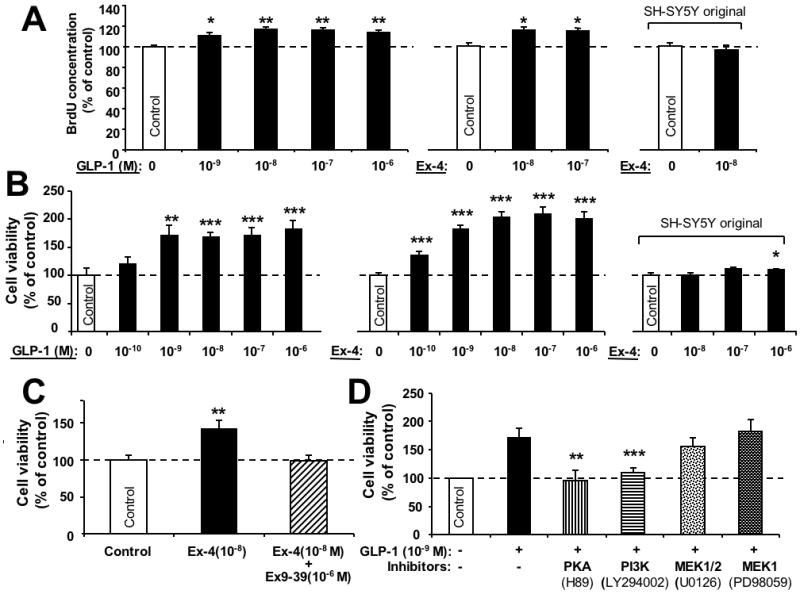
GLP-1R agonists promote cell proliferation in human neuroblastoma SH-hGLP-1R#9 cells. (A) Cell proliferation (BrdU): Increasing concentrations of GLP-1(10-9, 10-8,10-7,10-6 M) and Ex-4 (10-8, 10-7) elevated cellular BrdU concentrations after 24 h treatment versus respective controls. By contrast, Ex-4 (10-8 M) was without effect on BrdU in the original SH-SY5Y cells (right-hand side). (B) Similarly, increasing concentrations of GLP-1(10-10, 10-9, 10-8,10-7,10-6 M) and Ex-4 (10-10, 10-9, 10-8,10-7,10-6 M) increased cell viability after 24 h treatment (MTS assay). Such Ex-4 concentrations were without significant effect in the original SH-SY5Y cells (right-hand side) except at the highest concentration of 10-6 M. (C) Stimulation of cell viability by Ex-4 was abolished by simultaneous treatment of cells with the selective GLP-1R antagonist, Ex-9-39. Specifically, cells were treated with vehicle, Ex-4 (10-8M) and Ex-4 (10-8M) + Ex-9-39 (10-6 M) for 24 h, and cell viability was then determined by MTS assay. (D) Inhibition of GLP-1 (1 nM)-induced cell proliferation by specific pathway inhibitors. SH-hGLP-1R#9 cells were incubated with 10 μM H89 (PKA), 10 μM LY294002 (PI3K), 5 μM U0126 (MEK1/2) or 20 μM PD98059 (MEK1) for 20 min prior to GLP-1 treatment. After 24 h incubation with GLP-1, cell viability was evaluated by MTS assay and results are presented as a percentage of controls, n=6. All data are presented as mean ± S.E.M. Statistical evaluation, (A,B,C,D): Dunnett's t-test, p= *<0.05, **<0.01. ***<0.001, versus respective control.
Determination of which signaling pathways are essential for GLP-1/Ex-4-mediated neurotrophic actions was undertaken by utilizing specific pathway inhibitors. Following serum starvation, SH-hGLP-1R#9 cells were incubated with PKA inhibitor H89 (10 μM), PI3K inhibitor LY294002 (10 μM) or MEK inhibitor U0126 (5 μM) (MEK1/2) and PD98059 (20 μM) (MEK1) for 20 min; thereafter, GLP-1 (1 nM) was added and cell viability was assessed by MTS assay at 24 h. As illustrated in Figure 3D, inhibition of PKA (H89) and of PI3K (LY294002) markedly attenuated GLP-1 mediated neurotrophic effects, whereas inhibition of MEK1/2 by either U0126 (MEK1/2) or PD98059 (MEK1) did not significantly impact cell viability. Together, these results suggest that both PKA and PI3K pathways play physiological roles in GLP-1 induced neural cell proliferation, whereas MEK1/2 has little or no role.
