

This study reveals that components of the yeast ERAD-L pathway can discriminate between two subtly different forms of the same toxin substrate. Although precytosolic requirements are similar for both toxin structures, there is a divergence in fate on the cytosolic face of the ER membrane.
Abstract
We report that a toxic polypeptide retaining the potential to refold upon dislocation from the endoplasmic reticulum (ER) to the cytosol (ricin A chain; RTA) and a misfolded version that cannot (termed RTAΔ), follow ER-associated degradation (ERAD) pathways in Saccharomyces cerevisiae that substantially diverge in the cytosol. Both polypeptides are dislocated in a step mediated by the transmembrane Hrd1p ubiquitin ligase complex and subsequently degraded. Canonical polyubiquitylation is not a prerequisite for this interaction because a catalytically inactive Hrd1p E3 ubiquitin ligase retains the ability to retrotranslocate RTA, and variants lacking one or both endogenous lysyl residues also require the Hrd1p complex. In the case of native RTA, we established that dislocation also depends on other components of the classical ERAD-L pathway as well as an ongoing ER–Golgi transport. However, the dislocation pathways deviate strikingly upon entry into the cytosol. Here, the CDC48 complex is required only for RTAΔ, although the involvement of individual ATPases (Rpt proteins) in the 19S regulatory particle (RP) of the proteasome, and the 20S catalytic chamber itself, is very different for the two RTA variants. We conclude that cytosolic ERAD components, particularly the proteasome RP, can discriminate between structural features of the same substrate.
INTRODUCTION
Misfolded and orphan polypeptides in the endoplasmic reticulum (ER) of eukaryotic cells are detected and dispatched to the cytosol for proteasomal elimination via ER quality control pathways known collectively as ER-associated degradation (ERAD). Usually, they are extracted from the ER membrane by a p97/Cdc48p complex (Bays et al., 2001; Ye et al., 2001; Jarosch et al., 2002a; Rabinovich et al., 2002; Rouiller et al., 2002; Elkabetz et al., 2004), polyubiquitylated by membrane-bound dislocation complexes (Carvalho et al., 2006; Denic et al., 2006), and degraded by 26S proteasomes. Although a large number of components required for ERAD in yeast and mammalian cells has now been identified for a range of substrates (Meusser et al., 2005; Nakatsukasa and Brodsky, 2008), it is not clear whether a given cell will handle different forms of the same soluble substrate in exactly the same way.
To investigate this, we have used the protein cytotoxin ricin. Native holotoxin normally traffics to the ER lumen of target mammalian cells (Spooner et al., 2006) where it becomes reduced to its cell-binding B chain (RTB) and a structurally native cytotoxic A chain (RTA; Bellisola et al., 2004; Spooner et al., 2004). RTA is then thought to masquerade as a misfolded protein in order to co-opt an unknown number of ERAD components (Wesche et al., 1999; Lord et al., 2005). Subsequently, RTA is dislocated to the cytosol where it depurinates 28S rRNA, inhibiting protein synthesis (Endo et al., 1987). A dearth of lysyl residues in RTA (Hazes and Read, 1997; Deeks et al., 2002) reduces polyubiquitylation potential, presumably hampering targeting to proteasomes. Cytosolic RTA then disengages from ERAD, refolding to a catalytic conformation, assisted by Hsc70 (Spooner et al., 2008). These observations led us to probe the requirements for uncoupling from ERAD by comparing native RTA with a structurally defective form (RTAΔ; Simpson et al., 1999), which is known to be a model glycosylated, ubiquitylated ERAD substrate whose fate depends on the Png1p–Rad23p complex that acts between Cdc48p and the proteasome (Kim et al., 2006).
The extreme sensitivity of mammalian cells to ricin makes identification of the ERAD components exploited by this toxin very difficult. Because there are many similarities between yeast and mammalian ERAD (Kawaguchi and Ng, 2007) and because yeast ribosomes are less sensitive to RTA toxicity than their mammalian counterparts, we expressed these RTA variants in the yeast ER lumen by targeting the nascent proteins with a Kar2 signal peptide. Using biochemical analyses with both toxin forms and survival assays with native RTA, we identified a requirement for cycling via the Golgi complex and for the Hrd1p-Hrd3p ERAD dislocation complex. Although these precytosolic requirements are similar for both toxins, we unexpectedly find that the Cdc48p and proteasome requirements are strikingly different, demonstrating that structural features of an ERAD substrate can influence its ultimate fate.
