

Abstract
MAPK pathway activation is a frequent event in human cancer and is often the result of activating mutations in the BRAF and RAS oncogenes. Targeted inhibitors of BRAF and its downstream effectors are in various stages of preclinical and clinical development. These agents offer the possibility of greater efficacy and less toxicity than current therapies for tumors driven by oncogenic mutations in the MAPK pathway. Early clinical results with the BRAF-selective inhibitor PLX4032 suggest that this strategy will prove successful in a select group of patients whose tumors are driven by V600EBRAF. Relief of physiologic feedback upon pathway inhibition may, however, attenuate drug response and contribute to the development of acquired resistance. An improved understanding of the adaptive response of cancer cells to MAPK pathway inhibition may thus aid in the identification of those patients most likely to respond to targeted pathway inhibitors and provide a rational basis for tailored combination strategies.
BACKGROUND
One of the central regulators of growth factor-induced cell proliferation and survival in both normal and cancer cells is the RAS protein. Activation of RAS leads to activation of several effector pathways, the best characterized of which are the RAF/MEK/ERK pathway (“the classical MAPK pathway”), the PI3 kinase pathway and the Ral-GEFs (1–6). Constitutive MAPK pathway activation can result from activating mutations in RAS, BRAF and MEK1, loss of the tumor suppressor NF1 (7), or upstream activation mediated by mutations, amplification, or ligand-mediated activation of cell surface receptors (Figure 1). All three RAS family genes (KRAS, NRAS and HRAS) have been shown to be somatically mutated in human cancer, most commonly as a result of single point mutations at codons 12, 13 and 61 (8–11). These mutations impair GTP hydrolysis and thus promote formation of constitutively activated GTP-bound RAS. Somatic point mutations in BRAF occur in approximately 8% of human tumors, most frequently in melanoma, colorectal and thyroid cancers (12, 13). BRAF mutations are found, with rare exceptions, in a mutually exclusive pattern with RAS mutations, suggesting that these genetic alterations activate common downstream effectors of transformation.
Figure 1. The RAS-RAF-MEK-ERK signaling pathway.
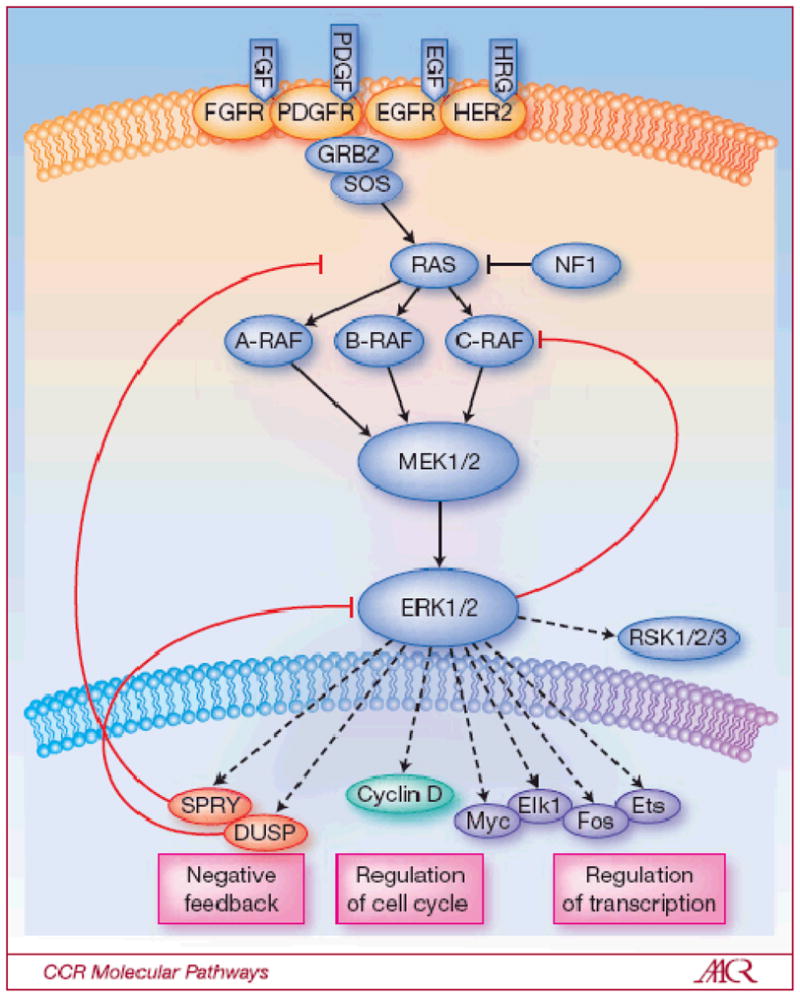