Stimulation of GLP-1R signaling protects SH-SY5Y cells against H2O2-induced oxidative stress
To examine whether stimulation of GLP-1R signaling can protect SH-SY5Y cells against H2O2 induced oxidative stress, following overnight serum-starvation, SH-hGLP-1R#9 cells were incubated with 1 μM Ex-4 or vehicle (PBS) for 2 h, exposed to H2O2 (final concentrations: 50, 100, 150, 200 and 250 μM for 24 h) and cell viability was determined by MTS assay. A significant H2O2-induced decline in cell viability was evident at 100 μM and greater concentrations (Fig. 4A). Ex-4 alone, in accord with Figure 3, induced a significant elevation in cell survival (+37%) and, notably, Ex-4 pretreatment significantly ameliorated H2O2-induced toxicity, even at the highest H2O2 concentration (250 μM), where Ex-4 enhanced cell viability by 27% (from 46% of control (250 μM H2O2 alone) to 73% of control (250 μM H2O2+ Ex-4)) (Figure 4A). Quantifying LDH levels in culture media samples was undertaken as an additional assessment of cell viability. As illustrated in Figure 4B, pretreatment of SH-hGLP-1R#9 cells with 100 nM Ex-4 significantly suppressed 100 μM H2O2-induced elevation in LDH, whereas 10 nM insulin (positive control) completely reversed elevated LDH levels. Together, these results demonstrate that GLP-1 and Ex-4 are cytoprotective against H2O2 insult to SH-hGLP-1R#9 cells.
Figure 4.
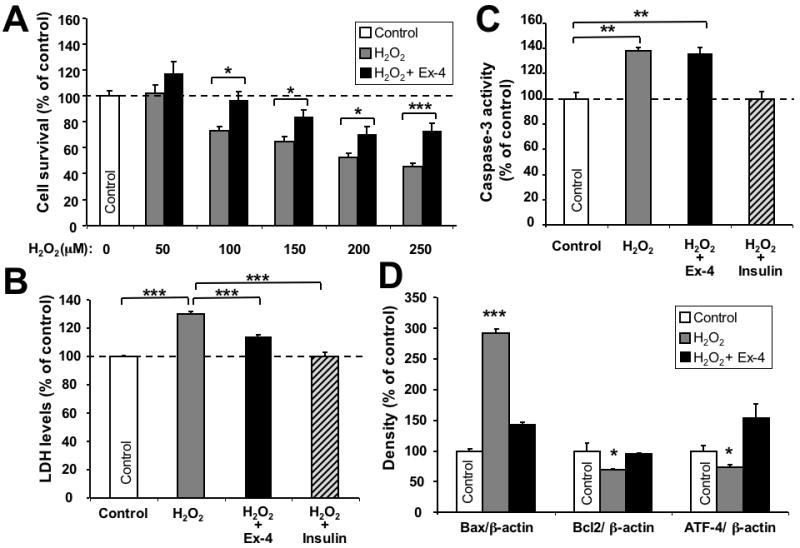
Neuroprotective effect of GLP-1 against H2O2-induced oxidative stress. (A) Ex-4 protected SH-hGLP-1R#9 cells from a wide concentration range (50, 100, 150, 200 and 250 μM) of H2O2-induced cell death. Cells were pretreated with 10-6 M Ex-4 for 2 h and, thereafter, exposed to different concentrations of H2O2 for 24 h. 100 μM H2O2 and greater concentrations induced significant cell death (Dunnett's t-test vs. no H2O2, p<0.05), and Ex-4 ameliorated H2O2-induced effects; (B) Ex-4 (10-7 M) pretreatment prevented H2O2 (100 μM) -induced elevation in LDH levels; insulin (10-8 M) was used as a positive control. (C) Caspase-3 activity assay: Ex-4 (10-7 M) pretreatment for 2 h proved unable to reverse H2O2-induced caspase-3 activity, whereas insulin pretreatment was capable of doing so. (D) Western blot analysis of Bax, Bcl-2 and ATF-4 protein levels. β-actin was used as an internal standard to normalize protein levels. Data are presented as mean ± S.E.M. and as a percentage of controls. Statistical evaluation, (A): unpaired student's t-test and one-way ANOVA; (B,C,D): Dunnett's t-test, p= *<0.05, **<0.01. ***<0.001, N=6.
