

Abstract
Limited proteolysis of APOBEC-1 Complementation Factor (ACF) and computational secondary structure modeling were used to guide the construction of a well-folded, truncation protein spanning residues 1-320 and containing three RNA Recognition Motifs (RRMs). ACF320 bound preferentially to apoB mRNA and supported APOBEC-1 dependent editing at 40% of the activity of full length ACF. Live cell FRET and immunoprecipitation assays revealed that ACF320 formed homomultimers in situ that were bridged by RNA. Our study predicted that the C to U editosome may be assembled on the mooring sequence of apoB mRNA as a dimer of ACF bound to a dimer of APOBEC-1.
Introduction
ACF was discovered as an RNA binding protein that facilitated site-specific targeting of apoB mRNA for C to U editing (1,2). The basis for this requirement has been proposed to be the maintenance of apoB mRNA in a single-stranded conformation that enables recruitment of the cytidine deaminase, APOBEC-1, for site-selective cytidine deamination of nucleotide 6666 (3,4). ACF has three consecutive RNA Recognition Motifs (RRMs). RRMs are a well-described structural fold consisting of 4 antiparallel β-strands packed by two alpha helices in a β1α1β2β3α2β4 topology (reviewed (3,5)). One RRM was sufficient to bind to a minimum of 2 nucleotides, demonstrated by CBP20 (6,7) and Nucleolin RRM2 (8), so their appearance in multiple copies is thought to represent a way to increase specificity and/or affinity for their substrate.
ACF RRM1 starts at amino acid (a.a.) 58, but the C-terminal boundary of the third RRM (RRM3) ends at either a.a. 293 (9) or 303 (10). The minimal portion of ACF that retains RNA binding activity remains to be determined and includes a.a. up to either 304, 380 or 391 (3,9,10). Deletion mapping of ACF showed that the most significant loss in RNA binding activity occurred when RRM2 was deleted (10) but point mutations of conserved residues with RRM1 also inhibited ACF RNA binding (9).
ACF truncated to a.a. 304 bound RNA but was not evaluated for APOBEC-1 binding or complementation of editing activity (3). So far ACF binding to APOBEC-1 only has been demonstrated for ACF truncations 1-380 and 1-391 (9,10). In vitro apoB mRNA editing activity was reduced by truncating the C-terminus of ACF beyond a.a. 380 and was lost when any one of the three RRMs had been deleted (3,9). Portions of RRMs 2 and 3 are implicated in ACF interaction with APOBEC-1, specifically a.a. 144-257 (9,10). Phosphorylation of serine 154 within RRM2 enhanced ACF binding to APOBEC-1 and ACF complementation of RNA editing (11,12). Residues C-terminal to RRM3 may also be required for APOBEC-1 interaction as ACF truncated to less than a.a. 1-377 fail to bind APOBEC-1 (9). All three RRMs may be required for complementation of editing activity in living cells (9,10,13,14) and that the C-terminal portion of ACF may modulate their function.
We show in this communication that the N-terminal 320 amino acids of ACF retained RNA binding selectivity and supported complementation of editing activity and in addition demonstrate a novel RNA bridged multimerization of ACF. The data support a model for the C to U editosome in which ACF dimers are bridged by the mooring sequence.
Materials and Methods
Recombinant ACF Cloning, Expression, and Purification
Rat ACF64 was cloned and expressed as previously described (15). ACF was dialyzed into 50mM Tris pH 8.0 with 1 mM CaCl2 and digested with 3.5 U of TPKC-trypsin (Worthington Biochemicals, Lakewood, NJ) for 20 minutes at room temperature and resolved by SDS-PAGE and either stained with Coomassie blue or western blotted. Identities of tryptic peptides were confirmed by western blot analysis using peptide-specific, rabbit polyclonal antibody raised against the N-terminus or C-terminus of ACF (12) and by comparison to the predicted trypsin cleavage sites using Peptide Cutter (http://www.expasy.ch/tools/peptidecutter/). Secondary structure boundaries were predicted using PHYRE (www.sbg.bio.ic.ac.uk/). A cDNA fragment encoding amino acids 1-320 was PCR amplified ACF320, cloned into pet28a mod and sequence verified (15). Ni2+ affinity purification of E. coli expressed protein was conducted as described (15). For HPLC ACF320 was concentrated and chromatographed on an S-100 sephacryl column (GE Healthcare, Waukesha, WI).