The classical MAP kinase pathway is activated in human tumors by several mechanisms including the binding of ligand to receptor tyrosine kinases (RTKs), mutational activation of an RTK, by loss of the tumor suppressor NF1, or by mutations in RAS, BRAF and MEK1. Phosphorylation and thus activation of ERK regulates transcription of target genes which promote cell cycle progression and tumor survival. The ERK pathway contains a classical feedback loop in which the expression of feedback elements such as SPRY and DUSP family proteins are regulated by the level of ERK activity. Loss of expression of SPRY and DUSP family members due to promoter methylation or deletion is thus permissive for persistently elevated pathway output. In the case of tumors with V600EBRAF expression, pathway output is enhanced by impaired upstream feedback regulation.
Activating BRAF mutations are found clustered within the P-loop (exon 11) and activation segment (exon 15) of the kinase domain, and a single point mutation, V600E, accounts for approximately 90% of cases (12, 14). Structural analysis of the V600E mutation suggests that it disrupts the interaction between the P-loop and the activation segment, which normally locks the kinase in the inactive conformation (15). “Impaired activity” mutants have also been reported. In contrast to the V600E mutant, these mutations activate the pathway in a RAF1-dependent manner (16).
CLINICAL-TRANSLATIONAL ADVANCES
Several strategies for inhibiting MAPK signaling are now being evaluated as cancer therapies. Clinically effective direct inhibitors of RAS have yet to be identified. Promising therapeutic approaches include targets that function as synthetic lethals in RAS mutant tumors (17) and inhibitors of downstream effectors such as RAF and MEK.
RAF kinase Inhibitors
Sorafenib (Nexavar) was the first RAF kinase inhibitor to enter human clinical testing. Sorafenib is now FDA approved for use in renal cell carcinoma and hepatocellular carcinoma. Although this compound was initially developed as a selective inhibitor of RAF, later studies revealed other biologically relevant targets, including VEGFR2/3, PDGFR, Flt-3, c-kit, and FGFR-1 (18). Sorafenib has virtually no activity as a single-agent in melanoma, the tumor type with highest frequency of BRAF mutations (19). Phase 2 trials combining sorafenib with chemotherapy showed early promise in melanoma but the activity of this combination regimen did not correlate with BRAF mutational status. Furthermore, a phase 3 trial of sorafenib in combination with carboplatin and paclitaxel in patients with advanced melanoma failed to meet its primary endpoint of improvement in overall survival (20). Overall, the data suggest that the primary mechanism of activity of sorafenib in renal cancer is likely anti-angiogenic and that RAF inhibition contributes minimally to its activity in patients with advanced cancer.
