

Abstract
Aberrant cell cycle activity and DNA damage are emerging as important pathological components in various neurodegenerative conditions. However, their underlying mechanisms are poorly understood. Here, we show that deregulation of HDAC1 activity by p25/Cdk5 induces aberrant cell cycle activity and double-strand DNA breaks leading to neurotoxicity. In a transgenic model for neurodegeneration, p25/Cdk5 activity elicited cell cycle reentry and double-strand DNA breaks that preceded neuronal death. Inhibition of HDAC1 activity by p25/Cdk5 was identified as an underlying mechanism for these events, and HDAC1 gain-of-function provided potent protection against DNA damage and neurotoxicity in cultured neurons and an in vivo model for ischemia. Our findings outline a novel pathological signaling pathway which illustrates the importance of maintaining HDAC1 activity in the adult neuron. This pathway constitutes a molecular link between aberrant cell cycle activity and DNA damage and is a potential target for therapeutics against diseases and conditions involving neuronal death.
INTRODUCTION
In a variety of conditions involving neuronal death such as ischemia and Alzheimer’s disease (Hayashi et al., 2000; Rashidian et al., 2007; Vincent et al., 1996; Yang et al., 2001), neurons engage in aberrant cell cycle activities, expressing cell cycle markers such as Ki-67 and PCNA, and synthesizing DNA (Yang et al., 2001). This is remarkable considering that neurons are terminally differentiated and remain quiescent for decades prior to the onset of these events. While the underlying mechanisms are poorly understood, these activities may play an early and contributory role in neuronal death (Busser et al., 1998; Herrup and Busser, 1995). For example, overexpression of cell cycle activity-inducing proteins such as SV40 large T antigen, c-myc, c-Myb, or E2F-1 can cause neuronal death in vitro and in vivo (al-Ubaidi et al., 1992; Konishi and Bonni, 2003; Liu and Greene, 2001), while pharmacological inhibitors of CDKs or other cell cycle components can exert neuroprotective effects (Padmanabhan et al., 1999).
DNA damage may also be involved in numerous conditions involving neuronal death (Adamec et al., 1999; Ferrante et al., 1997; Hayashi et al., 1999; Kruman et al., 2004; Robison and Bradley, 1984). For example, oxidative damage to neuronal DNA is observed in rodent models of ischemia (Hayashi et al., 1999). Accumulation of reactive oxygen species results in DNA damage, cell cycle activity, and neurodegeneration in mutant mice with disrupted apoptosis-inducing factor (AIF) (Klein et al., 2002). In addition, congenital syndromes with DNA repair gene mutations, such as ataxia telangiectasia and Werner’s syndrome, display a progressive neurodegeneration phenotype, demonstrating the importance of maintaining DNA integrity in the adult brain (Rolig and McKinnon, 2000). DNA damage is involved in the aging of the human brain (Lu et al., 2004), which suggests that DNA damage may play a role in age-dependent neurodegenerative diseases as well.
Regulating histone acetylation is an integral aspect of chromatin modulation and gene regulation that plays a critical role in many biological processes including cell proliferation and differentiation (Roth et al., 2001). Recent reports have detailed the importance of histone acetylation in CNS functions such as neuronal differentiation, memory formation, drug addiction, and depression (Citrome, 2003; Tsankova et al., 2006). Histone deacetylases (HDACs) remove acetyl groups from histones, resulting in chromatin compaction and decreased accessibility to DNA for interacting molecules such as transcription factors (Cerna et al., 2006). Of these, histone deacetylase 1 (HDAC1) was the first mammalian protein identified to have histone-directed deacetylase activity (Taunton et al., 1996). HDAC1 plays important roles in regulating the cell cycle and is required in the transcriptional repression of cell cycle genes such as p21/WAF, E2F-1, and cyclins A and E (Brehm et al., 1998; Lagger et al., 2002; Rayman et al., 2002; Stiegler et al., 1998). The association of HDAC1 with promotor regions of specific genes is linked to their transcriptional repression (Brehm et al., 1998; Gui et al., 2004; Rayman et al., 2002).
The serine/threonine kinase Cdk5 and its activating subunit p35 play important roles in both the developing and adult central nervous system (Dhavan and Tsai, 2001). In numerous neurodegenerative states including postmortem Alzheimer’s disease brains and animal models for stroke/ischemia (Lee et al., 2000; Nguyen et al., 2001; Patrick et al., 1999; Smith et al., 2003; Swatton et al., 2004; Wang et al., 2003), neurotoxic stimuli induce calpain mediated cleavage of p35 into p25, the accumulation of which elicits neurotoxicity in cultured neurons and in vivo (Lee et al., 2000; Patrick et al., 1999).
We have previously generated a bitransgenic mouse model (CK-p25 mice) which expresses a p25-GFP fusion protein under the control of the Calmodulin Kinase II promoter in an inducible, postdevelopmental, and forebrain-specific manner (Cruz et al., 2003). Upon induction of p25, neurodegenerative events occur in a rapid and orderly manner, as astrogliosis is observed after 4 weeks of induction, and neuronal loss and cognitive impairment is appreciable after 6 weeks of induction (Cruz et al., 2003; Fischer et al., 2005). Thus, this model may provide a tractable system for investigating mechanisms for neuronal death relevant to conditions involving p25, such as stroke/ischemia.
Here, we examined the gene expression profile in p25 transgenic mice induced for a short period to gain insights into early and instigating mechanisms involved in neurodegeneration. We observed that following p25 induction, neurons aberrantly express cell cycle proteins and form double-strand DNA breaks at an early stage prior to their death. p25 interacted with and inactivated HDAC1, and inactivation of HDAC1 through siRNA knockdown or pharmacological inhibition resulted in double-strand DNA breaks, aberrant cell cycle protein expression, and neuronal death. Importantly, restoring HDAC1 activity by overexpressing wild type HDAC1, but not the deacetylase activity-deficient mutant, rescued against p25-mediated double-strand DNA breaks and cell death. Furthermore, we provide evidence that this pathway is relevant in an in vivo model for stroke. Our findings suggest that the inactivation of HDAC1 by p25 may be involved in the pathogenesis of neurodegenerative diseases.
RESULTS
Gene expression profile of CK-p25 transgenic mice
In CK-p25 mice induced for two weeks, widespread forebrain neuron-specific expression of p25-GFP is observed (Supplementary Figure 1), without signs of neurotoxicity or reactive astrogliosis (Fischer et al., 2005). We carried out microarray analyses of CK-p25 mouse forebrains (Affymetrix, Supplemental procedures) at this period of induction in hopes of elucidating the initiating mechanisms accounting for the neurodegeneration observed in later weeks. A total of 225 genes (292 total probes) were found to be significantly altered in the induced transgenics compared to uninduced controls (Table S1). Surprisingly, genes involved in cell cycle or DNA damage repair/response (Gene Ontology database, http://www.geneontology.org/) were highly represented (Table S2), totaling 65 genes (84 total probes) with overlap between the annotation groups. Representative genes from these groups are summarized in Table 1. 63 of the 65 genes were upregulated, including cell cycle/proliferation genes such as Cyclins A, B, and E, E2F-1, Ki67 and PCNA, which have previously been shown to be upregulated in postmortem AD brains and rodent stroke models. In addition, a number of DNA damage response genes, in particular genes involved in the DNA double-strand breaks response such as Rad51, BRCA1, and Checkpoint 1, were found to be highly upregulated. Collectively, these findings suggest the aberrant expression of cell cycle proteins and a response to double-strand DNA breaks in the brains of CK-p25 mice.
Table 1. Genes involved in cell cycle and DNA damage response are upregulated in the forebrains of induced CK-p25 mice.