To characterize this protective response, components of the apoptotic pathway were analyzed in cells exposed to oxidative insults after being pretreated with PBS, Ex-4 or insulin. The activity of caspase-3, a key enzyme in the apoptosis process, was assessed in SH-hGLP-1R#9 cells exposed to 100 μM H2O2 for 24 h with or without pretreatment with 100 nM Ex-4 or 10 nM insulin. As shown in Figure 4C, H2O2 induced a 1.4-fold elevation in caspase-3 activity. Whereas, Ex-4 did not impact H2O2-induced caspase-3 activity, insulin pretreatment, in line with the literature, fully reversed the elevation. The levels of the pro- and anti-apoptotic proteins, Bax and Bcl2, were quantified under the same conditions. H2O2 increased protein levels of Bax by 3-fold, which was markedly ameliorated by Ex-4 pretreatment (Figure 4D). By contrast, Bcl-2 levels were slightly but significantly decreased by H2O2 (-29%), and were fully restored to control levels by Ex-4 pretreatment. Interestingly, activating transcription factor-4 (ATF-4) protein, which declined following H2O2 insult (-26%), was elevated in Ex-4 pretreated cells beyond control levels (+87%) (Figure 4D), indicating the involvement of ER stress in this H2O2 model and an anti-ER stress effect of GLP-1R agonists.
Stimulation of GLP-1R signaling protects SH-SY5Y cells against 6-OHDA-induced cell death and apoptosis
6-OHDA, a neurotoxin widely used to selectively kill dopaminergic neurons, was utilized to investigate neuroprotective actions of GLP-1/Ex-4 in SH-hGLP-1R#9 cells, a cell type known to possess dopaminergic markers (Fasano et al., 2008). As illustrated in Figure 5A, 30 μM 6-OHDA (24 h) induced a 30% reduction in cell viability that was fully ameliorated by GLP-1 (10-9 to 10-6 M 2 h pretreatment), as assessed by MTS assay. In parallel studies, 30, 50 and 100 μM 6-OHDA (24 h) generated a dose-dependent rise in LDH levels in the media of SH-hGLP-1R#9 cell cultures, inducing elevations of 19%, 64 and 85%, respectively, of untreated controls. Ex-4 (100 nM, 2 h pretreatment) significantly reduced LDH levels in basal (without 6-OHDA), as well as 6-OHDA 30 and 50 μM exposed cells, but not following 6-OHDA 100 μM exposure (Figure 5B). Taken together, utilizing two different methods to establish cell viability, a neuroprotective action of GLP-1 and Ex-4 was demonstrated against 6-OHDA toxicity. To characterize the mechanism of GLP-1 mediated neuroprotection, caspase-3 activity was quantified 24 h after 6-OHDA insult in control as well as vehicle, insulin and Ex-4 treated cells. As illustrated in Figure 5C, 30 μM 6-OHDA induced a 3.2-fold increase in caspase-3 activity in SH-hGLP-1R#9 cells. Ex-4 as well as insulin pretreatment significantly attenuated this rise by 65% and 58%, respectively, versus the 6-OHDA alone group. This Ex-4 mediated action on caspase-3 activity was completely abolished by treating cells with the PI3K inhibitor LY294002, and was partly reduced by the PKA inhibitor H89 (Figure 5C), suggesting that the PI3K pathway plays an essential role in GLP-1R mediated cell survival. As additionally shown in Figure 5C (right-hand side), the 100 μM 6-OHDA insult induced a dramatic 7.7-fold elevation in caspase-3 levels, which Ex-4 (100 nM) and insulin (10 nM) pretreatment proved incapable of ameliorating.
Figure 5.