Live Cell Quenched FRET (FqRET)
EGFP acts as a fluorescence donor and REACh2, a variant of YFP, is an acceptor that quenches EGFP fluorescence when in close proximity (16). EGFP-V5-ACF320 was transfected into HEK 293T cells alone or together with either REACh2-HA-ACF320 or HA-ACF320-REACh2 chimeras at the indicated ratios. Empty pIRES vector (Clonetech, Mountain View CA) was used to maintain equivalent input of transfected DNA. 24 hours following transfection EGFP fluorescence was imaged through a 20× objective with an Olympus IX 70 inverted fluorescence microscope and the gray value for each cell was determined with ImageJ software (National Institutes of Health). The average gray value was calculated by counting all of the cells in 5 random fields after subtracting the average gray value of REACh2 constructs as the background. The statistical significance of EGFP quenching was determined using an unpaired student's t-test with n = 109-115.
UV Cross-linking
Increasing amounts of ACF320 and ACF64 (0.3 to 40 pmols) were reacted at 30 °C for 1 hour with a fixed amount of in vitro transcribed, 32P ATP and CTP labeled apoB RNA (150 fmols) containing the editing site at nucleotide 6666 (448 nt long) (17). For the RNA excess competition studies, non-specific, unlabeled, xef competitor RNA (Ambion, Austin, TX) was transcribed using mMessage mMachine (Ambion) and added to the assembly reactions in 100-fold molar excess to apoB RNA. UV cross-linked reactions were digested with RNase A and T1 (Roche, Indianapolis, IN) for one hour and then precipitated with acetone and resolved by SDS-PAGE. Cross-linking was quantified by PhosphorImager analysis of the PAGE gels as described previously (18).
Immunoprecipitation Assays
Whole cell extracts from HEK 293T cells co-transfected with ACF320-V5-EGFP and ACF320-HA-EGFP were prepared 48 hours following transfection using Reporter Lysis Buffer (Promega) per manufacture's protocol supplemented with EDTA-free complete protease inhibitor cocktail (Roche). Equivalent microgram amounts of protein from each extract were resolved by SDS-PAGE to evaluate the expression of each ACF construct. Immunoprecipitation was conducted overnight at 4 °C using 1 μg of αV5 (Invitrogen) on protein A agarose (Roche) with precleared extract that had or had not been digested with 25 μg of RNase A (Sigma Aldrich). Bound protein was eluted from 500 mM NaCl washed beads using SDS PAGE sample treatment buffer at 37 °C. Eluants were acetone precipitated, resolved by SDS-PAGE and then western blotted with either HA or V5 epitope reactive antibodies. For co-immunoprecipitations, V5 epitope tagged acf64 and acf320 cDNA cloned into pcDNA3 were co-transfected into HEK 293T cells with pcDNA3 HA-tagged apobec-1 cDNA(12). Whole cell extracts were prepared 48 hours following transfection. Immunoprecipitations and protein detection were conducted as described above.
ApoB RNA Editing Assays
HEK 293T cells were co-transfected with APOBEC-1 and human apoB RNA editing reporter alone or in combination with ACF64 or ACF320 and a using FuGene6 (Roche). Following transfection (48 h), 50% of each culture was harvested for cell extract preparation and western blotting analysis and the rest was placed into TRI-Reagent (MRC, Cincinnati, OH) for RNA extraction. ApoB RNA was amplified from 2 μg of total cellular RNA (19) and purified PCR products were primer extended with a 32P radio-labeled DD3 primer in a poison primer extension assay and the relative proportion of edited RNA quantified by PhosphorImager scanning densitometry (19).
