

Abstract
Purpose of review
Endothelin is important in the development of cardiorenal pathology. This review discusses recent developments in understanding endothelin’s role in hypertension and chronic kidney disease (CKD).
Recent findings
Endothelin-1 production is increased in hypertension and CKD. Endothelin-1 stimulates vasoconstriction, inflammation and fibrosis, thereby promoting hypertension, atherosclerosis and CKD. These effects are closely linked to angiotensin II and reactive oxygen species. In preclinical studies, endothelin receptor antagonists were effective in treating hypertension (particularly with endothelial dysfunction) and CKD. In pre-clinical studies, endothelin A-selective, as opposed to combined endothelin A and B, receptor blockers have generally been more efficacious. Few clinical trials have been conducted in hypertension and/or kidney disease, partly due to concerns over side effects of testicular toxicity and fluid retention. Endothelin blockade reduces blood pressure in patients with resistant hypertension, with additional beneficial metabolic effects. Endothelin antagonism improves proteinuria in CKD (diabetic or not), particularly in patients taking inhibitors of angiotensin II action.
Summary
Endothelin is a promising target in the treatment of resistant hypertension and CKD, with additional potential benefits on atherosclerosis and the metabolic syndrome. The nature and mechanisms of drug side effects require elucidation before the potential of this new class of drugs can be fully realized.
Keywords: Endothelin-1, blood pressure, sodium excretion, kidney disease
Introduction
Endothelin-1 (ET-1) was first identified in 1988 as an endothelial cell-derived peptide with the greatest vasoconstrictor potency of any known endogenous compound. After over 22,000 publications dealing with endothelins, it is apparent that ET-1 exerts multiple biologic effects, including regulation of vascular tone, renal sodium and water excretion, cell growth and proliferation, extracellular matrix accumulation, and others. Such biologic complexity is due to several factors, including: 1) ET-1 is produced by, and binds to, almost every cell type in the body; 2) the two mammalian ET receptors (ETA and ETB) can mediate different biologic effects within the same cell as well as between different cell types; 3) ET-1 functions primarily in an autocrine or paracrine manner (it is mainly secreted abluminally), permitting localized microenvironmental effects; 4) a large variety of factors modulate ET-1 production, including vasoactive mediators, cytokines, growth factors, inflammatory substances and others (Figure 1); and 5) the biologic effects of ET-1 can differ depending upon the amount of ET-1 present. This review focuses on the role of ET-1 in vascular and renal pathology. As will be evident, this peptide has emerged as a key target for drug therapy of hypertension and chronic kidney disease (CKD).
Figure 1.
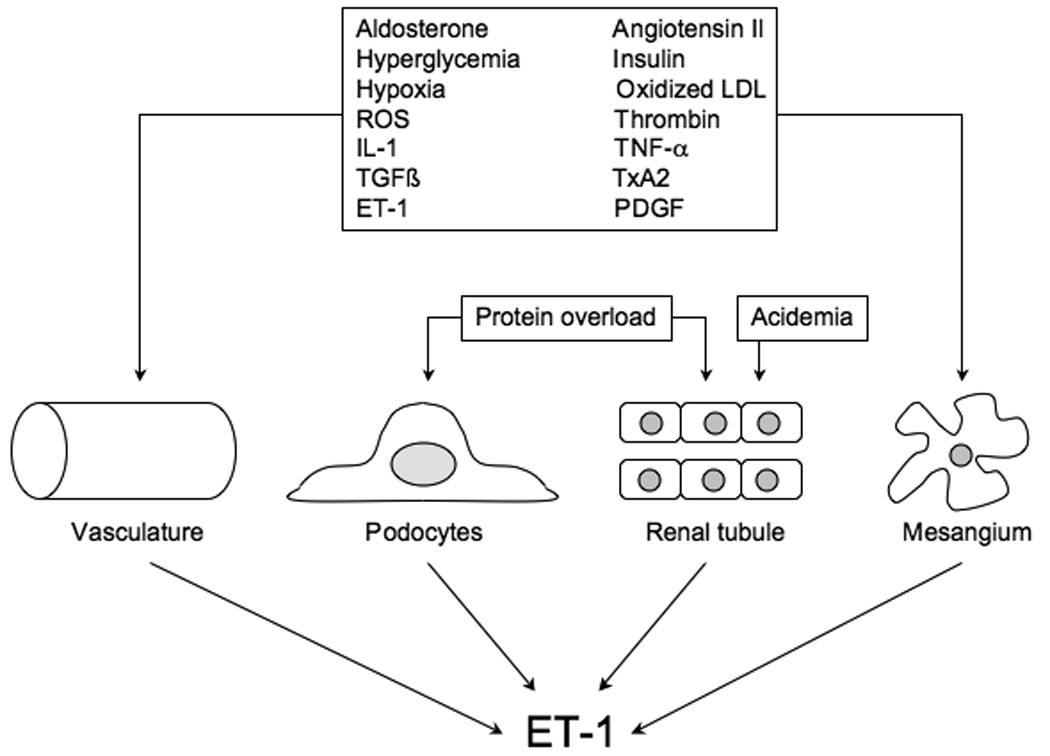
Regulation of ET-1 production in the vasculature and the kidney. IL-1 – interleukin-1, LDL – low density lipoproteins, PDGF – platelet derived growth factor, ROS – reactive oxygen species, TGF – transforming growth factor. TNF – tumor necrosis factor, TxA2 – thromboxane A2.