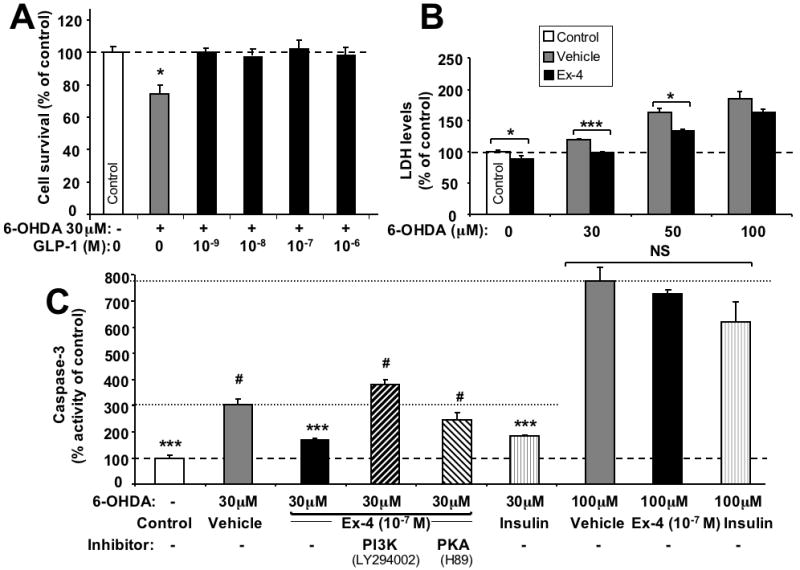
Neuroprotective effect of GLP-1 against 6-hydroxydopamine- (6-OHDA)-induced cell death. (A) Increasing concentrations (10-9, 10-8,10-7,10-6 M) of GLP-1 pretreatment for 2 h protected SH-hGLP-1R#9 cells from 30 μM 6-OHDA induced cell death: MTS assay at 24 h after 6-OHDA exposure. (B) Ex-4 (10-7 M) pretreatment for 2 h prevented 6-OHDA (30 and 50 μM, but not 100 μM) induced elevations in LDH levels in SH-hGLP-1R#9cells. (C) Caspase-3 activity assay: SH-hGLP-1R#9 cells exposed to 30 μM 6-OHDA induced a 3.2-fold elevation in caspase-3 activity. Ex-4 (10-7 M) pretreatment for 2 h significantly ameliorated 6-OHDA induced caspase-3 activity, and insulin (10-8 M), used as a positive control, showed a similar effect. However, following 20 min preincubation of cells with a PKA (10 μM H89) or PI3K inhibitor (10 μM LY294002) prior to Ex-4 addition, the effect of Ex-4 on caspase-3 activity was largely (in the case of H89) or completely (in case the of LY294002) abolished. A 100 μM 6-OHDA insult significantly elevated caspase-3 levels by 7.7-fold and, in accord with B, neither Ex-4 (10-7 M) nor insulin (10-8 M) were able to ameliorate this action. Data are presented as mean ± S.E.M. and as a percentage of controls. Statistical evaluation, (A): unpaired student's t-test and one-way ANOVA; (B,C,D): Dunnett's t-test, p= *<0.05, **<0.01. ***<0.001, # or NS: not significantly different from one another (p>0.05), N=6.
Discussion
The neurotrophic and neuroprotective actions of GLP-1 were first described by Perry et al., (2002a,b) in cellular and animal models. In the former, GLP-1R agonists induced neurite outgrowth in rat PC12 cells and, to a lesser extent, in human SK-N-SH cells (Perry et al., 2002b). In accord with this, recent studies by Hamilton and Holscher (2009) indicate that expression of the GLP-1R is highly selective to a subpopulation of neurons and, in addition to the cell body, it is expressed on dendrites and on or near synapses. Binding of GLP-1 or Ex-4 to the GLP-1R leads to its interaction with the GTP-binding protein Gs, resulting in the activation of adenylyl cyclase and increased production of intracellular cAMP in the pancreas (Li et al., 2003), which has likewise been confirmed in PC 12 cells (Perry et al., 2002b). GLP-1R-induced neurite extension appears to be mediated by pathways involving PI3 kinase and ERK-MAPK, with PKA playing a lesser role (Perry et al., 2002b). Whereas GLP-1R stimulation has demonstrated neuroprotective activity in both immortal and primary neuron cultures against a wide array of insults, including Aβ, Fe2+-, and glutamate-induced toxicity (Perry et al., 2003; Li et al., 2009; Li et al., 2010), the pathways mediating these anti-apoptotic actions in neurons remain to be fully elucidated.