MATERIALS AND METHODS
Yeast Strains and Plasmids
A yeast gene knockout collection derived from strain BY4741 (MATa hisΔ1, leu2Δ0, met15Δ0, ura3Δ0) was sourced from Open Biosystems (Huntsville, AL; Table 1). Other strains used are also listed in Table 1. Cultures were grown in rich YPDA media or synthetic media containing standard ingredients and 2% glucose (SD medium), 2% raffinose (SR medium), or 2% raffinose + 2% galactose (SRG medium). Where appropriate, medium lacking uracil and leucine was utilized. Yeast transformations were achieved by using the lithium acetate/single-stranded DNA/polyethylene glycol method as previously described (Gietz and Woods, 2002).
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source/Reference |
|---|---|---|
| BY4741 | MATa hisΔ1, leu2Δ0, met15Δ0, ura3Δ0 | Open Biosystems |
| cdc48-1 | cdc48-1, prc1-1, ura3-52, ade2-101, lys2-801, can1-100 | Ye et al. (2001) |
| cdc48-3 | cdc48-3, prc1-1, leu2-3,-112, ura3-52, ade2-101 | Ye et al. (2001) |
| SNY1025 | MATα Δscj1::TRP1 ura3 leu2 trp1 his3 lys2 suc2 | Heiligenstein et al. (2006) |
| SNY1028 | MATα Δjem1::LEU2 ura3 leu2 trp1 his3 lys2 suc2 | Heiligenstein et al. (2006) |
| SNY1026-7A | MATα Δjem1::LEU2 Δscj1::TRP1 ura3 leu2 trp1 his3 lys2 suc2 | Heiligenstein et al. (2006) |
| KRY167 | ade2-1 ura3-1 his3-11,15 leu2-3,112 trp1-1 can1-100 prc1-1 (pep4::URA3) | Gillece et al. (1999) |
| JN284 | MATα ura3-52, leu2 (-3,-112), his7-2, ise1 | Gaber et al. (1989); Vogel et al. (1993) |
| YPH 499 | MATa ura3-52 leu2-D1 his3-D200, trp1-D63 lys2-801 ade2-101 | Ghislain et al. (1993) |
| CMY 763 | MATa cim3-1, ura3-52 leu2-D1 his3-D200 | Ghislain et al. (1993) |
| CMY 765 | MATa cim5-1, ura3-52 leu2-D1 his3-D200 | Ghislain et al. (1993) |
| DHY9 | MATα ura3-52 lys2-801 ade2-101 trp1-Δ1 his3-Δ200 leu2-Δ1 | Springer et al. (2000) |
| RSY188 | DHY9, Δemp24::HIS Δerv25::HIS3 Δerp1::TRP1 Δerp2::HIS3 Δerp3::loxP Δerp4::loxP Δerp5::loxP Δerp6::loxP-Kanr-loxP | Springer et al. (2000) |
| RSY263 | MATα sec12-4 ura3-52 | Kaiser and Schekman (1990) |
| RSY281 | MATα sec23-1 ura3-52 his4-619 | Kaiser and Schekman (1990) |
| SUB62 | MATa his3-Δ200 lys2-801 leu2-3,112 trp1-1 ura3-52 | Rubin et al. (1998) |
| Rpt2RF | MATa his3-Δ200 lys2-801 leu2-3,112 trp1-1 ura3-52, rpt2RF | Rubin et al. (1998) |
| Rpt4R | MATa his3-Δ200 lys2-801 leu2-3,112 trp1-1 ura3-52, rpt4R | Rubin et al. (1998) |
| Cl3-ABYS-86 | MATα pra1-1 prb1-1 prc1-1 cps1-3 ura3Δ5 leu2-3, 112 his− | Heinemeyer et al. (1991) |
| pre1-1 | Cl3-ABYS-86, pre1-1 | Heinemeyer et al. (1991) |
Construction of ERAD Substrates and Phenotype Complementation
RTAΔ is a misfolded protein in which a stretch of five amino acids from within the active site has been deleted. For screening yeast strains using a viability assay, RTAE177A was used. This protein contains a minor active site change and exhibits only sixfold reduced activity (Kim et al., 1992). This slight attenuation of activity was a safeguard to permit survival should there be a less than full cotranslational coupling of ER import by the Kar2p signal peptide in situations where retrotranslocation was completely blocked. In experiments where RTA stability was measured, the less potent RTAE177D was used (Chaddock and Roberts, 1993; Allen et al., 2007). Structure determination has shown that this toxin shows only a subtle change within the active site cleft (Allen et al., 2007) and can therefore be regarded, like RTAE177A, as having an essentially native conformation. However, the protein has a 50–70-fold reduced catalytic activity (Chaddock and Roberts, 1993), and this permits greater growth of yeast cells and easier