Endothelin in the control of blood pressure
ET-1 affects many systems that impact blood pressure, including central and peripheral nerves, circulating hormones, the vasculature, the heart and the kidneys [1]. Vascular smooth muscle ETA and ETB activation causes vasoconstriction, while endothelial cell ETB activation is vasodilatory mainly due to nitric oxide release. ETB serve a clearance function, hence ETB blockade raises plasma, and presumably tissue, ET-1 concentrations.
ETB activation inhibits sodium transport in the nephron. The collecting duct ET system is particularly important; principal cells synthesize and bind unusually high levels of ET-1 [2], while collecting duct-specific disruption of ET-1 causes salt-sensitive hypertension [3]. Recent studies indicate that the natriuretic and antihypertensive effect of collecting duct-derived ET-1 is partly mediated by nitric oxide [4]. Some perplexing findings relate to studies in mice with collecting duct-specific knockout of ET receptors. Collecting duct ETB knockout mice have salt-sensitive hypertension [5], while collecting duct ETA knockout mice are normotensive [6]. However, collecting duct knockout of both ETA and ETB causes greater hypertension and sodium retention than in mice with only ETB disruption [7]. This suggests that, under certain circumstances, collecting duct ETA may exert a natriuretic effect. This conclusion is supported by recent studies in which renal medullary infusion of ET-1 into female rats lacking ETB increased urinary sodium excretion [8]. The mechanisms are unknown by which nephron ETA exerts a natriuretic effect, however further clarification of this pathway is of clinical relevance. To my knowledge, every ET receptor antagonist used in humans and experimental animals, whether a combined ETA/ETB blocker or purportedly ETA-selective, causes hemodilution and edema, strongly suggestive of fluid retention [9]. Indeed, such fluid retention may have been partly responsible for the failure of ET antagonists to benefit patients with congestive heart failure [1]. A recent phase III trial studying the effect of avosentan (relatively ETA selective) on renal function in patients with diabetic nephropathy was discontinued due to excessive fluid retention [10]. A follow-up study determined that avosentan dose-dependently reduced urinary sodium excretion in normal individuals [11]. While it remains to be determined if avosentan blocked ETB, particularly at the higher doses, the above results indicate that blocking renal ETB, and probably also ETA, reduces sodium excretion. Determining the mechanisms responsible for ET antagonist-induced fluid retention, and development of appropriate interventions, is clearly important to the future clinical utility of this class of drugs.
Endothelin in hypertension: pre-clinical studies
Numerous studies in animals or humans have implicated ET in the pathogenesis and/or maintenance of hypertension [1, 12, 13] (figures 1 and 2). In the setting of hypertension, ET-1 increases vasoconstriction and promotes vascular remodeling. The increased vasoconstriction relates to elevated ET-1 production, neurohumoral potentiation and/or downregulation of vasodilators such as nitric oxide. Both ETA and ETB have been implicated in the elevated vasoconstrictor response to ET-1 in hypertensive patients. Interestingly, ET-1-dependent vascular tone increases with aging, even in normotensive individuals (although the older patients had higher blood pressure than younger ones); this effect was via ETA activation and could be reversed by regular aerobic exercise [14]. The enhanced role for ET-1 in vascular tone with aging may relate to the well documented increase in reactive oxygen species; ET-1 can increase, as well as be increased by, oxidative stress [15, 16]. In this regard, it is notable that plasma ET-1 levels inversely correlate with plasma antioxidant status in elderly individuals [17].