Microarray analysis was carried out as described in Experimental Procedures, and genes were organized into functional groups based on gene ontology. Genes with multiple gene ontology annotations (e.g. PCNA) were arbitrary placed into one of the groups. ‘Fold Δ’ indicates fold change in expression levels for p25 compared to controls at 2 weeks of induction. The entire list of genes with significantly altered expression (Table S1) and cell cycle/DNA damage related genes (Table S2) are shown in the Supplementary Data section.
| Function | Gene name | Accession | Fold Δ |
|---|---|---|---|
| Cell cycle related genes | Cyclin A2 | X75483 | 3.78 |
| Cyclin B1 | NM_007629 | 10.58 | |
| Cyclin E1 | NM_007633 | 1.94 | |
| Cyclin E2 | AF091432 | 4.9 | |
| Cdc28 | NM_016904 | 2.4 | |
| Cdc28 regulatory subunit 1 | NM_025415 | 5.11 | |
| Cdc2a (cdk1) | NM_007659 | 8.47 | |
| Cdc20 | BB041150 | 4.09 | |
| Cell division associated 1 (Nuf2R) | AK010351 | 2.16 | |
| Polo-like kinase 4 | AI385771 | 2.9 | |
| Geminin | NM_020567 | 2.27 | |
| Mcm2 | NM_008564 | 5.01 | |
| Mcm3 | C80350 | 6.27 | |
| Mcm4 | BC013094 | 3.06 | |
| Mcm6 | NM_008567 | 6.04 | |
| Mcm7 | NM_008568 | 3.78 | |
| DNA primase, p49 subunit | J04620 | 3.19 | |
| DNA primase, p58 subunit | NM_008922 | 1.67 | |
| p21/WAF | AK007630 | 2.55 | |
| Proliferating cell nuclear antigen (PCNA) | BC010343 | 2.47 | |
| Ki-67 proferation antigen | X82786 | 16.14 | |
| E2F-1 | NM_007891 | 5.04 | |
| Transcription factor DP-1 | BG075396 | 1.73 | |
| DNA damage responsive genes | Rad51 | NM_011234 | 31.77 |
| Rad51 associated protein | BC003738 | 9.93 | |
| Topoisomerase II alpha | BM211413 | 6.02 | |
| DNA methyltransferase (cytosine-5) 1 | NM_010066 | 1.67 | |
| Flap endonuclease | BB393998 | 2.65 | |
| MutS homolog 6 | U42190 | 1.66 | |
| Ligase I | NM_010715 | 3.99 | |
| DNA polymerase epsilon | NM_011132 | 37.8 | |
| DNA polymerase delta 1, catalytic subunit | BB385244 | 1.93 | |
| Pmaip1 | NM_021451 | 5.23 | |
| Deoxyuridine triphosphatase | AF091101 | 1.65 | |
| Ribonucleotide reductase M2 | BB758819 | 5.65 | |
| Replication protein A1 | BB491281 | 1.64 | |
| Replication protein A2 | AK011530 | 2.13 | |
| Uracil DNA glycosylase | BC004037 | 3.24 | |
| Chromatin assembly factor 1b | NM_011132 | 5.81 | |
| BRCA1 | U31625 | 5.45 | |
| Checkpoint 1 | C85740 | 8.06 | |
| Mad2-like 1 | NM_019499 | 2.59 |
p25 induction results in aberrant expression of cell cycle proteins
We examined the expression various cell cycle proteins in CK-p25 mouse brains to confirm their aberrant upregulation as suggested by the microarray analyses. We confirmed by semiquantitative RT-PCR that mRNA levels of various cell cycle genes identified in the microarray were upregulated in CK-p25 mouse hippocampi compared to WT controls (Figure 1A). In addition, protein levels of PCNA, E2F-1, and Cyclin A were upregulated compared to WT controls (Figure 1B). There was no change in levels of glial fibrillary acidic protein (GFAP), consistent with the absence of neurodegeneration at this period of induction. Immunostaining demonstrated robust increases in cell cycle progression/proliferation markers Ki-67 and PCNA immunoreactivity in p25-expressing adult neurons which were identified by the GFP signal (Figures 1C and 1D). Importantly, only neurons expressing p25-GFP were found to have increased levels of cell cycle markers, while no neurons expressed these markers in WT mice. Some nonneuronal cells stained positively for these cell cycle markers (e.g. in the subventricular zone) in both CK-p25 and WT brains (data not shown), reflecting normal cell cycle activity in nonpostmitotic cells. In addition, a subset of p25-GFP neurons incorporated bromodeoxyuridine (BrdU), indicating DNA synthesis activity (data not shown). On the other hand, p25-GFP expressing neurons were not immunoreactive for the mitotic marker phospho(pS10)-Histone H3, indicating the absence of mitotic cell cycle activity (Figure 1E). Our results imply that p25 induction results in aberrant expression of cell cycle proteins in neurons and aberrant cell cycle activity.
Figure 1. Cell cycle markers are aberrantly upregulated following p25 induction.

(A) Hippocampi from 2 week-induced CK-p25 mice and WT mice were subjected to total RNA extraction, reverse transcription, and semi-quantitative PCR analyses as described. A representative blot of mRNA levels of various cell cycle genes from CK-p25 and WT hippocampi at 20, 25, and 30 cycles of semiquantitative RT-PCR are shown on the left. Quantifications from multiple animals are displayed in the histogram on right. Band quantifications were normalized to actin levels. Data is displayed as average ±S.E as fold increase over WT, with WT samples set to 1. P values (*p<0.05) were calculated from multiple animals by two-tailed, unpaired Student’s t-test. (B) Forebrains from 2-week induced CK-p25 mice and WT controls were analyzed for PCNA, cyclinA, and E2F-1 protein levels. Glial fibrillary acidic protein (GFAP), or BetaIII-tubulin, used as loading control, were unchanged. (C) Ki-67, a cell cycle progression marker, is upregulated in p25-expressing neurons in CK-p25 brains (top panels), but not in neurons of WT controls (bottom panels). CA1 region is shown. (D) Proliferating cell nuclear antigen (PCNA), a proliferation/S-phase marker, is induced in p25-expressing neurons in CK-p25 brains (top panels), but not in neurons of WT controls (bottom panels). CA1 region is shown. (E) p25-expressing neurons in CK-p25 brains are not immunoreactive for the mitotic marker phospho(pS10)-Histone H3 (top panels). Subventricular zone (SVZ) of the same CK-p25 brain is shown as a positive control for mitotic cells which display phospho-Histone H3 immunoreactivity. Scale bar = 50 μm.
p25 induction results in double-strand DNA breaks
The microarray analyses also suggested that p25 expression induced many genes involved in the double-strand DNA break response. To determine whether double-stranded DNA breaks occur in the CK-p25 mice, brains from 2-week induced mice were examined using the double-strand break marker phospho-serine 129 histone H2AX (γH2AX). Robust γH2AX immunoreactivity was detected both biochemically (Figure 2A) and by staining, revealing that γH2AX immunoreactivity was specific to p25-GFP expressing neurons (Fig. 2B). γH2AX staining was undetectable in the WT brain neurons. The double-strand DNA break response protein Rad51 was also found to be upregulated in CK-p25 brains (Fig. 2A).
Figure 2. Double-strand DNA damage occurs following p25 induction.

(A) Western blots from induced CK-p25 mice forebrain lysates show increased levels of γH2AX and Rad51 compared to WT controls. Asterisk indicates nonspecific band. Quantification of γH2AX levels (±S.D.) from multiple WT controls (n=5) and CK-p25 mice (n=5) induced between 2 and 12 weeks are shown in top panel. (B) Staining of vibratome sections with γH2AX reveals immunoreactivity specifically in the nuclei of p25-GFP-expressing neurons in two-week induced CK-p25 mice (top panels) but not in neurons of WT controls (bottom panels). CA1 region is shown. Inset is a magnification of γH2AX signal showing punctate nuclear staining. Scale bar = 50μM. (C) Primary cortical neurons were infected with increasing titers of herpesvirus expressing p25 (p25-HSV) or lacZ-HSV control and analyzed for γH2AX levels by Western blot. (D) Primary cortical neurons infected with p25-HSV and fixed 8 hours post-infection display robust immunoreactivity with γH2AX (right panels), compared to control uninfected neurons (left panels). p25 overexpression was verified with p35 antibody (top panels). Top and bottom panels are from different fields. (E) Comet assays were carried out on primary neurons infected with p25-HSV or lacZ-HSV for 10 hours. Representative micrographs of comet assay fields are shown in the left and middle panels for p25-HSV infected and lacZ-HSV infected neurons, respectively. Comet tails indicate DNA with breaks, resulting in increased migration towards the direction of the current (left to right). Right panel shows quantification of the percentage of neurons with comet tails from three separate experiments. Results are displayed as fold change to control (lacZ-HSV infected) neurons. P-values (**p<0.005) were calculated from multiple experiments by two-tailed, unpaired Student’s t-test.