The limited activity of sorafenib in tumors with BRAF mutation prompted the development of second-generation RAF inhibitors with greater selectivity for mutant BRAF and greater potency for the target in vivo. PLX4032/R7204 (and its close analogue PLX4720, Plexxikon/Roche) (21, 22) cause potent pathway inhibition and antiproliferative effects, but in contrast to sorafenib, do so only in cell lines harboring V600EBRAF (23). In the recent phase 1 clinical trial of PLX4032, high (30–50 μM) steady state serum levels of the drug were tolerated with modest toxicity. This resulted in profound inhibition of ERK signaling in the tumor. Profound antitumor activity was observed: a 78% response rate by RECIST (Response Evaluation Criteria in Solid Tumors) and tumor shrinkage in almost all patients (24, 25). Notably, toxicities included skin rash and the development of squamous cell carcinomas in approximately one third of patients. The average duration of response on the Phase 1 trial was approximately 9 months and a phase 3 trial comparing PLX4032 to dacarbazine as first-line therapy for patients with metastatic melanoma whose tumors harbor V600EBRAF is currently ongoing.
Testing for BRAF mutations became a prerequisite for study entry early in the trials of PLX4032, and all responses observed were in patients whose tumors harbored V600EBRAF. Notably, PLX4032 inhibits ERK activation only in cells with BRAF mutations, while in cells with wild-type BRAF, including those with RAS mutations, PLX4032 treatment results in a paradoxical induction of ERK phosphorylation (26–28). The mechanistic explanation for this observation is that binding of PLX4032 to one member of a CRAF homodimer inhibits the bound protomer but results in transactivation of the drug-free protomer (28). The paradoxical activation of ERK by PLX4032 is not observed in cells harboring V600EBRAF mutations, as in this context, RAS activity is low and dimer formation is not required for activation. Notably, the paradoxical activation of ERK signaling is not unique to PLX4032 but is also observed with sorafenib and other ATP-competitive inhibitors of RAF (26–28). These results suggest that the clinical activity of PLX4032 will be restricted to tumors harboring activating mutations of BRAF. Furthermore, PLX4032 may enhance the growth of tumors in which the ERK pathway is activated by RAS mutation or upstream receptor kinases. Inhibitors further downstream such as those targeting MEK or ERK may thus be preferable in tumors in which ERK activity is the result of RAS or receptor tyrosine kinase activation. Finally, the toxicities of PLX4032 such as the development of squamous cell carcinomas may be attributable to ERK activation.
Several additional small molecule inhibitors of RAF are also in early clinical testing. XL281 (Exelixis) has shown modest biological activity and modulation of MAPK pathway activity in tumor tissue: clinical benefit (PR or SD) was observed in 43% (13/30) of patients in the dose-escalation phase of a recent Phase 1 study (29). RAF265 (Novartis), which inhibits all three isoforms of RAF, as well as mutant BRAF, and has anti-angiogenic activity through inhibition of VEGFR-2 (30, 31), is also being investigated in Phase I clinical trials in malignant melanoma.
MEK inhibitors
CI-1040 (Pfizer Oncology), an allosteric inhibitor of MEK, was the first selective MEK inhibitor to advance into clinical testing (32, 33). In contrast to PLX4032, CI-1040 inhibits ERK activity in all cells irrespective of their mutational status. Cells with BRAF mutations are selectively sensitive to CI-1040 but in contrast to PLX4032, a subset of RAS mutant cell lines, and those with wild-type RAS and BRAF also exhibit MEK-dependence and CI-1040 sensitivity (34–36). Modest anti-tumor activity was observed in the phase 1 trial of CI-1040 (37), but clinical activity was disappointing in the phase 2 setting and development of CI-1040 was halted in favor of a second-generation compound PD0325901 (Pfizer Oncology) (38). PD0325901 exhibits 50–100-fold greater potency, improved oral bioavailability, and increased metabolic stability compared to CI-1040 (39, 40). RECIST responses were observed in three patients with melanoma on the phase 1 clinical trial of PD0325901, but further clinical development of this agent was not pursued due to concerns over neurological toxicity (41–43). In contrast to the recent trials of PLX4032, trials of CI-1040 and PD0325901 did not restrict eligibility to patients whose tumors harbored mutational activation of the ERK pathway and therefore the activity of these agents in the BRAF mutant class has yet to be clearly defined.