Analytical Ultracentrifugation
Ultracentrifugation was conducted at the University of Connecticut on S-100 column purified ACF320 in 20 mM HEPES, 10 mM NaCl, 1 mM DTT, pH 8.0. Sedimentation velocity analysis was conducted at 20 °C and 55,000 RPM using interference optics with a Beckman-Coulter XL-I analytical ultracentrifuge. Interference scans were acquired at 60 second intervals for 5½ hours. Data were collected at four protein concentrations. Normalized g(S*) distribution plots were created using DcDt+ version 2.1.0 (20,21) and c(S) distribution plots were created using Sedfit, version 11.71 (22).
Results
ACF64 RRMs Reside Within an Trypsin-Resistant Domain
Recombinant ACF64 expressed in E. coli was purified and digested with limiting amounts of trypsin. Coomassie stained SDS PAGE showed three highly abundant proteolytic cleavage products with estimated molecular masses of approximately 40, 34 and 27 kDa (denoted by arrowheads in Figure 1A) (Figure 1A). Western blotting with peptide-specific polyclonal antibodies that recognized epitopes within either the extreme C- (a.a. 508-522) or N-terminus (a.a. 1-14) of ACF (12,23) indicated that the 40 kDa species corresponded to a C-terminal deletion while the smaller species were N-terminal deletions (Figure 1B). As the minimal amino terminal portion of ACF that contains all three RRMs was predicted to have a maximum molecular weight of ∼34 kDa (9,10), the 40 kDa tryptic fragment must have contained all three RRMs. Peptide Cutter software predicted that the C-terminal tryptic cleavage of ACF to be at K364. PHYRE (Protein Homology/analog Y Recognition Engine) secondary structure homology modeling of ACF took into consideration the domain boundaries from the crystal structures of HuD (24), polyA binding protein (25) and sex lethal (26). K364 was predicted to lie within a 40-50 amino acid disordered region adjacent to an α-helix ending with a.a. 320 (www.sbg.bio.ic.ac.uk/).
Figure 1. Expression of Monomeric ACF320.

(A) Left, Coomassie staining of SDS-PAGE resolved affinity purified ACF64 with and without limited TPKC-trypsin digestion. Arrowheads denote prominent bands reactive in western blot analysis. (B) Western blot analysis of trypsin digested ACF64 probed with either N- or C-terminal ACF peptide reactive antibodies. (C) Coomassie staining of SDS-PAGE resolved ACF320 purified via a N-terminal 4His tag following expression in E. coli. (D) Sephacryl S-100 column (GE Healthcare) fractionation of pooled fractions 3 through 6 from ‘B. Solid line represents OD280 and dashed lines OD260, measured in mAU units in the y-axis. Molecular mass distribution is on the x-axis as determined through calibration with protein standards. Fractions ranging from 40-25 in ‘C’ were pooled and concentrated to 2.8 mg/mL and submitted for AU. (E) Four concentrations of ACF320 were analyzed ranging from 0.12 to 1.28 mg/mL and the sedimentation coefficient distribution, g(s*), values were determined using DcDt+, version 2.1.0 (20,21) and represented graphically. (F) The continuous sedimentation coefficient distribution c(s) of samples in ‘D’, values determined using Sedfit version 11.71 and represented graphically.
RNA-Depleted ACF320 is a Monomeric Protein
An ACF fragment lacking the disorder region and consisting of a.a. 1-320 (ACF320) was cloned and expressed in E. coli with an N-terminal 4 His tag. ACF320 was purified using Ni2+ affinity resin binding and resolved by SDS PAGE with an apparent molecular mass of 36.7 kDa (Figure 1C). Fractions were pooled and analyzed by size exclusion chromatography. ACF320 eluted at its predicted monomeric molecular mass (Figure 1D). The predominant monomeric nature of ACF320 also was suggested by dynamic light scatter (DLS) where samples appeared mono-dispersed with a hydrodynamic radius of 2.7-2.9 nm, consistent with a globular protein of 33-39 kDa.