visualization of toxin by SDS-PAGE. For simplicity, both RTAE177A and RTAE177D (essentially native) toxins are referred to as RTA throughout. PCR was used to fuse the yeast Kar2p signal sequence with the open reading frames of RTAE177A (Kim et al., 1992) and RTAE177D (Allen et al., 2007). Fusions were cloned under the transcriptional control of the GAL1 promoter in a yeast expression vector derived from pRS316 (Sikorski and Hieter, 1989). A fusion gene encoding Kar2-RTAΔ with a deletion of five amino acids in the active site that renders it both inactive and misfolded (Simpson et al., 1999) was similarly cloned downstream of the GAL1 promoter. In addition, the open reading frame of RTAE177D lacking a signal sequence was cloned downstream of the GAL1 promoter for cytosolic expression. RTA variants lacking lysyl residues—RTAE177A(1K) and RTAE177A(0K)—were created using a QuickChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). RTAE177A(1K) was constructed by changing Lys239 to Arg using the following oligonucleotides: 5′-gccgatgatatattccccagacaatacccaattataaac-3/5′-gtttataattgggatttgtctggggaatatatcatcggc-3′. RTAE177A(0K) was generated by changing Lys4 in RTAE177A(1K) to Arg using the oligonucleotides 5′-caaagacgtaatggttccagattcagtgtgtacgatgtg-3′/5′-cacatcgtacacactgaatctggaaccattacgtctttg-3′. Generation of a plasmid for the expression of a catalytically inactive Hrd1p involved altering Cys399 to Ser (Bordallo and Wolf, 1999) and cloning both the wild-type and mutant versions in an integrating vector with a LEU2 selective marker.
Pulse-Chase Analysis
Yeast cells carrying plasmids that expressed Kar2p-RTA fusions under control of the GAL1 promoter were grown overnight at 30°C in SR medium. Cells (1.48 × 108) were harvested and washed before being incubated (30 min, 30°C) in inducing SRG medium lacking methionine. After addition of 280 μCi·ml−1 [35S]methionine/cysteine the cells were incubated (20 min, 30°C), after which excess unlabeled methionine and cysteine were added. Chase samples were taken at time zero and various time points thereafter (see figure legends). Metabolically labeled RTA was immunoprecipitated from cell lysates as described previously (Simpson et al., 1999). Products were analyzed by reducing SDS-PAGE and autoradiography and were quantified using TotalLab software (Newcastle upon Tyne, United Kingdom). Where temperature-sensitive strains were used, cells were grown at the permissive temperature for growth and shifted to the restrictive temperature for 2 h before the start of the radioactive pulse. Where required, cells were incubated in medium containing inhibitors of protein trafficking (300 μM brefeldin A [BFA]), proteasomal degradation (20 μM clasto-lactacystin β-lactone [cLβ-l]) or N-glycosylation (25 μg·ml−1 tunicamycin) for 30 min before pulse-chase analysis, and the inhibitors were maintained throughout the pulse chase. To trap any ubiquitylated intermediates, expression of ER-targeted RTA or RTAΔ was induced by growth in galactose medium for 2 h in the presence or absence of cLβ-l. Cells were then pulse-labeled for 20 min, and cell lysates were prepared as before but with the addition of 10 mM N-ethyl maleimide in the initial lysis buffer.
Growth Sensitivity to Ricin A Chain
Yeast cells expressing a fusion of the Kar2 signal peptide and RTAE177A were grown overnight in liquid SR medium lacking uracil. Tenfold serial dilutions of cells from 106 to 101 were plated on S-galactose plates (2% galactose) lacking uracil to induce expression and on S-glucose plates (2% glucose) as controls. Plates were incubated at 30°C for 4 d and examined for growth differences.