Figure 2.
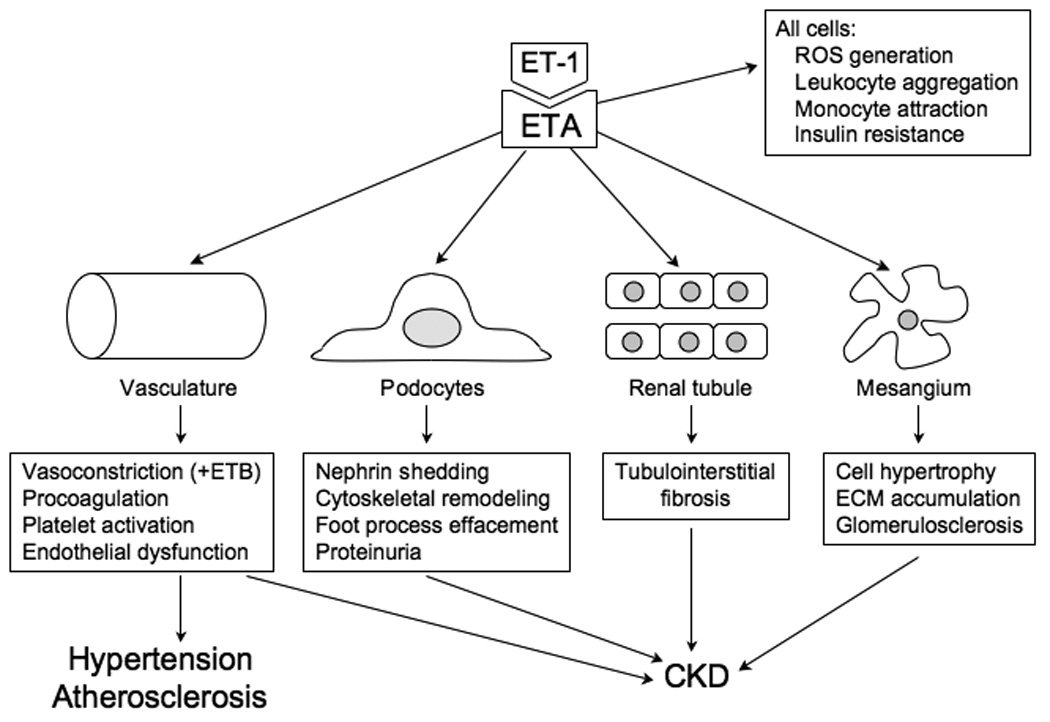
Effect of ET-1 on vascular and renal cells, resulting in hypertension, atherosclerosis and chronic kidney disease (CKD). While ETA likely mediates most of these effects of ET-1, vascular smooth muscle ETB can mediate vasoconstriction. ECM – extracellular matrix, ETA – ETA receptor, ROS – reactive oxygen species.
Other findings suggest a role for ET-1 in uric acid and fructose feeding associated hypertension. Rats fed a high fructose diet developed hypertension that was ameliorated by bosentan, a nonselective ET receptor antagonist [18]; bosentan also reduced plasma angiotensin II (AII) levels. While the effect of targeting the ET system in uric acid related hypertension have not been examined, in vitro studies found that uric acid stimulates ET-1 gene expression in smooth muscle cells [19] and cardiomyocytes [20]. In keeping with the findings described above for aging, antioxidants suppressed uric acid-stimulated ET-1 synthesis by both cell types. Further, mutation of the AP-1 binding site in the ET-1 gene prevented uric acid induction of ET-1 production; this finding is consistent with the known effect of reactive oxygen species to activate jun and fos that bind AP-1 [20].
Perhaps the greatest pathophysiological impact of ET-1 in hypertension relates to its effects on vascular remodeling. ET-1, via ETA activation, and in cooperation with AII, promotes vascular cell hypertrophy, hyperplasia, inflammatory cell infiltration, and fibrosis. In animals, ETA or nonselective ET receptor blockade reduces atherosclerosis [1, 12]. No long-term clinical trials have been conducted to date on the effect of ET antagonism on atherosclerosis; such studies will be of great importance.