Chromatographically purified, RNA-depleted ACF320 was evaluated by analytical ultracentrifugation (AU) over a broad range of ACF320 concentrations. An overlay of the normalized g(s*), which is the sedimentation coefficient distribution derived using the time derivative of the concentration distribution, over four concentrations of A320 input (0.12, 0.33, 0.72 and 1.28 mg/mL) demonstrated one peak with an estimated value of 2.8S (Figure 1E). The appearance of only one major peak with increasing protein concentration was consistent with most of ACF320 existing as monomer that did not interact with itself. The presence of a ‘tail’ in the g(s*) curves toward higher S values however was characteristic of a small population of larger complexes.
The AU data were fit to a two species non-interacting model in which the second species was restricted to twice the molecular weight of the main peak. The major peak represented ∼95% of the sample with a predicted molecular weight of 37.3-35.9 kDa. The secondary peak accounted for most of the remaining 5% of the population. A graphical representation of the c(s), calculated by removing the effects of diffusion from the g(s*) calculation, enabled a clearer visualization of the minor species (∼5%) consistent with dimeric form of ACF320 (Figure 3F). We concluded that ACF320 was predominantly a monomer in the absence of RNA.
Figure 3. ACF320 Specifically Binds ApoB RNA.
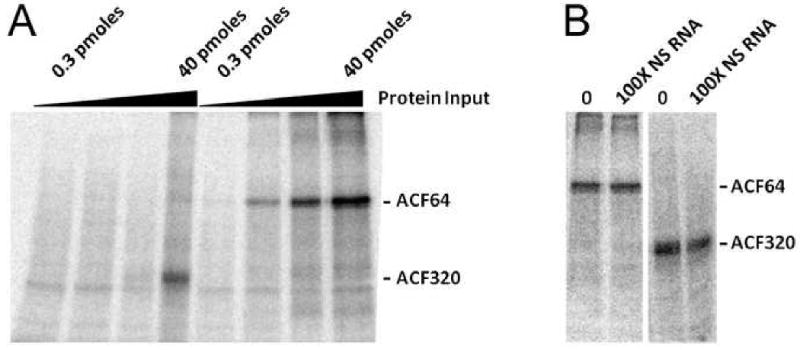
(A) UV cross-linking of ACF to RNA was conducted using increasing molar amounts (0.3, 1.5, 7.5, and 40 pmols) of purified ACF64 or ACF320 by irradiating reactions with 254 nM light, for 5 minutes, on ice and then digested with RNase A. Samples were resolved by SDS-PAGE and visualized by autoradiography. Results are representative of 3 trials. (B) 40 pmols of ACF64 or ACF320 were cross-linked as in ‘A’ in the presence or absence of 100-fold molar excess of non-specific (NS) as unlabeled competitor RNA. Results are representative of two trials.
ACF320 Self-Associates in vivo via an RNA-Dependent Interaction
Whether ACF320 was a monomer in living cells was evaluated using quenched FRET (FqRET) (16). EGFP (fluorescence donor) was expressed as an N-terminal fusion to V5-ACF320 while REACh2 (fluorescence quencher) was cloned as either an N or C-terminus fusion with HA-ACF320. HEK 293T cells were co-transfected with donor and quencher chimeras at a fixed amount of N-terminal EGFP-V5-ACF320 plasmid and increasing amounts of either REACh2-HA-ACF320 or HA-ACF320-REACh2 plasmids. HEK 293T cells were used for these experiments as they do not express endogenous ACF and are easily transfected. Cells co-transfected with EGFP tagged ACF320 and C-terminally REACh2 tagged ACF320 yielded maximally 5% quenching (Figure 2A) which was not statistically significant (p=0.60, n=111). Western blot analysis of whole cell extracts confirmed expression of V5-tagged EGFP construct and the HA-tagged REACh2 constructs (Figure 2 B,D). In contrast, co-transfection of HEK 293T cells with EGFP-ACF320 and REACh2-ACF320 plasmid resulted in maximally 22% reduction of the fluorescence (mean gray scale value of 1396+/-102 vs. 1090+/-84) (Figure 2 C,D) and was statistically significant (p=0.018, n=113). Given that the maximum reduction of fluorescence observed for covalently linked FqRET partners was 50% (16), the data supported the possibility that ACF320 formed dimers wherein the N-termini of the subunits were maintained in a closer proximity than the N- and C-termini, suggesting a parallel, head-to-head orientation of ACF320 monomers. That self-association of ACF320 was not mediated by protein/protein interactions but rather involved the formation of a protein-RNA-protein bridge was suggested by the finding that HA-tagged ACF320 co-immunoprecipitated with V5 epitope tagged ACF320 only when extracts from transfected cells (Figure 2E) were not digested with RNase A (Figure 2F).