We examined whether p25-mediated induction of double-strand breaks could be recapitulated in cultured primary neurons using herpes simplex virus (HSV)-mediated overexpression of p25. Expression of p25 in primary neurons also resulted in robust generation of γH2AX (Figure 2C and 2D). To provide physical proof of DNA damage, primary neurons overexpressing p25 were analyzed for DNA strand breaks using single cell gel electrophoresis (comet assay). We observed that nuclei of p25 overexpressing neurons displayed a ~1.8-fold higher incidence of comet tails indicative of DNA containing single- or double-strand breaks (Figure 2E). These results demonstrate that expression of p25 induces DNA strand breaks in neurons.
Double-strand DNA damage and cell cycle activity are tightly associated and precede neuronal death
Co-staining with γH2AX and Ki-67 in CK-p25 mice revealed that neurons undergoing aberrant expression of cell cycle proteins also exhibited double-strand DNA breaks, and vice versa, at high concurrency (92.3 ± 2.7% S.D.), suggesting that the two events are tightly linked (Figure 3A). In CK-p25 mice induced for 8 weeks (a period when massive neurodegeneration is evident (Cruz et al., 2003)), both the DNA damage marker γH2AX and the cell cycle progression marker Ki-67 were each associated with pyknotic nuclei (Figure 3B). Over 70% of CA1 neurons in CK-p25 mice that were positive for both p25-GFP and γH2AX, or both p25-GFP and Ki-67 had pyknotic nuclei compared to only 34% for neurons positive for p25-GFP alone (Figure 3B). Experiments in cultured neurons (Figure S2) and in vivo (Cruz et al., 2003) indicate that p25-expressing neurons die primarily through apoptosis as indicated by immunoreactivity to cleaved caspase-3 and cleaved PARP-1. Interestingly, in CK-p25 mice subjected to p25 expression for 2 weeks followed by suppression of p25 expression for 4 weeks (by feeding a doxycycline diet), we observed that γH2AX signal was abrogated (Figure 3C), while no signs of neuronal loss were observed (Fischer et al., 2005). This indicates that the degree of γH2AX formation observed by 2 weeks is reversible, and that γH2AX formation in CK-p25 mice precedes and is not secondary to cell death. Furthermore, we observed that the pan-caspase inhibitor z-VAD-fmk had no effect on p25-mediated γH2AX formation, further excluding DNA damage as a downstream consequence of apoptotic events (Figure S3).
Figure 3. Double-strand DNA breaks and aberrant cell cycle activity are concomitant and precede neuronal death.

(A) Double immunoflourescence staining for Ki-67 (green) and γH2AX (red) carried out in 2 week induced CK-p25 mice revealed that cell cycle reentry and DNA double-strand breaks occur concurrently. Representative images of CA1 region is shown in left panels, and quantification of neurons immunoreactive for both γH2AX and Ki-67, γH2AX only, or Ki-67 only from multiple 2 week induced CK-p25 mice are shown in the histogram. a: γH2AX+Ki-67 vs γH2AX only, p<0.001; b: γH2AX+Ki-67 vs Ki-67 only, p<0.001. One way ANOVA with Neuman-Keuls multiple comparison test. (B) γH2AX and Ki-67 are closely associated with dying neurons at 8 weeks of p25 induction. A representative image showing association of γH2AX and Ki-67 with pyknotic nuclei (first, second, and third panels). Fourth panel is a magnification of the boxed region in third panel. Quantification of cell death (pyknotic nuclei) in p25-GFP and γH2AX immunoreactive neurons, p25-GFP and Ki-67 immunoreactive neurons, or neurons immunoreactive for p25-GFP but not γH2AX or Ki-67 are shown from multiple 2-week induced and 8-week induced CK-p25 mice. a: p25 only vs p25+ γH2AX, p<0.01; b:p25 only vs p25+Ki-67, p<0.01. One way ANOVA with Neuman-Keuls multiple comparison test. (C) CK-p25 mice were induced for 2 weeks (top panels) and sacrificed, or induced for 2 weeks followed by 4 weeks of suppression through doxycyline diet prior to sacrifice. Sections were examined for GFP and γH2AX signals. It was previously determined that 2 week induction of p25 followed by 4 weeks of suppression did not result in neuronal loss (Fischer et al., 2005). Scale bar = 100 μM.
Collectively, our results demonstrate that cell cycle and DNA damage events are tightly correlated with each other, and that they precede cell death in neurons with p25 accumulation.
p25 interacts with and inhibits HDAC1
The tight association of cell cycle protein expression and DNA damage in CK-p25 mice suggested that a common mechanism may underlie these events. As both gene transcription and susceptibility to DNA damage are known to be tightly linked to the chromatin state, we considered the involvement of HDACs in the induction of aberrant neuronal cell cycle expression and DNA damage by p25/Cdk5. Inhibition of HDACs can induce gene transcription, and also increase sensitivity DNA to DNA damaging agents, both through increased accessibility (Cerna et al., 2006).
Of particular interest was HDAC1, based on its reported role in transcriptional repression of cell cycle related genes such as p21/WAF, cyclins A, D, and E, and cdc25A (Brehm et al., 1998; Lagger et al., 2002; Stiegler et al., 1998). We determined that in forebrains of CK-p25 mice induced for 2 weeks, p25 interacted with HDAC1 in vivo (Figure 4A). Interaction with HDAC1 was observed with both p25 and p35 co-transfected in 293T cells (Figure 4B). Interestingly, HDAC1 had an over 12-fold higher degree of interaction with p25, compared to the physiological, non-cleaved p35 (Figure 4B) which does not exert neurotoxicity. The preferential binding of HDAC1 with the pathological molecule p25 raised the interesting possibility that the p25-HDAC1 interaction may have deleterious consequences.
Figure 4. p25 interacts with HDAC1 and inhibits its activity.

(A) Forebrains from 2-week induced CK-p25 and WT control mice were homogenized and lysates immunoprecipitated with HDAC1 antibody as described, and probed for p25-GFP and HDAC1. (B) Flag-tagged HDAC1 was overexpressed with GFP-p25 or p35 in HEK293T cells, immunoprecipitated with flag-conjugated beads as described, and probed for p25-GFP or p35-GFP. (C) Flag-tagged full length HDAC1 or various truncation mutations were overexpressed with GFP-p25 and immunoprecipitated with flag-conjugated beads as described. The catalytic domain is indicated in brown. (D) Left panel: HEK293T cells were transfected with vector or with p25/Cdk5. 15 hours later, endogenous HDAC1 was immunoprecipitated then assayed for histone deacetylase activity as described. Averages from multiple experiments are displayed as fold change over control (vector only). Right panel: hippocampi from 2 week-induced WT and CK-p25 mice were dissected and assayed for endogenous HDAC1 activity as described. P-values (**p<0.005, *p<0.05) were calculated from multiple experiments by two-tailed, unpaired Student’s t-test. (E) p25/Cdk5 inhibits the transcriptional repressor activity of HDAC1. HDAC1-Gal4 construct was cotransfected with blank vector or p25/Cdk5 then measured for luciferase reporter activity as described. Values were normalized to protein levels of Gal4 constructs, and are expressed as relative light units (HDAC1-Gal4 only = 1). (F) Primary cortical neurons were infected with p25-HSV or GFP-HSV then subjected to fractionation as described. Lamin A and Histone 3 are used as markers for the nuclear and chromatin fractions, respectively. Band densitometry quantifications from multiple experiments (±S.D.) are shown in the histogram on the right. (G) HEK293T cells were transfected with blank vector or p25 and Cdk5, cross-linked, then subjected to chromatin immunoprecipitation using HDAC1 or acetylated Histone 3 at Lysine 9 (acetylH3K9) antibody. Immune complexes were subjected to semi-quantitative PCR amplification using primers towards the core promoter regions of E2F-1 and p21/WAF.