Endothelin in hypertension: Clinical trials
Only a few clinical trials have been completed using ET receptor blockade in systemic hypertension. The relative paucity of such trials relates, at least in part, to the side effect profile of this class of drugs [9]. First, ETA or ETB blockade is absolutely contraindicated in pregnancy due to teratogenic effects. Second, initial studies with bosentan were associated with hepatotoxicity, albeit modest (newer ET antagonists, particularly ETA blockers, do not appear to have this complication). Third, although the data has unfortunately never been published other than in company literature, ET antagonists caused testicular toxicity in some experimental animals and reduced sperm counts in some patients. Finally, as discussed above, ET blockade is associated with fluid retention. Thus, given the already broad array of antihypertensive agents, the clinical utility of ET antagonists will most likely be limited to a specific subset of hypertensive patients in whom the benefits outweigh the risks. Such patients most likely have resistant hypertension, particularly those with atherosclerosis. A recent phase III randomized trial (termed DORADO) using darusentan (relatively ETA selective) in 379 patients found a dose-dependent reduction in systolic (−18.1 mmHg) and diastolic (−10.7 mmHg) pressure after 14 weeks of treatment in patients with resistant hypertension (on at least three other antihypertensive agents, one of which was a diuretic) [13]. Another randomized trial involving 72 normotensive patients found that atrasentan (ETA selective) reduced mean blood pressure by 12 mmHg after 6 months in individuals with multiple cardiovascular risk factors and non-obstructive coronary artery disease [21]. In addition, compared to placebo, atrasentan decreased fasting glucose, glycosylated hemoglobin, triglyceride, lipoprotein A and uric acid levels. In a subgroup not treated with angiotensin converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB), atrasentan decreased serum creatinine concentration. Finally, no progression of angiographic coronary artery disease was detected in either atrasentan or placebo groups. Side effects were minimal, however testicular toxicity was not assessed. Thus, ET blockers may fill an important clinical niche, particularly in patients with resistant hypertension, metabolic syndrome and atherosclerosis. No trials have compared ETA with nonselective ET receptor antagonists in such patients, however the pre-clinical data suggests that ETA blockers hold the greatest promise in this regard.
Endothelin in chronic kidney disease - pre-clinical studies
Many studies have demonstrated that the ET system is important in the pathophysiology of chronic kidney disease (CKD) [22–24] (Figures 1 and 2). First, renal ET-1 production is increased in virtually every form of CKD in humans and experimental animals. In hypertensive patients, plasma ET-1 also correlates with renal dysfunction [25]. Similarly, plasma ET-1 levels directly correlated with serum creatinine and degree of albuminuria in patients with diabetic nephropathy [26]. Second, like in the vasculature, ET-1 stimulates fibrosis, cell proliferation and inflammation in the kidney; the peptide can exert pathologic effects on every glomerular cell type and tubule cells. Third, ET receptor antagonists reduce renal injury and disease progression in multiple models of CKD, whether due to diabetes, autoimmune, high renin- or mineralocorticoid-dependent hypertension, reduced renal mass, and other causes.
Multiple mechanisms are responsible for elevated renal ET-1 production in CKD. Renal ET-1 synthesis is stimulated by cytokines, growth factors, chemokines, vasoactive factors, hormones, reactive oxygen species, cholesterol and other substances [22] (Figure 1). Protein overload stimulates proximal tubule ET-1 production, an event that may be of critical importance in renal disease progression [22, 27]. In addition, ET-1 can induce nephrin loss from podocytes [28], thereby potentially impairing slit diaphragm function. High dietary protein, through induction of metabolic acidosis, decreases GFR in rats with reduced renal mass [29]. The high protein diet caused primarily tubulointerstitial injury, a finding in keeping with the known effect of metabolic acidosis to induce nephron ET-1 production [30]. An interesting recent study found that chromogranin A, which is released by sympathetic nerves and chromaffin cells, stimulates glomerular endothelial cell ET-1 release which, in turn, enhanced TGF-β 1 production by mesangial cells [31]. This group found that plasma chromogranin directly correlates with plasma ET-1 and inversely with GFR. Furthermore, a chromogranin promoter haplotype predicted the rate of GFR decline. Taken together, the above findings indicate that a truly impressive array of factors can enhance renal ET-1 production. Since excessive renal ET-1 is clearly pathogenic in the kidney, it begs the question as to why the peptide is induced by so many different mechanisms. While the reasons are not fully known, the very fact that ET-1 is produced by virtually every renal cell type, and that such synthesis can be under unique control in each cell type, sets the stage for this potent injurious peptide to be overproduced by a wide variety of conditions and stimuli.