RESULTS
ER Import Is a Prerequisite for the Degradation of Ricin A Chain
Pulse-labeled ER-directed RTAΔ has nonglycosylated (g0) and singly (g1), and doubly glycosylated (g2) forms: in contrast, ER-targeted RTA occurs primarily as g0 with a minor fraction of g1 (Figure 1A), suggesting that ER-directed RTA folds rapidly (Allen et al., 1995). Both disappeared during a 1-h chase with unlabeled amino acids (Figure 1B), not by turnover in vacuoles, nor by secretion, because disappearance was unaltered in a yeast strain lacking vacuolar proteinase A (Pep4p) that is required for activation of many vacuolar proteases (Figure 1C), and both were absent from extracellular medium (not shown). In marked contrast, RTA expressed cytosolically remained stable (Figure 1B) unlike a previously characterized cytosolic CPY*-GFP fusion (Medicherla et al., 2004). Thus ER import is a prerequisite for RTA degradation.
Figure 1.
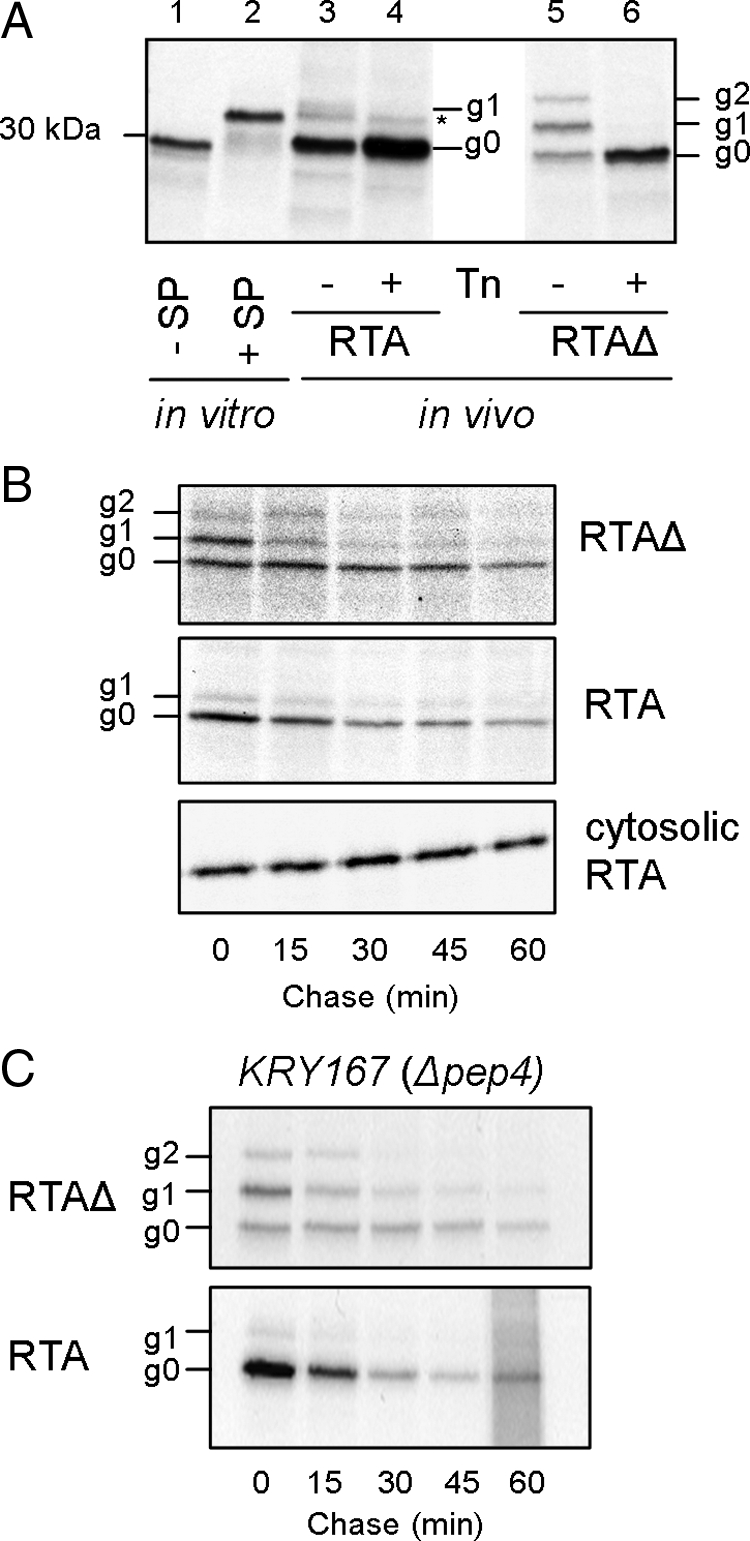
RTA is degraded after import into the ER lumen. Two RTA variants were immunoprecipitated from detergent soluble extracts prepared after a pulse of radiolabeled [35S]methionine/cysteine with or without a subsequent chase of unlabeled amino acids for the times indicated. Samples were analyzed by SDS-PAGE and fluorography, and fluorograms were quantified using TotalLab software. (A) Immunoprecipitated pulse-labeled RTA (lanes 3 and 4) and RTAΔ (lanes 5 and 6). Lanes 1 and 2, in vitro–translated mature RTA (−SP) and Kar2p signal peptide-RTA fusion (+SP). g0, nonglycosylated; g1, singly glycosylated; g2, doubly glycosylated; Tn, tunicamycin; *, unprocessed RTA. (B) Pulse-chase kinetics of ER-targeted RTA and cytosolic RTA. (C) Pulse-chase kinetics of the two ER-targeted RTAs in KRY167 (a pep4 deletion strain).
Misfolded and Native RTAs Require the Hrd1-Hrd3p Complex for Dislocation
RTA severely hampers yeast cell growth when expressed within the ER lumen in a form that is competent to acquire a native conformation after its retrotranslocation to the cytosol (Allen et al., 2007). This effect on viability is due to A chain–catalyzed depurination of the EF-2–binding site on the ribosome and the consequent inhibition of protein synthesis (Endo et al., 1987), even though most of the dislocated toxin is proteolysed in the