Because HDAC1 is exclusively localized in the nucleus (de Ruijter et al., 2003), we examined the localization of p25-GFP in the CK-p25 mouse (Figure S4). While abundant p25-GFP signal was observed in the cytoplasm, a lower but significant nuclear pool of p25-GFP was also observed, providing a basis for p25/HDAC1 interaction. This is consistent with previous reports of nuclear localization of p35 or p25 (O’Hare et al., 2005).
We further characterized the p25/HDAC1 interaction by identifying the interaction domains. To this end, we generated multiple HDAC1 fragments spanning the entire protein, the C terminal region, the N terminal region containing the catalytic domain, or a small N-terminal region within the catalytic domain. By examining the ability of these fragments to coimmunoprecipitate full length p25, we mapped the interaction domain of p25 and HDAC1 to an N-terminal region within the histone deacetylase catalytic domain (Figure 4C).
The interaction of p25 with the catalytic domain of HDAC1 suggested that p25/Cdk5 may affect the enzymatic activity and/or the function of HDAC1. Indeed, overexpression of p25 and Cdk5 in 293T cells resulted in a significant decrease in endogenous HDAC1 activity (Figure 4D). Importantly, inhibitory effects on endogenous HDAC1 activity were confirmed in vivo in hippocampi from 2 week-induced CK-p25 mice compared to WT controls (Figure 4D). Similar effects on HDAC1 activity were observed in primary neurons infected with p25-HSV (data not shown). To determine whether this was linked to increased HDAC1 repressor activity, we coexpressed p25 and Cdk5 with HDAC1-Gal4 in a luciferase reporter system. Fusion of HDAC1 with Gal4 significantly repressed Gal4 transcriptional activity (Nagy et al., 1997) (lane 2 vs. 1, Figure 4E); however, co-expression with p25 increased HDAC1-Gal4-induced reporter activity 7.9-fold, indicating decreased repression by HDAC1 (lane 3). Importantly, this effect was not observed with p35/Cdk5 or with p25/dominant negative Cdk5 (lanes 4 and 5), indicating that the inhibitory effect on HDAC1 transcriptional repression was specific to p25 and not p35, and that it required Cdk5 activity.
Inhibition of HDAC catalytic activity results in the loss of HDAC1 association with the p21/WAF1 promotor region (Gui et al., 2004). Therefore, we investigated whether p25/Cdk5 could inhibit the association of HDAC1 from the promotor of p21/WAF1 and other cell cycle related genes. First, we examined whether overexpression of p25 could affect the overall chromatin association of HDAC1 in primary neurons. We observed that HSV-mediated overexpression of p25 led to a decrease in chromatin-bound HDAC1, and an increase in the nucleoplasmic, non-chromatin-bound fraction of HDAC1 (Figure 4F). Next, we carried out HDAC1 chromatin immunoprecipitation experiments in 293T cells transfected with p25/Cdk5 or a vector control to examine the association of HDAC1 with the core promotor regions of p21/WAF1 and E2F-1. Overexpression of p25/Cdk5 resulted in a loss of HDAC1 association with p21/WAF1 and E2F-1 promoters, as well as a concomitant increase in associated histone acetylation, suggesting a loss of histone deacetylation in this locus (Figure 4G). As HDAC1 association and local histone deacetylation of specific promotor regions is linked with their repression, our result suggested that p25/Cdk5-mediated loss of HDAC1 activity and association with promotors of cell cycle related genes may account for the aberrant expression of cell cycle related genes observed in the CK-p25 mice.
Collectively, our results demonstrate that p25/Cdk5 inhibits multiple facets of HDAC1 function, including histone deacetylase activity, transcriptional repressor activity, and association with chromatin and specific promotor regions.
Inhibition of HDAC1 induces DNA damage, cell cycle activity, and death
Our findings raised the possibility that p25/Cdk5 may cause both cell cycle reentry and DNA damage through inhibition of HDAC1 activity. We examined the effects of siRNA-mediated knockdown or pharmacological inhibition of HDAC1. Knockdown of HDAC1 with a previously published sequence (Supplemental methods) resulted in a significant increase in double-strand DNA breaks and cell death compared to the random sequence control (Figure 5A). In addition, treatment of primary neurons with 1μM of the class I HDAC inhibitor MS-275, which results in over 70% inhibition of HDAC1 activity with lesser effects on HDAC3 and HDAC8 (Hu et al., 2003), was sufficient to increase double-strand DNA breaks (8.1-fold increase) and stimulate the aberrant expression of Ki-67 (1.8-fold increase) compared to controls (Figure 5B). These results demonstrate that inhibition of HDAC1 in neurons can induce double-strand DNA breaks and cell cycle activity in neurons.
Figure 5. Loss of HDAC1 or pharmacological inhibition of HDAC1 results in DNA damage, cell cycle reentry, and neurotoxicity.

(A,B) Primary cortical neurons were transfected with either HDAC1 siRNA or random sequence siRNA, along with GFP at a 7:1 ratio to label transfected neurons. Cells were fixed at 24h, 48h, and 72h post-transfection and immunostained for γH2AX. GFP-positive neurons were scored for γH2AX immunoreactivity and for cell death based on nuclear condensation and neuritic integrity, as described. (A) Representative micrographs. HDAC1 siRNA or control (random sequence) siRNA transfected neurons are indicated by arrows. The HDAC1 siRNA transfected neurons display neuritic breakage. The inset is a magnification of the γH2AX staining of the neuron indicated by arrow and asterisk, showing γH2AX foci of varying sizes. Percentage of γH2AX and cell death are shown as averages from multiple sets ±S.D. It was noted that transfection of control siRNA per se appeared to cause a low but significant baseline level of γH2AX immunoreactivity and cell death. (B) Primary cortical neurons were treated with 1μM of the HDAC1 inhibitor MS-275 for 24h, fixed, and immunostained for γH2AX and Ki-67. Controls were treated with equal amounts of vehicle (DMSO). Total numbers of γH2AX and Ki-67 positive neurons were quantified over 20 microscope fields (field diameter ~900 μm). Scale bar = 100 μm. (C) Wild-type mice were injected intraperitoneally with 50mg/kg MS-275 (n=3) or saline (n=3) daily for 10 days, then sacrificed and examined for γH2AX. MS-275 injection resulted in a dramatic induction of γH2AX within the CA1 (bottom panels), whereas saline injection did not induce γH2AX (top panels). Scale bar = 100μM.
Furthermore, daily intraperitoneal injection of high doses of MS-275 (50 mg/kg) for 10 days in WT mice resulted in a dramatic induction of γH2AX in CA1 neurons (Figure 5C) as well as other brain regions (Figure S5), which was not observed with saline injection. In contrast to previous studies using the non-selective HDAC inhibitors sodium butyrate and trichostatin A (Fischer et al., 2007; Levenson et al., 2004), MS-275 also impaired associative learning capability in WT mice in a dose dependent manner, as examined using a contextual fear conditioning paradigm (Figure S6). These results support that loss of HDAC1 activity can cause DNA damage, neurodegeneration, and neurologic defects in vivo.
HDAC1 gain-of-function rescues against DNA damage and neuronal death in cultured neurons and in vivo
Having demonstrated that inhibition of HDAC1 induces DNA double-strand breaks and aberrant cell cycle activity, we examined whether HDAC1 gain-of-function by overexpression can attenuate p25-mediated DNA damage and neurotoxicity. To this end, we overexpressed HDAC1 or control constructs followed by viral expression of p25 at a high rate of infection (>80%). Overexpression of HDAC1, but not HDAC2, decreased the percentage of neurons positive for p25-induced γH2AX by 37.9% compared to GFP control (Figure 6A). We also examined whether co-expression of HDAC1 could rescue against cell death induced by transfection with p25-GFP. Co-expression of HDAC1, but not a catalytically dead mutant (HDAC1 H141A), rescued against p25-mediated neuronal death by 59.8% compared to the control (Figure 6B). Similar results were obtained when examining apoptosis as determined by cleaved caspase-3 immunoreactivity (Figure S7). These results demonstrate that restoring HDAC1 activity can rescue against p25-mediated DNA damage and death.