To characterize potential mechanisms, SH-SY5Y cells were studied. This is a subcloned line of neuroblastic, N-type cells from the parent SK-N-SH cell line. SH-SY5Y cells express relatively low levels of GLP-1R mRNA and protein, as assessed by RT-PCR and Western blot analysis (Fig. 1). Hence, neuroprotective actions have sometimes been marginal when studying GLP-1 effects in these cells, making it difficult to clarify underlying mechanisms. On the other hand, although robust GLP-1-dependent neuroprotective responses have been described in primary neurons from discrete brain regions, such as hippocampus (Perry et al., 2003), cerebral cortex and ventral mesencephalon (Li et al., 2009), such primary cultures contain a mixture of GLP-1R-expressing and –non expressing cells of a variety of phenotypes. Thus, faced with the unavailability of a stable homogeneously high GLP-1R-expressing neuronal cell line, we established one. Our SH-hGLP-1R#9 line over-expressed the hGLP-1R protein by 2-fold and, although morphologically similar, appeared more homogeneous than parental SH-SY5Y cells. Furthermore and in line with the response of primary neuron cultures, following stimulation with a GLP-1R agonist, intracellular cAMP levels were robustly elevated in SH-hGLP-1R#9 cells, reaching a maximal level at 15 min and gradually declining to basal levels thereafter. This indicates that the over-expressed GLP-1R was functional in SH-hGLP-1R#9 cells. By contrast, the cAMP response proved to be weaker in parent SH-SY5Y cells (up to 44% of SH-hGLP-1R#9 cells). Hence, in the present study, utilizing SH-hGLP-1R#9 cells that featured enhanced GLP-1R signaling, we evaluated the mitogenic and neuroprotective effects of GLP-1R agonists and explored responsible underlying mechanisms.
In addition to inducing glucose-dependent insulin secretion, a key GLP-1R agonist action is the stimulation of β-cell proliferation and neogenesis, and inhibition of β-cell apoptosis that, together, lead to an increased β-cell population. Hence, Ex-4, as a diabetic drug, not only may effectively lower glycemia, but also has the potential of augmenting β-cell mass (Baggio & Drucker 2007). In neurodegenerative disorders, such as AD and PD, that, similar to type 2 diabetes, feature oxidative stress and a progressive cellular loss (Reddy et al., 2009), it is of interest to know whether stimulation of GLP-1R signaling in the brain can, likewise, preserve and restore neuronal cells. In our cellular system, treatment with GLP-1 or Ex-4 significantly elevated BrdU incorporation (+20%), a marker of cell proliferation, and increased cell viability by more than two-fold over 24 h. This proved to be concentration-dependent, was achieved at levels as low a 1× 10-10 M (100 pmol/L), and inhibited by a selective GLP-1R antagonist (Ex-9-39), in line with a GLP-1R mediated action. From a physiological perspective, plasma GLP-1 levels are at their lowest during the fasted state, lie in the range of 5–10 pmol/L, and increase rapidly after eating, reaching 15–50 pmol/L. A 6-week subcutaneous infusion of GLP-1 (4.8 pmol kg-1 min-1) that achieved substantial clinical improvements in type 2 diabetics was associated with GLP-1 plasma levels of 60–70 pmol/L (Zander et al., 2002; Drucker & Nauck 2006), and a clinically effective dose of Ex-4 (Kolterman et al., 2005) is associated with plasma concentrations of 450 pg/ml (1× 10-10 M). Together, these clinical studies suggest that the doses utilized in our cell culture studies are of physiological relevance. Our further studies suggest that this trophic action is primarily mediated through PKA and PI3K pathways as selective inhibition of either PKA or PI3K signaling by H89 or LY294002, respectively, abolished this effect. By contrast, inhibition of the MEK1/2 signaling pathway by PD98059 or U0126 had no significant effect, suggesting that MEK1/2 signaling plays a minor or no role in GLP-1R agonist mediated neuronal proliferation and survival.