cytosol (Simpson et al., 1999). Two major membrane-associated E3 ubiquitin ligase complexes (Hrd1p–Hrd3p and Doa10p) play roles in ERAD dislocation (Carvalho et al., 2006; Denic et al., 2006). Open Biosystems yeast knockout strains with deletions of genes encoding proteins associated with these complexes were transformed with RTA and examined by viability assay on both noninducing (glucose) and inducing (galactose) plates (Figure 2A, Table 1) and by monitoring RTA stability by pulse-chase analysis (Figure 2B). Genetic deletions that conferred growth advantages correlated with acquired stability on pulse-chase. RTAΔ was also stabilized in Δhrd1 but not in Δdoa10 yeast strains (Figure 2C), as shown in a previous study (Kim et al., 2006). The need for Hrd3p, Usa1p, and Der1p is consistent with the critical involvement of Hrd1p, because these cofactors form a near stoichiometric complex even though Der1p itself is only involved in the turnover of a subset of luminal (or ERAD-L) substrates (Knop et al., 1996; Taxis et al., 2003; Hitt and Wolf, 2004; Carvalho et al., 2006). We also note relatively minor roles for Usa1p, which is required for optimal activity of the Hrd1-Hrd3 complex (Carroll and Hampton, 2010); the membrane-integral Cue1p, which recruits (Biederer et al., 1997) and activates Ubc7p (Bazirgan and Hampton, 2008); and Ubx2p, which couples the Hrd1 complex to the cytosolic Cdc48 complex (Wilson et al., 2006).
Figure 2.

RTA variants dislocate via the Hrd1p–Hrd3p–Der1p complex. (A) Viabilities of yeast strains harboring RTA were assessed by spotting dilutions of cells on noninducing (glucose, left) and inducing (galactose, right) media after growth at 30°C for 5 d. WT, wild-type BY4741. (B) Typical immunoprecipitates of pulse-chase samples revealed by SDS-PAGE/fluorography (gel slices). The associated mean quantitations (±1 SD) of native RTA in selected strains are presented graphically, where n = 4 (WT, Δhrd1, Δhrd3, Δubx2, and Δubc7), 3 (Δder1), or 2 (Δusa1, Δcue1, and Δdoa10). Overlapping error bars have been omitted for clarity. (C) Pulse-chase analysis of RTAΔ in WT (left, top), Δhrd1 (middle), and Δdoa10 strains (bottom) and their quantitations (right, graph). In all cases quantitations reflect the sum of all RTA bands in each lane (graphs).