AZD6244 (AstraZeneca) is a third, ATP non-competitive, allosteric inhibitor of MEK1/MEK2. AZD6244 completed phase 2 testing in melanoma, colorectal and lung cancers. In a phase 2 randomization trial of AZD6244 vs. temozolomide in patients with melanoma, antitumor activity with AZD6244 was observed, with partial responses in six patients, five of whom had tumors harboring V600E BRAF mutations (44, 45). There was, however, no significant difference between the treatment arms for the primary endpoint of progression free survival. Similar results were observed in phase 2 trials of AZD6244 in non-small cell lung and colon cancer in which the drug was compared to pemetrexed and capecitabine, respectively (46, 47). In summary, the three randomized phase 2 trials of AZD6244 suggested that activity with this agent was comparable to, but not superior to, disease-specific standard chemotherapy. However, these trials did not enrich for patients whose tumors were most likely to respond to MEK inhibition as predicted by preclinical data. Ongoing studies are now testing the efficacy of AZD6244 in trials in which study entry is restricted to only those patients whose tumors harbor activating mutations in BRAF and/or RAS.
Physiological feedback and drug response
Given the diversity of mutations that induce ERK pathway activation, expression of phosphorylated ERK has been postulated as a potential biomarker of ERK pathway activation and thus MEK inhibitor sensitivity. Several reports demonstrate, however, that the expression of phosphorylated ERK does not correlate with MAP kinase pathway dependence and MEK-inhibitor sensitivity (34, 36, 48). The explanation for this lack of correlation is that the level of phosphorylated ERK is a poor surrogate for MAPK pathway activity (49). Physiologic activation of RAS/RAF signaling is balanced by inhibitory regulators of the pathway which include the sprouty (SPRY) proteins, the MAP kinase phosphatases (MKPs or DUSPs), KSR-1, and RKIP (2, 50–53), and by scaffolding proteins such as 14-3-3 which regulate RAF cellular localization and stability (54, 55). Pathway activity is also regulated by cross-talk with parallel signaling pathways, such as by AKT phosphorylation of inhibitory sites on RAF (56) and through PI3K-dependent feedback (57). In non-transformed cells, activation of the ERK pathway is balanced by inhibitory signals, which dampen or limit the duration of its activity. In tumor cells, however, this normal feedback is often disabled, either through mutation or decreased expression of feedback regulators (58–62). This disruption of normal pathway feedback allows for unhindered ERK pathway activation and is likely a prerequisite for ERK-dependent transformation.
The ERK pathway is thus a classical feedback loop in which negative regulators of the pathway are transcriptionally controlled by ERK. Specifically, ERK activation leads to increased expression of DUSPs and SPRYs that in turn downregulate pathway activity in normal cells. In BRAF mutant tumors, the upstream feedback at the level of the RAF is disrupted, at least in part by the inability of Sprouty family proteins to bind to and inhibit mutant BRAF (63, 64). Feedback at the level of ERK, mediated by the MAPK phosphatases, remains intact and therefore, in BRAF mutant cells, steady state levels of phosphorylated ERK are not dramatically elevated, despite high levels of MEK phosphorylation and high levels of ERK pathway output (49). These findings provide a mechanistic basis for the lack of correlation between phosphorylated ERK expression and ERK pathway output. They also suggest assays of phosphorylated MEK expression and PCR based methods to detect elevated expression of the transcriptional output of ERK may be useful surrogates for high MAPK pathway activity.