Although the specific mechanisms remain to be fully elucidated, it is clear that oxidative stress plays an important role in neurodegenerative diseases. In the current study, H2O2 and 6-OHDA were utilized to impair function and provoke neuronal cell death, as both agents can induce the production of ROS, leading to oxidative damage. Pretreatment with GLP-1 and Ex-4, however, significantly protected SH-hGLP-1R#9 cells from both insults, ameliorating the loss of cell viability, as determined by MTS assay, and preserving membrane integrity as assessed by LDH assay. This neuroprotective action promotes anti-apoptotic signaling pathways that involve two major components of the Bcl-2 family: Bax and Bcl-2. GLP-1R stimulation suppressed the expression of pro-apoptotic protein, Bax, and stimulated the expression of anti-apoptotic protein, Bcl-2; thereby, favorably modifying the Bax/Bcl-2 ratio, an indicator of cell apoptosis. Interestingly, the neuroprotective effect of GLP-1R agonists was associated with a decrease in caspase-3 activities under 6-OHDA challenge but not under H2O2 challenge, suggesting diverse signaling pathways in these two models, albeit that H2O2 has been implicated as a key mediator of 6-OHDA-induced apoptosis at high concentrations (150 μM) (Hanrott et al., 2006). However, used as a positive control, insulin proved capable of suppressing the induction of caspase-3 activities in both the H2O2 and 6-OHDA models.
Expression of mutant proteins disrupts protein folding in the endoplasmic reticulum (ER), causes ER stress, and activates a signaling network termed the unfolded protein response (UPR). Like oxidative stress, ER stress is likewise associated with neurodegenerative diseases. Although the mechanisms underlying this interaction are largely unknown, several reports support the notion that ER stress is linked to oxidative stress and these stresses, together, may integrate into other common cellular signaling pathway (Malhotra and Kaufman 2007). At the cellular level, neuronal death or apoptosis may be mediated by oxidative stress, ER stress or both. Activating transcription factor-4 (ATF-4) is an indicator of the activation of an ER resident transmembrane protein kinase, known as PERK (Liang et al., 2006), which is one of the three arms of the UPR, and is required for expression of genes involved in resistance to oxidative stress (Harding et al., 2000). Recent studies (Yusta et al. 2006) have shown that GLP-1R agonists significantly potentiate the induction of ATF-4 in INS-1 cells and demonstrated that GLP-1R signaling can directly modulate the ER stress response leading to promotion of β-cell adaptation and survival. Interestingly, we found in SH-SY5Y cells that GLP-1 treatment significantly induced ATF-4 protein levels upon H2O2 challenge, indicating the involvement of an anti-ER stress action of GLP-1 in this in vitro model of neuroprotection.
In summary, we have established a neuroblastoma cell line, SH-hGLP-1R#9, that possesses enhanced GLP-1R signaling to aid characterize GLP-1-induced neurotrophic and protective actions. The GLP-1R agonists, GLP-1 and Ex-4, stimulated neuronal cell proliferation and protected cells from H2O2- and 6-OHDA-induced cell death at physiologically relevant doses, which were inhibited by selective GLP-1R antagonism. GLP-1R neurotrophic and protective actions were chiefly mediated via PKA and PI3K signaling pathways, and favorably altered the balance between apoptotic proteins to promote survival. Our data confirm that GLP-1R agonists have clinical potential in treating neuronal stresses relevant to neurodegenerative conditions; understanding the molecular cascades involved may allow optimization of their clinical utility in both acute and chronic neurological disorders.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. A portion of that support was through a R & D contract with MedStar Research Institute. The authors declare no conflicts of interest.