Endothelin in chronic kidney disease – metabolic syndrome and diabetes
ET-1 induced renal injury in the context of the metabolic syndrome and diabetes deserves special mention. Insulin, whether in the setting of frank diabetes or insulin resistance, stimulates both the production and the action of ET-1 in experimental animals and/or in humans [32]. The insulin-stimulated ET-1 production is due, at least in part, to phosphoinositide-3-kinase-dependent activation of glycogen synthase-3β [33]. Insulin potentiation of ET-1 action was recently shown to involve an interaction with AII since losartan blunted the enhanced ETA-mediated vasoconstriction in insulin-treated diabetic rats [34]. Such an interaction with AII was highlighted in recent studies in diabetic rats wherein renal damage actually regressed when avosentan and lisinopril were given together, but not separately [35]. These findings are further supported by the observation that ET antagonism reduced renal fibrosis in an animal model of severe AII-dependent hypertension [36].
Endothelin in chronic kidney disease – clinical trials
No long-term studies have assessed the effect of ET antagonists on CKD progression. However, ET blockers have a substantial antiproteinuric effect. The ASCEND trial, involving over 1300 patients, reported up to a 50% reduction in albuminuria after 3 or 6 months of avosentan therapy in patients with diabetic nephropathy on ACEI/ARB treatment [10]. Unfortunately, as discussed earlier, this trial was terminated due to fluid retention. A follow-up study by this group involving 286 patients with diabetic nephropathy on ACEI/ARB therapy, using lower doses of avosentan, noted up to a 30% decrease in albuminuria after 12 weeks of treatment [37]. A recent study found that acute ETA blockade decreased proteinuria by about 30% in non-diabetic CKD patients [38]; blood pressure was similarly lowered by nifedipine, however this increased proteinuria by 26%. In keeping with the concept of an interaction with AII, a follow-up analysis by this group found that the magnitude of proteinuria reduction was greatest in patients on combined ACEI/ARB therapy [39]. Thus, while only proteinuria has been assessed to date, ET antagonism has exciting potential as a therapy in CKD.
Endothelin in chronic kidney disease – which receptor(s) to target?
Several studies have compared the effects of ETA-selective with combined ETA/B antagonists in various models of CKD, including hypertension, high dietary protein and reduced renal mass, as well as acutely in hypertensive patients with CKD [23]. In almost every instance, ETA, as compared to combined ETA/B, blockade has achieved superior results (GFR, fibrosis and/or proteinuria) [23, 40]. Recent studies have supported these findings. Induction of diabetes in rats genetically deficient in ETB causes severe hypertension and progressive renal failure [41]. In a mouse model of polycystic kidney disease, ETB, but not ETA, antagonism accelerated the cystic kidney disease [42]; notably, the ETB blocker effect was prevented by ETA antagonism. In high dietary protein-induced CKD in rats with reduced renal mass, ETA, but not combined ETA/B antagonism, prevented the reduction in GFR [29]. In a rat model of high AII hypertensive renal injury, ETA blockade reduced glomerular injury to a greater extent than did combined ETA/B antagonism [43]. All of these findings are in keeping with the known effect of ETA, but not ETB, to mediate vasoconstriction, cell injury and fibrosis in the kidney [22]. Thus, the available data support using ETA-selective antagonists in CKD. Having said that, it must be recognized that most of the studies are in experimental animals; until ETA-selective and combined ETA/B antagonists are compared in patients with CKD, this issue remains unresolved. Nonetheless, my own bias is that, at least in initial studies on CKD progression, ETA-selective blockers should be used.