Figure 2. ACF320 Self-Association In vivo is Due to RNA-Bridging.

(A) FqRET pairs EGFP (donor) and REACh2 (acceptor) fused to ACF320 were co-transfected in HEK 293T fibroblasts. EGFP was always N-terminal while REACh2 was either C- (A) or N-terminal (C) shown graphically on the far left of each panel. Fluorescence microscopy of cells co-transfected with the indicated ratio of plasmids (indicated below the micrographs). The intensity of fluorescence in pseudocolor increases from blue to white as denoted by the scale left of the images. Percentage of quenching relative to the signal from cells transfected with EGFP-ACF320 alone, (‘*’) denotes a statistically significant quenching (p=0.018). Western blot analysis (B,D) of equal μg amounts lysate from transfected cells were reacted with anti- V5, anti-HA or anti-actin to validate expression of chimeric proteins and similar loading of total cellular protein for each sample. (C) Equal microgram aliquots of whole cell extracts prepared from 293T cells that had been co-transfected with V5-ACF320-EGFP and HA-ACF320-EGFP were resolved by SDS-PAGE and western blotted with V5 or HA reactivity antibodies. (D) Co-immunoprecipitation using a monoclonal antibody recognizing V5 was performed with or without RNase A digestion of the cell extract. Western blots were probed sequentially for HA and the V5 (top right panel) after stripping the blot. The result is representative of two independent experiments.
ACF320 binds preferentially to apoB RNA
The apoB RNA binding activities of full length ACF64 and ACF320 were determined using UV cross-linking of RNA-protein complexes assembled in vitro (18,27). RNA cross-linking signals increased when ACF64 was titrated (0.3 to 40 pmols) against a fixed amount (150 fmols) of radiolabeled apoB RNA (Figure 3A). A similar titration of ACF320 demonstrated binding to apoB RNA primarily at 40 pmols of input ACF320. The yield of cross-linked complex was comparable to that seen with 1.5 pmols ACF64 (based on PhosphorImager quantification). Nonspecific RNA competition certified a selectivity of ACF320 for the apoB RNA editing site. Nonspecific RNA, at >100× molar excess of apoB RNA, demonstrated that despite its reduced RNA binding activity, ACF320 at 40 pmols input protein preferentially UV cross-linking to apoB RNA, comparable to that seen with ACF64 at 1.5 pmol input of protein (Figure 3B).
ACF320 binds to APOBEC-1 and Complements apoB Editing
V5-ACF320 or V5-ACF64 were immunoprecipitated from extracts of HEK 293T cell that had been co-transfected with HA epitope tagged APOBEC-1 (Figure 4A). APOBEC-1 co-immunoprecipitated with both ACF64 and ACF320 though comparatively, the yield of APOBEC-1 co-immunoprecipitated with ACF320 was much less than that recovered with ACF64 (Figure 4B).
Figure 4. ACF320 Complements ApoB Editing Activity.