Figure 6. HDAC1 gain-of-function rescues against p25-mediated double-strand DNA breaks and neurotoxicity.

(A) Overexpression of HDAC1 rescues against p25 mediated formation of γH2AX. Primary cortical neurons were transfected with vector, HDAC1, or HDAC2 as described. At 12 hours posttransfection, neurons were infected with p25-HSV virus, fixed after 8 hours, and immunostained for γH2AX. HDAC-positive cells were scored for immunoreactivity towards γH2AX. (B) Overexpresson of HDAC1 rescues against p25-mediated neurotoxicity. Primary cortical were transfected with p25-GFP plus blank vector, flag-HDAC1, or catalytic dead flag-HDAC1-H141A mutant. At 24h posttransfection, cells were fixed and immunostained for flag. p25 transfected cells and p25/HDAC1 transfected cells were scored for cell death based on nuclear condensation and neuritic integrity as described. For (A) and (B), averages from multiple experiments ±S.D. are shown. Representative micrographs for HDAC1 are shown on left panels. Arrows indicate p25-positive neurons expressing or not expressing HDAC1. P-values (HDAC1 vs control, **p<0.005) were calculated from multiple experiments by two-tailed, unpaired Student’s t-test. Bar= 50 μM. (C) Adult Sprague-Dawley rats were subjected to unilateral middle cerebral artery occlusion (MCAO) as described. Paraffin sections from brains fixed at three hours post-MCAO show γH2AX immunoreactivity specifically within the ischemic area (left panels) but not in the contralateral area (right panels). Images are representative of multiple animals. Average numbers of γH2AX-positive cells per field (field diameter ~900μm) from multiple experiments ±S.D. are displayed. 20 fields were counted per experiment. P-values (**p<0.005) were calculated from multiple experiments by two-tailed, unpaired Student’s t-test. (D) Injection of blank vector (expressing GFP) into striatum results in efficient and widespread expression in striatal neurons. Injection of virus into the striatum of adult Sprague-Dawley rats was followed examination of GFP expression after 24 hours. Left pane bar = 100μM, right panel bar = 30μM. (E) HDAC1 expression protects against ischemia-induced neuronal death and γH2AX formation in vivo. Adult Sprague-Dawley rats were injected with virus in the striatum, subjected to bilateral transient forebrain ischemia after 24 hours, then examined 6 days later for Fluoro-Jade and γH2AX staining as described. Representative images from mice injected with HSV-HDAC1, HSV-HDAC1H141A, and blank HSV (Vector) are shown. Scale bar = 100μM. (F) Quantification of γH2AX+ cells from mice injected with saline, HSV-HDAC1, HSV-HDAC1H141A, vector, or mice subjected to sham procedure are shown. (G) Quantification of FJ+ cells from the same mice as (F). For (F) and (G), Data is presented as Mean ± SEM. P-values (*p<0.05; **p<0.005) were calculated from multiple experiments by two-tailed, unpaired Student’s t-test. Bar = 100μM.
Interestingly, treatment with the HAT inhibitor Garcinol also rescued against p25-mediated neurotoxicity (Figure S7). However, co-expression of HDAC1 plus Garcinol treatment did not have any additional protective benefit compared to HDAC1 coexpression or Garcinol treatment alone (Figure S7), suggesting that HDAC1 neuroprotection involves deacetylation of histones.
Next, we sought to examine whether the neuroprotective properties of HDAC1 could be recapitulated in an established in vivo model for stroke, i.e. rats subjected to transient forebrain ischemia. We and other groups have previously demonstrated the involvement of p25 in this model (Garcia-Bonilla et al., 2006; Wang et al., 2003; Wen et al., 2007). Also, p25 is upregulated in human postmortem brains following ischemic stroke (Mitsios et al., 2007). Furthermore, cell cycle markers such as Cyclin A, PCNA, and E2F-1, which were upregulated in our p25 mice (Figure 1), are upregulated in rodent models for stroke/ischemia (Rashidian et al., 2007).
Therefore, we examined whether γH2AX levels are upregulated as well in this model. Brains from rats subjected to unilateral transient forebrain ischemia for various periods were examined for γH2AX immunoreactivity. Increased γH2AX immunoreactivity was observed as early as three hours post-ischemia in the ischemic region (Fig. 6C). Significant levels of γH2AX were not observed in ipsilateral non-ischemic region (not shown) or the contralateral hemisphere (Fig. 6C).
We examined whether overexpression of HDAC1 conferred neuroprotection in this model. To this end, Sprague Dawley rats were injected with saline, blank HSV, HSV-HDAC1, or HSV-HDAC1H141A catalytic-dead mutant, into the striatum, which resulted in robust neuronal expression of constructs (Figure 6D). After 24 hours, rats were subjected to bilateral transient forebrain ischemia. Six days later, brain sections were stained with γH2AX and Fluoro-Jade to label degenerating neurons. We and other groups observe that Fluoro-Jade labels regions undergoing apoptotic neuronal death (Figure S8) (Zuch et al., 2000).
HSV-mediated overexpression of HDAC1 in the striatum resulted in a 38% reduction in γH2AX-positive neurons in the striatum compared to blank HSV, while the HDAC1H141A mutant did not confer neuroprotection (Figure 6E and 6F). In addition, the number of degenerating neurons, as labeled by FluoroJade, significantly decreased (33%) following HDAC1 expression (Figure 6E and 6G). Importantly, this demonstrates that increasing HDAC1 activity can protect neurons against ischemia-induced DNA damage and neurotoxicity in vivo.
DISCUSSIONS
The CK-p25 mouse is a model for neurodegeneration in which neurons predictably begin to die at around 5–6 weeks of induction (Cruz et al., 2003; Fischer et al., 2005). In our current study, using an unbiased approach of examining the gene expression profile at a specific time point of induction followed by validation, we determined that aberrant expression of cell cycle proteins and induction of double-strand DNA breaks are early events in p25-mediated neurodegeneration. Furthermore, we identified deregulation of HDAC1 activity as a mechanism involved in p25-mediated DNA double-strand break formation, cell cycle protein expression, and neuronal death. Collectively, our results outline a novel pathway in neurodegeneration by which the inactivation of HDAC1 by p25 leads to enhanced susceptibility of DNA to double-strand breaks, and the de-repression of transcription leading to aberrant expression of cell cycle related genes. In addition, our findings provide mechanistic insights into a common link between DNA damage and aberrant cell cycle activity in neurodegeneration. As cell cycle reentry, DNA damage, and p25 accumulation are emerging as important pathological components of various neurodegenerative conditions, this pathway may constitute a shared mechanism responsible for neuronal death in multiple conditions such as stroke/ischemia, Alzheimer’s Disease, and Parkinson’s Disease. The neurotoxic effects of p25 accumulation and downstream events appear to be reversible (Figure 3C), further advocating the therapeutic potential of targeting this pathway in conditions involving neuronal death. Our proposed pathway is summarized in Figure 7. It will be of interest to examine the protective properties of HDAC1 gain-of-function in various mouse models of neurodegenerative diseases such as AD, ALS, and PD, in future studies.
Figure 7. Schematic model.