Retrotranslocation of Native RTA Does Not Depend on an Active Hrd1p or on Lysyl Ubiquitylation
Because the Hrd1 complex is required, we examined the importance of the catalytic activity of this E3 ubiquitin ligase for RTA dislocation (Figure 3). Expression of either wt Hrd1p or the catalytically inactive Hrd1C399S (Bordallo and Wolf, 1999) in a Δhrd1 strain restored the toxicity of RTA, resulting in an inhibition of growth (Figure 3A). That Hrd1C399S lacked catalytic activity was also confirmed by its ability to stabilize CPY* (Figure 3B). The dislocation of RTA must therefore require structural features of Hrd1p rather than its catalytic activity. We further show that dislocation of the native toxin does not involve ubiquitylation of lysyl residues because changing one or both endogenous lysines to arginine does not alter the requirement for Hrd1p (Figure 3C). Eventual fate in the cytosol is also independent of canonical ubiquitylation because the turnover of native RTA is identical to that of RTA(0K) (Figure 3D). We conclude that both RTA variants serve as ERAD-L substrates in a process that, for the native toxin, is independent of its lysine content and does not require the catalytic activity of Hrd1p.
Figure 3.

RTA dislocation and fate are not mediated by ubiquitylation of lysyl residues. (A) Top, viabilities of WT and Δhrd1 strains expressing RTA on noninducing (glucose) and inducing (galactose) media. Lower panel: viabilities of WT and Δhrd1 strains expressing RTA after complementation, as appropriate, with wild type (WT) or catalytically inactive Ubc7p or Hrd1p on noninducing (glucose) and inducing (galactose) media. (B) Imunoprecipitates of pulse-chase samples of CPY* in the Open BioSystems CPY knockout strain (PRC1) overexpressing either wild-type HRD1 (top) or Hrd3C399S (middle), and associated quantitations (bottom, graph). (C) Viabilities of WT and the indicated mutant strains expressing RTA variants lacking one (RTA(1K)) or both (RTA(0K)) endogenous lysyl residues on noninducing (glucose) and inducing (galactose) media. (D) RTA fate is independent of lysine content. Typical immunoprecipitates of pulse-chase samples of RTA (top) and RTA(0K) (middle) and their quantitations (bottom, graph); n = 2.
Access to the Golgi Is Essential for Dislocation of Both Forms of RTA
Active ER–Golgi transport is required for a number of yeast ERAD substrates (Caldwell et al., 2001; Vashist et al., 2001; Haynes et al., 2002; Vashist and Ng, 2004). We therefore tested the effects of the fungal metabolite BFA that fuses endosomal compartments and merges the Golgi and ER, reversibly blocking early but not late protein transport steps in the yeast secretory pathway (Graham et al., 1993). In the drug-sensitive strain JN284 (Vogel et al., 1993), BFA treatment stabilized both RTA and RTAΔ, compared with cells treated only with the BFA vehicle ethanol (Figure 4A).
Figure 4.

RTA dislocation requires Golgi transport. (A) Pulse-chase kinetics of the indicated RTAs in the drug sensitive yeast strain JN284. Cells treated with solvent alone (top; EtOH, ethanol) and cells treated with 300 μM BFA dissolved in EtOH (middle) and their quantitations of the amount of immunoprecipitated RTA as measured by densitometry and expressed as a percentage of the total RTA present at the end of the pulse (bottom, graph). (B) Pulse-chase of RTAΔ in a sec12 mutant. Top, permissive temperature; bottom, restrictive temperature. (C) Pulse-labeled RTA (top) in sec12 (middle) and sec23 strains (bottom). (D) Top, viabilities of wild-type (WT) and Δerp2 cells expressing RTA on inducing medium (galactose); middle, viabilities of WT and Δerp2 strains expressing RTA after complementation, as appropriate, with vector or wt Erp2p on inducing (galactose-Ura-Leu); and bottom, viabilities of WT DHY9 and p24Δ8 yeasts expressing RTA on inducing (galactose) medium. (E) Pulse-chase of RTA in WT strain (top) and in Δerp2 (middle) and quantitations as described for A (bottom; n = 4 or 5 ±1 SD). (F) Pulse-chase of RTAΔ in wild-type strain (top) and in Δerp2 cells (bottom).