(A) equivalent microgram input from HEK 293T whole cell extract were western blotted simultaneously with V5 reactive antibody to detect ACF isoforms and with anti-HA antibodies to detect APOBEC-1 in transfected cells. (B) co-immunoprecipitation of HA tagged APOBEC-1 with V5 epitope pulldown of ACF64 or ACF320 from extracts in ‘C’ were western blotted with the respective antibodies. The image is representative of duplicate trials. Extracts from HEK 293T cells co-transfected with APOBEC-1, apoB RNA editing reporter and acf64 or acf320 were split into two aliquots. (C) One aliquot was western blotted for the ACF variants with the V5 epitope and APOBEC-1 with the HA epitope. (D) RNA was extracted from the second aliquot and poison-primer extension quantification of RNA editing. UAA represents edited transcript while CAA represents unedited message. Percent editing was calculated by ((UAA)/(CAA+UAA)) × 100, results and S.D. were calculated based on an n=3.
HEK 293T cells co-transfected with the apoB reporter, HA-tagged APOBEC-1 and V5-tagged ACF320 or ACF64 also were evaluated for apoB mRNA editing. All transfectants displayed similar expression levels of ACF and APOBEC-1 as observed by western blotting (Figure 4C). Only background levels of apoB RNA editing were observed in the absence of APOBEC-1 and in the absence of exogenous ACF64, APOBEC-1 only supported low levels of editing activity (6% +/-1) (Figure 4D). The highest level of editing complementation (39% +/-3) was observed when APOBEC-1 and full length ACF64 were co-expressed. ACF320 complementation of APOBEC-1 editing activity (17% +/- 2) was approximately half as efficient as that observed with ACF64.
Discussion
We used biochemical and computational methods to predict a well-folded polypeptide of ACF comprised of the three RRMs. The resulting protein (ACF320) was cloned and expressed as a monodisperse soluble protein. ACF320 bound to apoB RNA preferentially, and its interaction with APOBEC-1 was sufficient to complement C to U RNA editing. We show that ACF320 can be isolated as a monomeric subunit with limited protein-protein self-association. However, dimers and higher-order oligomers form in cells through RNA-bridging. Live cell FqRET analysis suggested that RNA-bridged ACF320 subunits might be organized such that their N-termini are proximal.
ACF320 was approximately 27-fold less efficient in binding apoB RNA compared with full length ACF64. These data were of interest because ACF320 was 57 amino acids smaller than the minimal truncation mutant previously shown to have both RNA-binding and APOBEC-1 complementation activities and yet it retained approximately >100-fold higher RNA binding activity than previously tested constructs (9,10). Prior studies predicted the C-terminal boundary of the three RRM domain in ACF to end at residue 391 based on an alignment only with GRY-RBP (9,10). Our studies suggested that C-terminal truncations beyond a.a. 391 would expose a disordered region (a.a. 331 to 377) that may have a negative impact on RNA binding. In this regard, ACF320 was designed to eliminate this disordered region but include an alpha helical region C-terminal to the end of RRM3 (a.a. 301 to 320).
Yeast two hybrid analyses suggested that ACF64 and its truncated variants (1,2,14,28-30) had the capacity for self association (14,31). The RNA dependence of ACF320 self-association makes it tempting to speculate that its mode of binding had structural and functional similarities to the RNA-induced, self-association seen with U1A. Monomers of U1A formed dimers in a parallel orientation when bound to PIE RNA (32). The helices C-terminal to U1A RRMs functioned to stabilization U1A binding to RNA. We propose that the mooring sequence RNA-bridged, head-to-head self-association of ACF may be critical for editosome function in that it orients dimers of APOBEC-1 (33-35) to a specific site for C to U editing.
Acknowledgments
The authors are grateful to Jenny Smith for assistance in figure preparation.