The mechanisms whereby MAP kinase pathway feedback is disrupted in BRAF mutant cells may also explain in part the exquisite dependence of BRAF mutant tumors on MEK-ERK signaling and the selectivity of PLX4032 for tumors cells harboring V600EBRAF. As discussed above, PLX4032 and other ATP-competitive inhibitors of RAF induce a paradoxical activation of ERK in tumors with RAS mutation and in those with wild-type BRAF and RAS (28). Activation of ERK by PLX4032 requires direct binding of the compound to the ATP-site of RAF and results from transactivation of the unbound protomer within RAF dimers (28). As RAS activation promotes RAF dimer formation, pathway activation is observed prominently in cells expressing mutant RAS. In contrast to the paradoxical activation of ERK observed in RAS mutant cells, PLX4032 does not induce ERK activation in cells with V600EBRAF. One possible explanation for this result is that RAS activity is low in cells harboring V600EBRAF due to the high levels of sprouty and other negative feedback regulators induced by the oncogene. Ectopic expression of mutant RAS in V600EBRAF cells can, however, induce resistance to PLX4032 (28), suggesting that the pattern of feedback dysregulation observed in V600EBRAF mutant cells may be critical in determining response to selective inhibitors of RAF.
Relief of upstream feedback within the MAPK pathway may also attenuate the response to selective inhibitors of RAF and MEK and contribute to drug resistance. In V600EBRAF tumor cells, MEK phosphorylation is high and not further induced by treatment with MEK inhibitors (49). This contrasts with the response of most tumor and normal cells, including those driven by receptor tyrosine kinase activation and RAS mutations, in which MEK inhibition induces a rapid and profound stimulation of MEK phosphorylation, presumably due to relief of feedback inhibition of MEK (49, 65, 66). This relief of physiological feedback inhibition upstream of MEK following treatment with MEK inhibitors may attenuate drug response in one of several ways. First, activation of RAF, RAS or upstream receptor tyrosine kinases upon downregulation of Sprouty and other feedback elements may lead to activation of parallel signaling pathways previously suppressed by the high levels of SPRYs and DUSPs. As many of these parallel pathways harbor the potential to redundantly regulate the same common downstream effectors of transformation regulated by ERK, such as D cyclins, their activation may attenuate drug response. Alternatively, the hyperphosphorylation of MEK following treatment with an inhibitor such as PD0325901 may promote a state of hyperactivation during times in which the drug is not present at sufficient concentrations to inhibit the enzyme. Such a scenario would occur if continuous drug exposure were not possible as a result of toxicity limitations.
Finally, feedback regulation of the ERK pathway is mediated not only by transcriptional events, but also via direct phosphorylation of CRAF by ERK. CRAF contains six inhibitory sites, the phosphorylation of which require ERK activation (67). CRAF mediated phosphorylation of MEK is thus observed following treatment with MEK inhibitors (49, 65, 66), in the setting of impaired ERK activation by dominant negative kinase suppressor of RAS (KSR) (68), and in cells overexpressing IMP (69). One translational implication of these findings is that the clinical activity of selective inhibitors of BRAF may be attenuated by relief of feedback inhibition of CRAF. Consistent with this possibility, overexpression of CRAF has been shown to be a mechanism of acquired resistance to the selective RAF inhibitor AZ628 (70).
Conclusion
Activation of the MAP kinase pathway is a frequent event in human cancer and pathway activity is often the result of activating mutations in RAS and BRAF. Agents that target RAF and its primary downstream effector MEK are in early stage clinical development, and in the case of the RAF inhibitor PLX4032 have demonstrated sufficient clinical activity to warrant progression to definitive Phase 3 trials. As the activity of these agents correlates with the mechanism responsible for pathway activation (BRAF mutation, RAS mutation or receptor tyrosine kinase activation), prospective genotyping of patients to enrich for those most likely to respond will be critical in the future development of drugs targeting this pathway. Relief of physiological feedback may attenuate the response of cells to selective inhibitors of RAF and MEK and may contribute to drug resistance. A further understanding of the role of feedback elements in promoting transformation and attenuating drug response may thus inform the development of combination strategies that maximize tumor response.