References
- Albani D, Polito L, Batelli S, et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J Neurochem. 2009;110:1445–1456. doi: 10.1111/j.1471-4159.2009.06228.x. [DOI] [PubMed] [Google Scholar]
- Banks WA, Goulet M, Rusche JR, Niehoff ML, Boismenu R. Differential transport of a secretin analog across the blood-brain and blood-cerebrospinal fluid barriers of the mouse. J Pharmacol Exp Ther. 2002;302:1062–1069. doi: 10.1124/jpet.102.036129. [DOI] [PubMed] [Google Scholar]
- Banks WA, Uchida D, Arimura A, Somogyvári-Vigh A, Shioda S. Transport of pituitary adenylate cyclase-activating polypeptide across the blood-brain barrier and the prevention of ischemia-induced death of hippocampal neurons. Ann N Y Acad Sci. 1996;805:270–277. doi: 10.1111/j.1749-6632.1996.tb17489.x. [DOI] [PubMed] [Google Scholar]
- Baggio L, Drucker D. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Bertilsson G, Patrone C, Zachrisson O, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson's disease. J Neurosci Res. 2008;86:326–338. doi: 10.1002/jnr.21483. [DOI] [PubMed] [Google Scholar]
- Dejda A, Sokołowska P, Nowak J. Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol Rep. 57:307–320. [PubMed] [Google Scholar]
- Drucker D, Nauck M. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- Fasano M, Alberio T, Colapinto M, Mila S, Lopiano L. Proteomics as a tool to investigate cell models for dopamine toxicity. Parkinsonism Relat Disord. 2008;14 2:S135–138. doi: 10.1016/j.parkreldis.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Gault V, Hölscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol. 2008;587:112–117. doi: 10.1016/j.ejphar.2008.03.025. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hanrott K, Gudmunsen L, O'Neill M, Wonnacott S. 6-hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cdelta. J Biol Chem. 2006;281:5373–5382. doi: 10.1074/jbc.M511560200. [DOI] [PubMed] [Google Scholar]
- Harding H, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harkavyi A, Abuirmeileh A, Lever R, Kingsbury A, Biggs C, Whitton P. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson's disease. J Neuroinflammation. 2008;5:19. doi: 10.1186/1742-2094-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolterman O, Kim D, Shen L, Ruggles J, Nielsen L, Fineman M, Baron A. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. 2005;62:173–181. doi: 10.1093/ajhp/62.2.173. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar-Am O, Youdim M, Blumenfeld Z. The neuroprotective effect of Activin A and B: implication for neurodegenerative diseases. J Neurochem. 2007;103:962–971. doi: 10.1111/j.1471-4159.2007.04785.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Duffy K, Ottinger M, et al. GLP-1 Receptor Stimulation Reduces Amyloid-beta Peptide Accumulation and Cytotoxicity in Cellular and Animal Models of Alzheimer's Disease. J Alzheimers Dis. 2010;19 doi: 10.3233/JAD-2010-1314. in press [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hansotia T, Yusta B, Ris F, Halban P, Drucker D. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem. 2003;278:471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy M, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Zhang W, McGrath B, Zhang P, Cavener D. PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem J. 2006;393:201–209. doi: 10.1042/BJ20050374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J, Kaufman R. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Perry T, Greig N. A new Alzheimer's disease interventive strategy: GLP-1. Curr Drug Targets. 2004;5:565–571. doi: 10.2174/1389450043345245. [DOI] [PubMed] [Google Scholar]
- Perry T, Haughey N, Mattson M, Egan J, Greig N. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002a;302:881–888. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]
- Perry T, Holloway H, Weerasuriya A, Mouton P, Duffy K, Mattison J, Greig N. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp Neurol. 2007;203:293–301. doi: 10.1016/j.expneurol.2006.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T, Lahiri D, Chen D, Zhou J, Shaw K, Egan J, Greig N. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002b;300:958–966. doi: 10.1124/jpet.300.3.958. [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri D, Sambamurti K, Chen D, Mattson M, Egan J, Greig N. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603–612. doi: 10.1002/jnr.10611. [DOI] [PubMed] [Google Scholar]
- Reddy V, Zhu X, Perry G, Smith M. Oxidative stress in diabetes and Alzheimer's disease. J Alzheimers Dis. 2009;16:763–774. doi: 10.3233/JAD-2009-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Butterfield D. Role of Oxidative Stress in the Progression of Alzheimer's Disease. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1222. [DOI] [PubMed] [Google Scholar]
- Uberti D, Piccioni L, Colzi A, Bravi D, Canonico P, Memo M. Pergolide protects SH-SY5Y cells against neurodegeneration induced by H(2)O(2) Eur J Pharmacol. 2002;434:17–20. doi: 10.1016/s0014-2999(01)01537-0. [DOI] [PubMed] [Google Scholar]
- Ulrich Cn, Holtmann M, Miller L. Secretin and vasoactive intestinal peptide receptors: members of a unique family of G protein-coupled receptors. Gastroenterology. 1998;114:382–397. doi: 10.1016/s0016-5085(98)70491-3. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin M, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Yusta B, Baggio L, Estall J, Koehler J, Holland D, Li H, Pipeleers D, Ling Z, Drucker D. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Zander M, Madsbad S, Madsen J, Holst J. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]