Funding: This work was supported in part by a Public Health Services Grant DK043739 and AI058789 awarded to HCS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lellek H, Kirsten R, Diehl I, Apostel F, Buck F, Greeve J. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J Biol Chem. 2000;275:19848–19856. doi: 10.1074/jbc.M001786200. [DOI] [PubMed] [Google Scholar]
- 2.Mehta A, Kinter MT, Sherman NE, Driscoll DM. Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol Cell Biol. 2000;20:1846–1854. doi: 10.1128/mcb.20.5.1846-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith HC. Apolipoprotein B mRNA editing: the sequence to the event. Semin Cell Biol. 1993;4:267–278. doi: 10.1006/scel.1993.1032. [DOI] [PubMed] [Google Scholar]
- 5.Maris C, Masse J, Chester A, Navaratnam N, Allain FH. NMR structure of the apoB mRNA stem-loop and its interaction with the C to U editing APOBEC1 complementary factor. RNA. 2005;11:173–186. doi: 10.1261/rna.7190705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calero G, Wilson KF, Ly T, Rios-Steiner JL, Clardy JC, Cerione RA. Structural basis of m7GpppG binding to the nuclear cap-binding protein complex. Nat Struct Biol. 2002;9:912–917. doi: 10.1038/nsb874. [DOI] [PubMed] [Google Scholar]
- 7.Mazza C, Segref A, Mattaj IW, Cusack S. Large-scale induced fit recognition of an m(7)GpppG cap analogue by the human nuclear cap-binding complex. Embo J. 2002;21:5548–5557. doi: 10.1093/emboj/cdf538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johansson C, Finger LD, Trantirek L, Mueller TD, Kim S, Laird-Offringa IA, Feigon J. Solution structure of the complex formed by the two N-terminal RNA-binding domains of nucleolin and a pre-rRNA target. J Mol Biol. 2004;337:799–816. doi: 10.1016/j.jmb.2004.01.056. [DOI] [PubMed] [Google Scholar]
- 9.Mehta A, Driscoll DM. Identification of Domains in APOBEC-1 Complementation Factor Required for RNA Binding and Apolipoprotein B mRNA editing. RNA. 2002;8:69–82. doi: 10.1017/s1355838202015649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanc V, Henderson JO, Kennedy S, Davidson NO. Mutagenesis of apobec-1 complementation factor reveals distinct domains that modulate RNA binding, protein-protein interaction with apobec-1, and complementation of C to U RNA-editing activity. J Biol Chem. 2001;276:46386–46393. doi: 10.1074/jbc.M107654200. [DOI] [PubMed] [Google Scholar]
- 11.Lehmann DM, Galloway CA, Macelrevey C, Sowden MP, Wedekind JE, Smith HC. Functional characterization of APOBEC-1 complementation factor phosphorylation sites. Biochim Biophys Acta. 2007;1773:408–418. doi: 10.1016/j.bbamcr.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann DM, Galloway CA, Sowden MP, Smith HC. Metabolic regulation of apoB mRNA editing is associated with phosphorylation of APOBEC-1 complementation factor. Nucleic Acids Res. 2006;34:3299–3308. doi: 10.1093/nar/gkl417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanc V, Kennedy S, Davidson NO. A novel nuclear localization signal in the auxiliary domain of apobec-1 complementation factor regulates nucleocytoplasmic import and shuttling. J Biol Chem. 2003;278:41198–41204. doi: 10.1074/jbc.M302951200. [DOI] [PubMed] [Google Scholar]
- 14.Sowden MP, Lehmann DM, Lin X, Smith CO, Smith HC. Identification of Novel Alternative Splice Variants of APOBEC-1 Complementation Factor with Different Capacities to Support ApoB mRNA Editing. J Biol Chem. 2004;278:197–206. doi: 10.1074/jbc.M307920200. [DOI] [PubMed] [Google Scholar]
- 15.Galloway CA, Sowden MP, Smith HC. Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques. 2003;34:524–526. 528, 530. doi: 10.2144/03343st04. [DOI] [PubMed] [Google Scholar]
- 16.Ganesan S, Ameer-Beg SM, Ng TT, Vojnovic B, Wouters FS. A dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc Natl Acad Sci U S A. 2006;103:4089–4094. doi: 10.1073/pnas.0509922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith HC, Kuo SR, Backus JW, Harris SG, Sparks CE, Sparks JD. In vitro apolipoprotein B mRNA editing: identification of a 27S editing complex. Proc Natl Acad Sci U S A. 1991;88:1489–1493. doi: 10.1073/pnas.88.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith HC. Analysis of protein complexes assembled on apolipoprotein B mRNA for mooring sequence-dependent RNA editing. Methods. 1998;15:27–39. doi: 10.1006/meth.1998.0603. [DOI] [PubMed] [Google Scholar]