References
- 1.McCormick F. Signal transduction. How receptors turn Ras on. Nature. 1993;363:15–6. doi: 10.1038/363015a0. [DOI] [PubMed] [Google Scholar]
- 2.Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–9. doi: 10.1016/s0955-0674(97)80060-9. [DOI] [PubMed] [Google Scholar]
- 3.Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–9. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 4.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–61. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 5.Catling AD, Schaeffer HJ, Reuter CW, Reddy GR, Weber MJ. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol Cell Biol. 1995;15:5214–25. doi: 10.1128/mcb.15.10.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–5. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 8.Chang EH, Furth ME, Scolnick EM, Lowy DR. Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature. 1982;297:479–83. doi: 10.1038/297479a0. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu K, Goldfarb M, Suard Y, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci U S A. 1983;80:2112–6. doi: 10.1073/pnas.80.8.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A. 1982;79:3637–40. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 12.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 13.Gorden A, Osman I, Gai W, et al. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003;63:3955–7. [PubMed] [Google Scholar]
- 14.Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 15.Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 16.Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20:963–9. doi: 10.1016/j.molcel.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 17.Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 19.Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–6. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–30. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 21.Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai J, Zhang J, Bremer R, Artis R, Hirth P, Bollag G. Development of a novel inhibitor of oncogenic B-Raf. Proc AACR. 2006 [Google Scholar]
- 23.Joseph E, Poulikakos P, Pratilas C, et al. PLX4032, a selective inhibitor of RAF kinase activity, inhibits BRAFV600E tumor proliferation and MAPK signaling in vitro and in vivo. AACR Meeting Abstracts 2008. 2008:3437. [Google Scholar]
- 24.Flaherty K, Puzanov I, Sosman J, et al. Phase I study of PLX4032: Proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J Clin Oncol. 2009;27(suppl) abstr 9000. [Google Scholar]
- 25.Chapman P, Puzanov I, Sosman J, et al. Early efficacy signal demonstrated in advanced melanoma in a phase I trial of the oncogenic BRAF-selective inhibitor PLX4032. European Journal of Cancer Supplements. 2009;7(3):5. [Google Scholar]
- 26.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 28.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz GK, Robertson S, Shen A, et al. A phase I study of XL281, a selective oral RAF kinase inhibitor, in patients (Pts) with advanced solid tumors. J Clin Oncol. 2009;(suppl) abstr 3513. [Google Scholar]
- 30.Ramurthy S, Subramanian S, Aikawa M, et al. Design and synthesis of orally bioavailable benzimidazoles as Raf kinase inhibitors. J Med Chem. 2008;51:7049–52. doi: 10.1021/jm801050k. [DOI] [PubMed] [Google Scholar]
- 31.Stuart DD, Aardalen KM, Lorenzana EG, et al. Characterization of a novel Raf kinase inhibitor that causes target dependent tumor regression in human melanoma xenografts expressing mutant B-Raf. AACR Meeting Abstracts 2006. 2006:1140-b. [Google Scholar]
- 32.Sebolt-Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 33.Ohren JF, Chen H, Pavlovsky A, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 34.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solit DB, Santos E, Pratilas CA, et al. 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography is a sensitive method for imaging the response of BRAF-dependent tumors to MEK inhibition. Cancer Res. 2007;67:11463–9. doi: 10.1158/0008-5472.CAN-07-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–93. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 38.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 39.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–47. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 40.Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–9. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007;1773:1248–55. doi: 10.1016/j.bbamcr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 42.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 43.Haura EB, Ricart AD, Larson TG, et al. A Phase II Study of PD-0325901, an Oral MEK Inhibitor, in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 44.Dummer R, Robert C, Chapman PB, et al. AZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients (pts) with advanced melanoma: An open-label, randomized, multicenter, phase II study. J Clin Oncol. 2008;26(suppl) abstr 9033; 2008. [Google Scholar]