Conclusion
In summary, ET-1 is an important pathogenic factor in hypertension, particularly in association with metabolic syndrome and/or atherosclerosis. In addition, ET-1 plays a substantial role in CKD progression. Numerous pre-clinical, as well the few completed clinical, studies suggest a substantial beneficial role for ET receptor antagonists, and particularly ETA blockers, in these disorders. However, in order to move forward, several key issues need to be addressed. First, the nature and mechanisms of ET antagonist-related adverse effects need elucidation. Second, appropriate doses of ET blockers need to be defined, including determination of ones that are truly ETA-selective. Third, consideration must be given to which agents would best be co-administered with ET blockers. Finally, given the great promise of these drugs, the pharmaceutical industry and academia must work together to conduct long-term studies on cardiovascular and renal outcomes.
Acknowledgements
The research from the author's laboratory was supported in part by NIH grant DK96392.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The author has no conflicts of interest.
References and recommended reading
- 1. Dhaun N, Goddard J, Kohan DE, et al. Role of endothelin-1 in clinical hypertension: 20 years on. Hypertension. 2008;52:452–459. doi: 10.1161/HYPERTENSIONAHA.108.117366. This is a good recent review on the role of ET-1 in hypertension
- 2.Kohan DE. Biology of endothelin receptors in the collecting duct. Kidney Int. 2009;76:481–486. doi: 10.1038/ki.2009.203. [DOI] [PubMed] [Google Scholar]
- 3.Ahn D, Ge Y, Stricklett PK, et al. Collecting duct-specific knockout of endothelin-1causes hypertension and sodium retention. J Clin Invest. 2004;114:504–511. doi: 10.1172/JCI21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider MP, Ge Y, Pollock DM, et al. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension. 2008;51:1605–1610. doi: 10.1161/HYPERTENSIONAHA.107.108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ge Y, Bagnall A, Stricklett PK, et al. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2006;291:F1274–F1280. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- 6.Ge Y, Stricklett PK, Hughes AK, et al. Collecting duct-specific knockout of the endothelin A receptor alters renal vasopressin responsiveness, but not sodium excretion or blood pressure. Am J Physiol Renal Physiol. 2005;289:F692–F698. doi: 10.1152/ajprenal.00100.2005. [DOI] [PubMed] [Google Scholar]
- 7. Ge Y, Bagnall A, Stricklett PK, et al. Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2008;295:F1635–F1640. doi: 10.1152/ajprenal.90279.2008. This study describes the role of ETA and ETB receptors in regulating sodium reabsorption and discusses how they may interact.
- 8. Nakano D, Pollock DM. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension. 2009;53:324–330. doi: 10.1161/HYPERTENSIONAHA.108.123687. This study makes the novel observation that renal medullary ETA receptors may cause a natriuresis.
- 9.Battistini B, Berthiaume N, Kelland N, et al. Profile of past and current clinical trials involving endothelin receptor antagonists: the novel "-sentan" class of drug. Exp Biol Med. 2006 [PubMed] [Google Scholar]
- 10. Viberti G. The ASCEND trial. 2008 Available from: www.speedel.com/assets/2008_SPP301ASCENDStudy.pdf. This link describes the first Phase III study evaluating the effect of ET receptor blockade on renal function in patients with diabetic nephropathy.
- 11. Smolander J, Vogt B, Maillard M, et al. Dose-dependent acute and sustained renal effects of the endothelin receptor antagonist avosentan in healthy subjects. Clin Pharmacol Ther. 2009;85:628–634. doi: 10.1038/clpt.2009.15. This study demonstrates that ET receptor blockade can cause renal sodium retention as well as the possible responsible mechanisms.