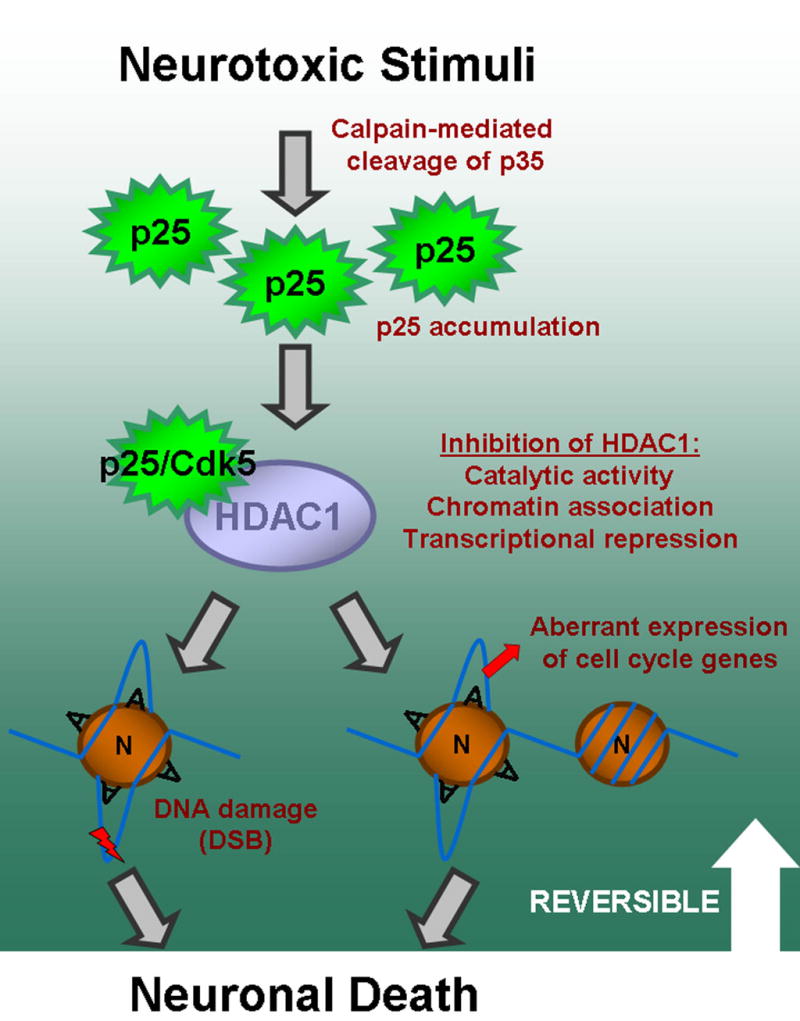
Proposed model for p25-mediated cell death involving inhibition of HDAC1 activity which leads to DNA double-strand breaks and aberrant cell cycle activity. Neurotoxic stimuli such as ischemia results in p25 accumulation. This accumulation results in interaction with and inhibition of multiple aspects of HDAC1 activity, as shown in Figure 4, in a manner that is dependent on Cdk5, as shown in Figure 4E. Inhibition of HDAC1 results in DNA damage and aberrant expression of cell cycle genes which is likely associated with local histone deacetylation (Figure 5, Figure 4G, Figure S7), and which ultimately leads to neuronal death (Figure 3). The neurotoxic effects of p25 accumulation and downstream effects appear to be reversible before a certain period of induction (Figure 3C). The circle labeled ‘N’ represents nucleosome, while ‘A’ represents acetylation of histone tails. The nucleosomes with ‘A’ represent acetylated nucleosomes and open chromatin loci, while nucleosome on far right represents a deacetylated nucleosome and closed chromatin locus.
HDAC1 inactivation by p25/Cdk5
We have demonstrated that p25 can inhibit multiple aspects of HDAC1 activity, including HDAC1 catalytic activity and association of HDAC1 with chromatin. This inhibition appears to be Cdk5 dependent (Figure 4E). How does p25/Cdk5 inhibit HDAC1? This may involve the posttranslational modification of HDAC1 by p25/Cdk5. HDAC1 catalytic activity and association with corepressors can be modulated by phosphorylation (Galasinski et al., 2002; Pflum et al., 2001). Alternatively, the p25/HDAC1 interaction may recruit p25/Cdk5 to HDAC1-containing corepressor complexes, where p25/Cdk5 phosphorylates and modulates co-repressors required for HDAC1 activity, such as mSin3a or SMRT/NcoR2 (de Ruijter et al., 2003; Nagy et al., 1997).
HDAC1 inactivation and cell cycle reentry
While aberrant cell cycle activity in neurons in neurodegenerative states has been extensively documented, the underlying mechanisms and purposes are unclear. Our model introduces loss of HDAC1 activity as a novel underlying mechanism, and suggests a simplified model of aberrant cell cycle activity as a pathological de-repression of multiple cell cycle genes that are normally suppressed in neurons. p25/Cdk5 inhibits the transcriptional repression activity of HDAC1 in a luciferase reporter system (Figure 4E), and induces the disassociation of HDAC1 from the promotor region of cell cycle proteins E2F-1 and p21/WAF (Figure 4G). Inhibition of HDAC1 in primary neurons resulted in upregulation of the cell cycle activity marker Ki-67 (Figure 5B). Thus, our model suggests that constitutive HDAC1, which is normally associated with and represses cell cycle related genes in postmitotic neurons, is inactivated by p25, leading to aberrant expression of cell cycle genes. The idea that aberrant cell cycle gene expression in neurons is a consequence of loss of HDAC1 repressional activity is consistent with the previously reported roles of HDAC1 as a transcriptional repressor for many cell cycle genes.
DNA damage induced by HDAC1 inactivation may also play a role in aberrant cell cycle activity, as it has been demonstrated that increased oxidative DNA damage in the ‘harlequin’ mouse mutants or drug-induced DNA damage in primary neurons can induce aberrant cell cycle activity (Klein et al., 2002; Kruman et al., 2004).
HDAC1 inactivation and DNA damage
Double-stranded DNA breaks also preceded neuronal death in our p25 model. Future studies should address specifically how HDAC1 inactivation results in double-strand DNA damage and cell cycle reentry. One likely scenario involves hypersensitization of chromatin to DNA damaging agents following loss of HDAC1 activity. In cancer cells, HDAC inhibitors can hypersensitize DNA to damaging agents such as UV and gamma-irradiation by increasing the acetylation state and thus the accessibility of chromatin (Cerna et al., 2006).
Interestingly, overexpression of p25 or HDAC1 inhibition/knockdown was sufficient to induce DNA damage in neurons and did not require additional genotoxic stimuli. Neurons are constantly subjected to DNA damaging events; for example, it has been estimated that the typical neuron of an aged mouse is subjected to 2,000,000 oxidative lesions per day (Hamilton et al., 2001). Therefore, enhanced accessibility to DNA damaging agents, combined with the relatively low levels of DNA repair factors present in neurons compared to proliferating cells (Gobbel et al., 1998; Nouspikel and Hanawalt, 2003), may result in a robust accumulation of DNA damage.
DNA damage, cell cycle reentry, and cell death
In this study, we report the formation of DNA double-strand breaks in the CK-p25 model as well as in a rodent model for stroke/ischemia. Both DNA double-strand breaks and cell cycle activity preceded and was later tightly associated with neurodegeneration (Figure 3B). Compared to single nucleotide lesions such as 8-oxoguanine lesions, DNA double-strand breaks are potentially lethal lesions that induce cell cycle-dependent checkpoint responses in proliferating cells resulting in cell death (Sancar et al., 2004). However, because neurons are postmitotic, DNA damage events per se may only have limited toxic consequences, with the exception of altered gene expression (Nouspikel and Hanawalt, 2003). Thus, DNA double-strand breaks and cell cycle events such as DNA replication may synergistically induce cell death in CK-p25 neurons, likely in a checkpoint-dependent manner. In support of this notion, the p53 DNA damage checkpoint protein is upregulated in CK-p25 mice, and knockdown of p53 results in reduction of neuronal death in p25-transfected neurons (Kim et al., 2007).
Role for HDAC1 in postmitotic neurons
As an important modulator of transcription, HDAC1 is undoubtedly involved in a variety of biological processes, and its involvement is well established in the regulation of the cell cycle in proliferating cells. Studies in the developing zebrafish retina demonstrate a role for HDAC1 in cell cycle exit and differentiation of retinal progenitors into neurons (Yamaguchi et al., 2005). Our study implicates HDAC1 in the maintenance and survival of adult neurons as well. Our findings suggest a function for HDAC1 in maintaining a state of ‘quiescence’ through transcriptional repression of cell cycle genes. We also demonstrate a role for HDAC1 in maintaining DNA integrity in adult neurons, a function that may be tightly linked to its regulation of the accessibility of DNA to damaging agents. Collectively, our results outline an important role within the CNS for HDAC1, the deregulation of which can lead to aberrant expression of cell cycle genes, DNA damage, and ultimately death in adult neurons.