- 19.Sowden M, Hamm JK, Spinelli S, Smith HC. Determinants involved in regulating the proportion of edited apolipoprotein B RNAs. Rna. 1996;2:274–288. [PMC free article] [PubMed] [Google Scholar]
- 20.Philo JS. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 21.Philo JS. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal Biochem. 2006;354:238–246. doi: 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 22.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sowden MP, Ballatori N, de Mesy Jensen KL, Hamilton Reed L, Smith HC. The editosome for cytidine to uridine mRNA editing has a native complexity of 27S: identification of intracellular domains containing active and inactive editing factors. J Cell Science. 2002;115:1027–1039. doi: 10.1242/jcs.115.5.1027. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Tanaka Hall TM. Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat Struct Biol. 2001;8:141–145. doi: 10.1038/84131. [DOI] [PubMed] [Google Scholar]
- 25.Deo RC, Bonanno JB, Sonenberg N, Burley SK. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell. 1999;98:835–845. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
- 26.Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 27.Harris SG, Sabio I, Mayer E, Steinberg MF, Backus JW, Sparks JD, Sparks CE, Smith HC. Extract-specific heterogeneity in high-order complexes containing apolipoprotein B mRNA editing activity and RNA-binding proteins. J Biol Chem. 1993;268:7382–7392. [PubMed] [Google Scholar]
- 28.Dur S, Krause K, Pluntke N, Greeve J. Gene structure and expression of the mouse APOBEC-1 complementation factor: multiple transcriptional initiation sites and a spliced variant with a premature stop translation codon. Biochim Biophys Acta. 2004;1680:11–23. doi: 10.1016/j.bbaexp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Henderson JO, Blanc V, Davidson NO. Isolation, characterization and developmental regulation of the human apobec-1 complementation factot (ACF) gene. Biochim Biophys Acta. 2001;1522:22–30. doi: 10.1016/s0167-4781(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 30.Dance GS, Beemiller P, Yang Y, Mater DV, Mian IS, Smith HC. Identification of the yeast cytidine deaminase CDD1 as an orphan C-->U RNA editase. Nucleic Acids Res. 2001;29:1772–1780. doi: 10.1093/nar/29.8.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith HC. Measuring editing activity and identifying cytidine-to-uridine mRNA editing factors in cells and biochemical isolates. Methods Enzymol. 2007;424:389–416. doi: 10.1016/S0076-6879(07)24018-2. [DOI] [PubMed] [Google Scholar]
- 32.Varani L, Gunderson SI, Mattaj IW, Kay LE, Neuhaus D, Varani G. The NMR structure of the 38 kDa U1A protein - PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat Struct Biol. 2000;7:329–335. doi: 10.1038/74101. [DOI] [PubMed] [Google Scholar]
- 33.Lau PP, Zhu HJ, Baldini HA, Charnsangavej C, Chan L. Dimeric structure of a human apo B mRNA editing protein and cloning and chromosomal localization of its gene. Proc Natl Acad Sci USA. 1994;91:8522–8526. doi: 10.1073/pnas.91.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teng BB, Ochsner S, Zhang Q, Soman KV, Lau PP, Chan L. Mutational analysis of apolipoprotein B mRNA editing enzyme (APOBEC1). structure-function relationships of RNA editing and dimerization. J Lipid Res. 1999;40:623–635. [PubMed] [Google Scholar]
- 35.Navaratnam N, Fujino T, Bayliss J, Jarmuz A, How A, Richardson N, Somasekaram A, Bhattacharya S, Carter C, Scott J. Escherichia coli cytidine deaminase provides a molecular model for ApoB RNA editing and a mechanism for RNA substrate recognition. J Mol Biol. 1998;275:695–714. doi: 10.1006/jmbi.1997.1506. [DOI] [PubMed] [Google Scholar]