- 12.Epstein BJ, Anderson S. Endothelin receptor antagonists as antihypertensives: the next frontier. Expert Rev Cardiovasc Ther. 2009;7:675–687. doi: 10.1586/erc.09.24. [DOI] [PubMed] [Google Scholar]
- 13.Prasad VS, Palaniswamy C, Frishman WH. Endothelin as a clinical target in the treatment of systemic hypertension. Cardiol Rev. 2009;17:181–191. doi: 10.1097/CRD.0b013e3181aa8f4a. [DOI] [PubMed] [Google Scholar]
- 14.Van Guilder GP, Westby CM, Greiner JJ, et al. Endothelin-1 vasoconstrictor tone increases with age in healthy men but can be reduced by regular aerobic exercise. Hypertension. 2007;50:403–409. doi: 10.1161/HYPERTENSIONAHA.107.088294. [DOI] [PubMed] [Google Scholar]
- 15.Camici GG, Sudano I, Noll G, et al. Molecular pathways of aging and hypertension. Curr Opin Nephrol Hypertens. 2009;18:134–137. doi: 10.1097/MNH.0b013e328326093f. [DOI] [PubMed] [Google Scholar]
- 16.Pollock DM, Pollock JS. Endothelin and oxidative stress in the vascular system. Curr Vasc Pharmacol. 2005;3:365–367. doi: 10.2174/157016105774329408. [DOI] [PubMed] [Google Scholar]
- 17.Skalska AB, Pietrzycka A, Stepniewski M. Correlation of endothelin 1 plasma levels with plasma antioxidant capacity in elderly patients treated for hypertension. Clin Biochem. 2009;42:358–364. doi: 10.1016/j.clinbiochem.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Tran LT, MacLeod KM, McNeill JH. Endothelin-1 modulates angiotensin II in the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;325:89–97. doi: 10.1007/s11010-008-0023-z. [DOI] [PubMed] [Google Scholar]
- 19.Chao HH, Liu JC, Lin JW, et al. Uric acid stimulates endothelin-1 gene expression associated with NADPH oxidase in human aortic smooth muscle cells. Acta Pharmacol Sin. 2008;29:1301–1312. doi: 10.1111/j.1745-7254.2008.00877.x. [DOI] [PubMed] [Google Scholar]
- 20.Cheng TH, Lin JW, Chao HH, et al. Uric acid activates extracellular signal-regulated kinases and thereafter endothelin-1 expression in rat cardiac fibroblasts. Int J Cardiol. 2008 doi: 10.1016/j.ijcard.2008.09.004. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 21. Raichlin E, Prasad A, Mathew V, et al. Efficacy and safety of atrasentan in patients with cardiovascular risk and early atherosclerosis. Hypertension. 2008;52:522–528. doi: 10.1161/HYPERTENSIONAHA.108.113068. This study shows that, in normotensive patients with coronary artery disease, ETA receptor antagonism improves metabolic parameters.
- 22. Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. NatClin Pract Nephrol. 2008;4:490–501. doi: 10.1038/ncpneph0891. This is an excellent recent review on the role of endothelin in proteinuric renal diseases.
- 23. Neuhofer W, Pittrow D. Endothelin receptor selectivity in chronic kidney disease: rationale and review of recent evidence. Eur J Clin Invest. 2009;39 Suppl 2:50–67. doi: 10.1111/j.1365-2362.2009.02121.x. This is an excellent recent review with a very detailed and well organized description of studies on the effect of ET receptor antagonists in kidney disease.
- 24.Longaretti L, Benigni A. Endothelin receptor selectivity in chronic renal failure. Eur J ClinInvest. 2009;39 Suppl 2:32–37. doi: 10.1111/j.1365-2362.2009.02119.x. [DOI] [PubMed] [Google Scholar]
- 25.Cottone S, Mule G, Guarneri M, et al. Endothelin-1 and F2-isoprostane relate to and predict renal dysfunction in hypertensive patients. Nephrol Dial Transplant. 2009;24:497–503. doi: 10.1093/ndt/gfn489. [DOI] [PubMed] [Google Scholar]
- 26.Zanatta CM, Gerchman F, Burttet L, et al. Endothelin-1 levels and albuminuria inpatients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2008;80:299–304. doi: 10.1016/j.diabres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 27.Zoja C, Benigni A, Remuzzi G. Cellular responses to protein overload: key event in renal disease progression. Curr Opin Nephrol Hypertens. 2004;13:31–37. doi: 10.1097/00041552-200401000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Collino F, Bussolati B, Gerbaudo E, et al. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am J Physiol Renal Physiol. 2008;294:F1185–F1194. doi: 10.1152/ajprenal.00442.2007. [DOI] [PubMed] [Google Scholar]
- 29. Phisitkul S, Hacker C, Simoni J, et al. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kidney Int. 2008;73:192–199. doi: 10.1038/sj.ki.5002647. This study shows that ET mediates acidosis-induced renal functional deterioration and that ETA, but not ETB, blockade is efficacious.