Therapeutic potential for HDAC1 gain-of-function
We have shown that inhibition of HDAC1 can lead to DNA damage, cell cycle gene expression, and neuronal death. In support of this idea, recent studies reporting the neuroprotective function of p130 and histone deacetylase-related protein (HDRP) demonstrated a requirement for association with HDAC1 for their pro-survival effects (Liu et al., 2005; Morrison et al., 2006). Furthermore, a recent phase I clinical trial of MS-275 in leukemia patients demonstrated neurologic toxicity manisfesting as unsteady gait and somnolence as a dose-limiting toxicity (DLT) (Gojo et al., 2006). In addition, numerous studies have demonstrated neurotoxic effects of high dose general HDAC inhibitor treatment (Boutillier et al., 2002, 2003; Kim et al., 2004; Salminen et al., 1998).
On the other hand, it is clear that HDAC inhibitors can have beneficial effects. We recently demonstrated that treatment with the nonselective HDAC inhibitor sodium butyrate enhanced synapse formation and long term memory recall (Fischer et al., 2007), and learning and memory benefits have been reported with Trichostatin A (Levenson et al., 2004; Vecsey et al., 2007). Along similar lines, studies have shown beneficial effects of HDAC inhibitors in patients or models of psychiatric disorders such as depression (Citrome, 2003; Tsankova et al., 2006). In addition, HDAC inhibitors such as phenylbutyrate had neuroprotective properties, within a therapeutic window, in models of Huntington’s disease (HD) (Hockly et al., 2003; Langley et al., 2005; McCampbell et al., 2001; Steffan et al., 2001). The use of HDAC inhibitors in HD models is based on the finding that Huntingtin inhibits the histone acetyltransferases CREB-binding protein (CBP) and p300/CBP associated factor (P/CAF), leading to a deficiency in levels of histone acetylation (Bates, 2001).
Thus, both beneficial and adverse signals can be triggered by histone deacetylase inhibition. These effects are likely based on discrepancy in functions and gene targets of the different HDACs. For example, it was recently shown that specific downregulation of the class II HDACs HDAC4 and HDAC5 by the antidepressant imipramine derepressed BDNF expression and suppressed depression-like behavior (Tsankova et al., 2006). Thus, inhibition of other HDAC(s) may result in benefits through derepression of beneficial gene expression, while HDAC1 inhibition elicits deleterious consequences such as derepression of cell cycle genes. We are currently in the process of elucidating the discrepancies in function and gene targets of the different HDACs which may account for the varying effects of HDAC inhibition.
Also, we cannot overlook the possibility that the beneficial versus deleterious effects of HDAC inhibition may also closely depend on the dosage and length of HDAC inhibition, as well as the context (e.g. state of hypoacetylation in Huntington’s Disease).
Our current study demonstrates for the first time the therapeutic potential for replenishing HDAC1 activity in certain neurodegenerative contexts such as ischemia (Figure 6). The previous studies with general HDAC inhibitors and our current study, collectively, illustrate the complex and broadly impacting nature of manipulating HDAC activity, and underline the importance of chromatin regulation in a variety of processes in the CNS. Importantly, our study exemplifies the catastrophic consequences of deregulating this process, and introduces a novel and unexpected avenue for therapeutic strategies in neurodegeneration.
Experimental Procedures
A detailed description of all procedures is included in the Supplemental section.
Mice
CK-p25 double transgenic mice were raised on a doxycycline containing diet then switched to a normal diet at 6~8 weeks of age to induce p25-GFP in a postnatal, forebrain neuron-specific manner as described (Cruz et al., 2003). Further details are provided in the supplemental section.
Microarray analyses
Total RNA was extracted from forebrains of 2 week induced CK-p25 Tg mice (n=3) and uninduced CK-p25 controls (n=3) using Trizol reagent (Sigma). RNA was subjected to further purification with RNEasy columns (Qiagen), reverse transcribed, biotin-labeled, and hybridized onto Mouse Genome 430A 2.0 Arrays (Affymetrix). Array data was analyzed using dCHIP and GeneChip Operating software (GCOS,m Affymetrix) as described in the Supplemental section.
Quantitative RT-PCR analyses
Hippocampi from 2 week-induced CK-p25 mice and WT controls were incubated in RNAlater solution (Ambion). Total RNA was isolated using TRIzol reagent (Invitrogen) according to manufacturer’s instructions. Reverse transcription of equal amounts of total RNA was carried out using Superscript III First-Strand Synthesis kit (Invitrogen) according to manufacturer’s instructions. Semiquantitive conditions were obtained for all primers, with sequence and cycle information provided in the Supplemental section. Band quantifications were normalized to those obtained for actin for each animal.
Comet assay
Primary rat cortical neurons at DIV 6~8 were infected with herpesvirus expressing p25 (p25-HSV) or lacZ (lacZ-HSV). After 10 hours, neurons were dissociated and embedded in a thin layer of agarose. Lysis, alkaline treatment, and single cell gel electrophoreses (comet assay) was carried out.
Immunohistochemistry
Mice were transcardially perfused, and brains were prepared as paraffin sections or free floating vibratome sections. Citrate buffer based antigen retrieval was required for paraffin sections. Primary antibodies used with paraffin sections werγH2AX(monoclonal from Upstate; polyclonal from Trevigen), Ki-67(Novocastra), PCNA(Oncogene Sciences), phospho(pS10)-Histone H3(Upstate), and GFP(monoclonal from Santa Cruz; polyclonal from Molecular probes). Primary antibodies used with vibratome sections were GFP(Aves Labs), γH2AX(Upstate), Cleaved Caspase-3(Asp175, Cell Signaling Technology), and Cleaved PARP-1(p25, Upstate), and NeuN(Chemicon).While the CA1 region of hippocampus is primarily shown in figures, similar results are seen in the cortex as well (not shown).
Immunoblot Analyses
CK-p25 and control forebrains were dissected and dounce homogenized in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors. Equal quantities of brain lysates were subjected to SDS-PAGE and Western blot analysis using antibodies to γH2AX(Trevigen), alpha-tubulin(Sigma), E2F-1(Santa Cruz), Cyclin A(Santa Cruz), p35(Santa Cruz), p27(Santa Cruz), GFAP(Sigma), and BetaIII-tubulin(Sigma).
Luciferase Assays
Hela cells were transfected with reporter, HDAC1-Gal4 fusion protein, and either blank vector or p25 plus Cdk5 expression vectors. At 15 hours post-transfection, cells were lysed with passive lysis buffer and luciferase assay was performed according to manufacturer’s instructions (Promega). Values were normalized to Gal4 protein levels as renilla reporters were also substantially repressed by HDAC1-Gal4.
Co-immunoprecipitation analyses
HEK293T cells were transfected, lysed, and subjected to immunoprecipitation with anti-flag-conjugated beads (Sigma), and analysed by SDS-PAGE as described in the Supplemental section. For in vivo analysis of p25/HDAC1 interaction, two week-induced CK-p25 mice and WT control forebrains were dounce homogenized in RIPA buffer, subjected to immunoprecipitation with anti-HDAC1(Abcam), and analyzed as described. Antibodies to HDAC1(Abcam), GFP(Santa Cruz), Flag(Sigma), and p35/p25(Sigma) were used for immunoblotting.
HDAC1 enzymatic activity assay
HEK293T cells were transfected with blank vector or with p25 and Cdk5 expression vectors with Lipofectamine 2000. Cells were lysed with IP buffer at15 hours post-transfection, and immunoprecipitated with anti-HDAC1(Abcam). Endogenous HDAC1 bound to beads were analyzed for histone deacetylase activity using the Histone deacetylase assay kit (Upstate) according to the manufacturer’s instructions. Histone deacetylase activity was normalized to input HDAC1 protein levels which were analyzed by western blot. For analyses of HDAC1 activity in vivo, hippocampi were dissected from 2-week induced CK-p25 mice and WT littermates, and dounce homogenized in IP buffer with high salt (400mM NaCl) to aid HDAC1 extraction. Lysates were immunoprecipitated (in IP buffer with final 200mM NaCl) and analyzed as described.
p25 and HDAC1 based toxicity assays in primary neurons
Optimization of transfection conditions was required to minimize toxicity and γH2AX induced by the transfection procedure itself, as described in the Supplemental section. For cell death rescue assays, primary mouse cortical neurons were transfected with p25-GFP plus blank vector or p25-GFP plus flag-HDAC1. At 24 hours post-transfection, neurons were fixed, stained, and transfected neurons were scored for cell death based on nuclear morphology and neuritic integrity, or scored for immunoreactivity towards cleaved caspase 3 (Cell Signaling Technology) in a blind manner. Only cells with a sufficient level of transfection so that entire neuritic outline was clearly visible were counted. For γH2AX rescue assays, primary rat cortical neurons were transfected with flag-HDAC1, flag-HDAC2, or GFP and at 12 hours post-transfection, infected with p25-HSV. Flag-HDAC1 or HDAC2, or GFP- positive neurons were blindly scored for γH2AX immunoreactivity.