- 45.Board RE, Ellison G, Orr MC, et al. Detection of BRAF mutations in the tumour and serum of patients enrolled in the AZD6244 (ARRY-142886) advanced melanoma phase II study. Br J Cancer. 2009;101:1724–30. doi: 10.1038/sj.bjc.6605371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tzekova V, Cebotaru C, Ciuleanu TE, et al. Efficacy and safety of AZD6244 (ARRY-142886) as second/third-line treatment of patients (pts) with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2008 May 20;26(suppl) abstr 8029); 2008; 2008. [Google Scholar]
- 47.Lang I, Adenis A, Boer K, et al. AZD6244 (ARRY-142886) versus capecitabine (CAP) in patients (pts) with metastatic colorectal cancer (mCRC) who have failed prior chemotherapy. J Clin Oncol. 2008 May 20;26(suppl) abstr 4114) 2008; 2008. [Google Scholar]
- 48.Leboeuf R, Baumgartner JE, Benezra M, et al. BRAFV600E mutation is associated with preferential sensitivity to mitogen-activated protein kinase kinase inhibition in thyroid cancer cell lines. J Clin Endocrinol Metab. 2008;93:2194–201. doi: 10.1210/jc.2007-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pratilas CA, Taylor BS, Ye Q, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–24. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol. 2004;5:441–50. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- 51.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–61. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 52.Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–88. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 53.Yeung K, Seitz T, Li S, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 54.Morrison D. 14-3-3: modulators of signaling proteins? Science. 1994;266:56–7. doi: 10.1126/science.7939645. [DOI] [PubMed] [Google Scholar]
- 55.Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–84. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- 56.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–4. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 57.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lo TL, Yusoff P, Fong CW, et al. The ras/mitogen-activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res. 2004;64:6127–36. doi: 10.1158/0008-5472.CAN-04-1207. [DOI] [PubMed] [Google Scholar]
- 59.Fong CW, Chua MS, McKie AB, et al. Sprouty 2, an inhibitor of mitogen-activated protein kinase signaling, is down-regulated in hepatocellular carcinoma. Cancer Res. 2006;66:2048–58. doi: 10.1158/0008-5472.CAN-05-1072. [DOI] [PubMed] [Google Scholar]
- 60.Sutterluty H, Mayer CE, Setinek U, et al. Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol Cancer Res. 2007;5:509–20. doi: 10.1158/1541-7786.MCR-06-0273. [DOI] [PubMed] [Google Scholar]
- 61.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807–15. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogawa K, Sun C, Horii A. Exploration of genetic alterations in human endometrial cancer and melanoma: distinct tumorigenic pathways that share a frequent abnormal PI3K/AKT cascade. Oncol Rep. 2005;14:1481–5. [PubMed] [Google Scholar]
- 63.Tsavachidou D, Coleman ML, Athanasiadis G, et al. SPRY2 is an inhibitor of the ras/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599E mutant. Cancer Res. 2004;64:5556–9. doi: 10.1158/0008-5472.CAN-04-1669. [DOI] [PubMed] [Google Scholar]
- 64.Brady SC, Coleman ML, Munro J, Feller SM, Morrice NA, Olson MF. Sprouty2 association with B-Raf is regulated by phosphorylation and kinase conformation. Cancer Res. 2009;69:6773–81. doi: 10.1158/0008-5472.CAN-08-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 66.Friday BB, Yu C, Dy GK, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res. 2008;68:6145–53. doi: 10.1158/0008-5472.CAN-08-1430. [DOI] [PubMed] [Google Scholar]
- 67.Dougherty MK, Muller J, Ritt DA, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 68.Therrien M, Michaud NR, Rubin GM, Morrison DK. KSR modulates signal propagation within the MAPK cascade. Genes Dev. 1996;10:2684–95. doi: 10.1101/gad.10.21.2684. [DOI] [PubMed] [Google Scholar]
- 69.Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature. 2004;427:256–60. doi: 10.1038/nature02237. [DOI] [PubMed] [Google Scholar]
- 70.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]