- 30.Wesson DE. Regulation of kidney acid excretion by endothelins. Kidney Int. 2006;70:2066–2073. doi: 10.1038/sj.ki.5001905. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Mahata M, Rao F, et al. Chromogranin A regulates renal function by triggering Weibel-Palade body exocytosis. J Am Soc Nephrol. 2009;20:1623–1632. doi: 10.1681/ASN.2008111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarafidis PA, Lasaridis AN. Insulin resistance and endothelin: another pathway for renal injury in patients with the cardiometabolic syndrome? J Cardiometab Syndr. 2008;3:183–187. doi: 10.1111/j.1559-4572.2008.00009.x. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Li JC. Stimulation of endothelin-1 gene expression by insulin via phosphoinositide-3 kinase-glycogen synthase kinase-3beta signaling in endothelial cells. Life Sci. 2008;82:512–518. doi: 10.1016/j.lfs.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi T, Nogami T, Taguchi K, et al. Diabetic state, high plasma insulin and angiotensin II combine to augment endothelin-1-induced vasoconstriction via ETA receptors and ERK. Br J Pharmacol. 2008;155:974–983. doi: 10.1038/bjp.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gagliardini E, Corna D, Zoja C, et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol Renal Physiol. 2009 doi: 10.1152/ajprenal.00340.2009. (Epub ahead of print) This study demonstrates that combined ET receptor blockade and ACEI therapy cause injury regression in diabetic nephropathy in animals. The study is important because it makes a good case for combined therapy with these agents.
- 36.Seccia TM, Maniero C, Belloni AS, et al. Role of angiotensin II, endothelin-1 and L-type calcium channel in the development of glomerular, tubulointerstitial and perivascular fibrosis. J Hypertens. 2008;26:2022–2029. doi: 10.1097/HJH.0b013e328309f00a. [DOI] [PubMed] [Google Scholar]
- 37. Wenzel RR, Littke T, Kuranoff S, et al. Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J Am Soc Nephrol. 2009;20:655–664. doi: 10.1681/ASN.2008050482. This is the only published chronic study in humans showing that ET receptor blockade decreases albuminuria in diabetic patients. This important information helps set the stage for subsequent trials looking at ET antagonists in CKD.
- 38. Dhaun N, Macintyre IM, Melville V, et al. Blood pressure-independent reduction in proteinuria and arterial stiffness after acute endothelin-A receptor antagonism in chronic kidney disease. Hypertension. 2009;54:113–119. doi: 10.1161/HYPERTENSIONAHA.109.132670. This is the only study to examine the effect of ET receptor blockade on proteinuria in humans with non-diabetic kidney disease. The finding of reduced proteinuria suggests that ET antagonists may be effective in many forms of CKD, not just diabetes.
- 39.Dhaun N, Macintyre IM, Melville V, et al. Effects of endothelin receptor antagonism relate to the degree of renin-angiotensin system blockade in chronic proteinuric kidney disease. Hypertension. 2009;54:e19–e20. doi: 10.1161/HYPERTENSIONAHA.109.138263. [DOI] [PubMed] [Google Scholar]
- 40.Ohkita M, Takaoka M, Matsumura Y. Drug discovery for overcoming chronic kidney disease (CKD): the endothelin ET B receptor/nitric oxide system functions as a protective factor in CKD. J Pharmacol Sci. 2009;109:7–13. doi: 10.1254/jphs.08r10fm. [DOI] [PubMed] [Google Scholar]
- 41.Pfab T, Thone-Reineke C, Theilig F, et al. Diabetic endothelin B receptor-deficient rats develop severe hypertension and progressive renal failure. J Am Soc Nephrol. 2006;17:1082–1089. doi: 10.1681/ASN.2005080833. [DOI] [PubMed] [Google Scholar]
- 42.Chang MY, Parker E, El Nahas M, et al. Endothelin B receptor blockade accelerates disease progression in a murine model of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:560–569. doi: 10.1681/ASN.2006090994. [DOI] [PubMed] [Google Scholar]
- 43.Vernerova Z, Kramer HJ, Backer A, et al. Late-onset endothelin receptor blockade in hypertensive heterozygous REN-2 transgenic rats. Vascul Pharmacol. 2008;48:165–173. doi: 10.1016/j.vph.2008.01.009. [DOI] [PubMed] [Google Scholar]