Transient forebrain ischemia
For experiments examining HDAC1-mediated rescue of transient forebrain ischemia, rats were subjected to bilateral transient forebrain ischemia as described in the Supplemental section. Briefly, virus particles were injected into the striatum of adult Sprague-Dawley rats. After 24h, rats were subjected to ischemia by bilaterally occluding common carotid arteries with aneurysm clips for 15min, after which cerebral blood flow was restored. After 6 days, mice were processed, subjected to γH2AX immunohistochemistry, then subjected Fluoro-Jade staining as previously described (Wang et al., 2003). Histological quantification of γH2AX immunoreactivity and Fluoro-Jade labeling in striatal neurons was carried out as described in Supplemental section. Averaged neuronal cell counts were obtained from six animals per group.
For co-labeling of Fluoro-Jade B with apoptosis markers, mice were subjected to middle cerebral artery occlusion transient forebrain ischemia as described, except unilaterally with contralateral hemisphere used as negative control. Cleaved caspase-3(Cell Signaling Technology) and Cleaved PARP-1(Upstate) were used as apoptotic markers.
Subcellular Fractionation
Chromatin fractionation was based on a Rat primary neurons at DIV5-7 were infected with GFP-HSV or p25-GFP-HSV. After 20 hours, neurons were subjected to chromatin fractionation as described in the Supplemental section. Cytosolic/Nuclear fractionation was carried out using the Nuclear Extract Kit(Active Motif) according to manufacturer’s instructions. Antibodies to GFP(Sigma), HDAC1(Abcam), and Neurofilament Heavy subunit (Covance) were used for immunoblotting.
Chromatin Immunoprecipitation
For chromatin immunoprecipitation experiments, 293T cells were transfected with the indicated constructs, fixed 14 hours after transfection with 1% formaldehyde, and processed according to manufacturer’s instructions (#17–195, Upstate). HDAC1(ChIP grade, Abcam) and acetyl-H3K9(Upstate) antibodies were used to immunoprecipitate endogenous HDAC1 or Histone 3 acetylated at Lysine 9, respectively. Core promoter regions of p21 and E2F-1 were amplified by PCR under semi-quantitative conditions as described in the Supplemental section.
Fear conditioning
Fear conditioning experiments were carried out as previously described (Kim et al., 2007), using a fear conditioning apparatus (TSE Systems).
Supplementary Material
Acknowledgments
We thank Drs. Bruce Yankner, John Whetstine, Sangki Park, Benjamin Samuels, Fei Lan, and Konstantinos Meletis for helpful discussions. L.-H.T. is an investigator of the Howard Hughes Medical Institute. This work is partially supported by a NIH PO1 grant (AG27916) to L.-H.T and by NIH/NINDS (R01 NS051383-01) and American Heart Association (05553413) to Y.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamec E, Vonsattel JP, Nixon RA. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999;849:67–77. doi: 10.1016/s0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W. Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci U S A. 1992;89:1194–1198. doi: 10.1073/pnas.89.4.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP. Huntington’s disease. Exploiting expression. Nature. 2001;413:691, 693–694. doi: 10.1038/35099656. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP. Constitutive repression of E2F1 transcriptional activity through HDAC proteins is essential for neuronal survival. Ann N Y Acad Sci. 2002;973:438–442. doi: 10.1111/j.1749-6632.2002.tb04679.x. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem. 2003;84:814–828. doi: 10.1046/j.1471-4159.2003.01581.x. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerna D, Camphausen K, Tofilon PJ. Histone deacetylation as a target for radiosensitization. Curr Top Dev Biol. 2006;73:173–204. doi: 10.1016/S0070-2153(05)73006-4. [DOI] [PubMed] [Google Scholar]
- Citrome L. Schizophrenia and valproate. Psychopharmacol Bull. 2003;37 Suppl 2:74–88. [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Jr, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–838. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Galasinski SC, Resing KA, Goodrich JA, Ahn NG. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem. 2002;277:19618–19626. doi: 10.1074/jbc.M201174200. [DOI] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Burda J, Pineiro D, Ayuso I, Gomez-Calcerrada M, Salinas M. Calpain-induced proteolysis after transient global cerebral ischemia and ischemic tolerance in a rat model. Neurochem Res. 2006;31:1433–1441. doi: 10.1007/s11064-006-9195-7. [DOI] [PubMed] [Google Scholar]
- Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan PH. Response of postmitotic neurons to X-irradiation: implications for the role of DNA damage in neuronal apoptosis. J Neurosci. 1998;18:147–155. doi: 10.1523/JNEUROSCI.18-01-00147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, et al. Phase 1 and pharmacological study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2006 doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Sakai K, Sasaki C, Zhang WR, Abe K. Phosphorylation of retinoblastoma protein in rat brain after transient middle cerebral artery occlusion. Neuropathol Appl Neurobiol. 2000;26:390–397. doi: 10.1046/j.1365-2990.2000.00264.x. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Sakurai M, Itoyama Y, Abe K. Oxidative damage and breakage of DNA in rat brain after transient MCA occlusion. Brain Res. 1999;832:159–163. doi: 10.1016/s0006-8993(99)01409-2. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. Embo J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kim EM, Kim NJ, Chang KA, Choi Y, Ahn KW, Lee JH, Kim S, Park CH, Suh YH. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J Neurosci Res. 2004;75:117–124. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Bonni A. The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J Neurosci. 2003;23:1649–1658. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R, Jr, Gorospe M, Mattson MP. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. Embo J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001;32:425–438. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Liu DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005;19:719–732. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci U S A. 2001;98:15179–15184. doi: 10.1073/pnas.261400698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsios N, Pennucci R, Krupinski J, Sanfeliu C, Gaffney J, Kumar P, Kumar S, Juan-Babot O, Slevin M. Expression of cyclin-dependent kinase 5 mRNA and protein in the human brain following acute ischemic stroke. Brain Pathol. 2007;17:11–23. doi: 10.1111/j.1750-3639.2006.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer’s disease. Bioessays. 2003;25:168–173. doi: 10.1002/bies.10227. [DOI] [PubMed] [Google Scholar]
- O’Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pflum MK, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–47741. doi: 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, te Riele H, Dynlacht BD. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison SH, Bradley WG. DNA damage and chronic neuronal degenerations. J Neurol Sci. 1984;64:11–20. doi: 10.1016/0022-510x(84)90051-0. [DOI] [PubMed] [Google Scholar]
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61:203–206. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O’Hare MJ, Callaghan S, Slack RS, Przedborski S, et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Stiegler P, De Luca A, Bagella L, Giordano A. The COOH-terminal region of pRb2/p130 binds to histone deacetylase 1 (HDAC1), enhancing transcriptional repression of the E2F-dependent cyclin A promoter. Cancer Res. 1998;58:5049–5052. [PubMed] [Google Scholar]
- Swatton JE, Sellers LA, Faull RL, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur J Neurosci. 2004;19:2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Tonou-Fujimori N, Komori A, Maeda R, Nojima Y, Li H, Okamoto H, Masai I. Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development. 2005;132:3027–3043. doi: 10.1242/dev.01881. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuch CL, Nordstroem VK, Briedrick LA, Hoernig GR, Granholm AC, Bickford PC. Time course of degenerative alterations in nigral dopaminergic neurons following a 6-hydroxydopamine lesion. J Comp Neurol. 2000;427:440–454. doi: 10.1002/1096-9861(20001120)427:3<440::aid-cne10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.