ER-to-Golgi transport requires COPII vesicles. In a mutant containing a temperature-sensitive lesion in Sec12p, a guanine nucleotide exchange factor of the COPII-associated GTPase Sar1p (Barlowe and Schekman, 1993), RTAΔ was stabilized at the restrictive temperature (37°C, Figure 4B), but not at the permissive temperature (24°C). Structurally native RTA was also stabilized at the restrictive temperature (Figure 4C) but not at the permissive temperature (24°C, not shown). Likewise, in a strain bearing a temperature-sensitive lesion in Sec23p, a GTPase-activating protein that stimulates Sar1p (Duden, 2003), RTA was rendered stable at the restrictive temperature (Figure 4C) but not at the permissive temperature (24°C, not shown).
Finally, for native RTA, we investigated the p24 mediators of ER-to-Golgi trafficking (Springer et al., 2000). A particularly striking growth advantage was seen in cells lacking Erp2p after induction of RTA expression (Figure 4D, top). Individual deletion of each of the other p24 family members (ERP1, ERP3-6, EMP24 and ERV25) and additional proteins localized to COPII-coated ER exit sites did not permit such survival (Table 2). Expression of functional Erp2p protein in the Δerp2 strain restored normal RTA toxicity (Figure 4D, middle). Curiously, deletion of all eight p24 family genes gave only a slight growth advantage compared with the congenic parental strain (Figure 4D, bottom). This is consistent with previous observations showing that individual p24 proteins act as negative regulators of COPII vesicle entry for different substrates (Springer et al., 2000). As expected, pulse-labeled RTA was stabilized in the Δerp2 strain (Figure 4E), as indeed was RTAΔ, pointing to a common requirement for Erp2p function for both these substrates (Figure 4F).
Table 2.
Growth characteristics of the gene deletion library members screened for survival after induced expression of ER–targeted RTA
| Gene deletion |
Growth advantage (fold)a | |
|---|---|---|
| Standard name | Systematic name | |
| Dislocation complex proteins | ||
| CUE1 | YMR264W | 10–100 |
| UBC7 | YMR022W | 10–100 |
| HRD1 | YOL013C | ∼1000 |
| DOA10 | YIL030C | nme |
| HRD3 | YLR207W | ∼1000 |
| USA1 | YML029W | ∼100 |
| DER1 | YBR201W | ∼1000 |
| ER-Golgi transport | ||
| ERP1 | YAR002C-A | nme |
| ERP2 | YAL007C | 10,000 |
| ERP3 | YDL018C | nme |
| ERP4 | YOR016C | nme |
| ERP5 | YHR110W | nme |
| ERP6 | YGL002W | nme |
| EMP24 | YGL200C | nme |
| ERV25 | YML012W | nme |
| ERV29 | YGR284C | nme |
| ERV41 | YML067C | nme |
| ERV46 | YAL042W | nme |
| ERV14 | YGL054C | nme |
| EMP46 | YLR080W | nme |
| EMP47 | YFL048C | nme |
| ER luminal chaperones | ||
| YOS9 | YDR057W | nme |
| MNL1 | YHR204W | nme |
| JEM1 | YJL073W | nme |
| CNE1 | YAL058W | nme |
| Cytosolic chaperones and cochaperones | ||
| HLJ1 | YMR161W | nme |
| ZUO1 | YGR285C | nme |
| APJ1 | YNL077W | nme |
| YDJ1 | YNL064C | nme |
| SSA1 | YAL005C | nme |
| SSA2 | YLL024C | nme |
| SSA3 | YBL075C | nme |
| SSZ1 | YHR064C | nme |
| SSE1 | YPL106C | nme |
| SNL1 | YIL016W | nme |
| STI1 | YOR027W | nme |
| HSC82 | YMR186W | nme |
| HSP82 | YPL240C | nme |
| CPR6 | YLR216C | nme |
| CPR7 | YJR032W | nme |
| SBA1 | YKL117W | nme |
| AHA1 | YDR214W | nme |
| HCH1 | YNL281W | nme |
| Proteasome and associated proteins | ||
| UBR1 | YGR184C | nme |
| RPN10 | YHR200W | nme |
| SEM1 | YDR363W-A | nme |
| UBP6 | YFR010W | nme |
| UMP1 | YBR173C | nme |
| PNG1 | YPL096W | nme |
| RAD23 | YEL037C | nme |
| RAD4 | YER162C | nme |
| DSK2 | YMR276W | nme |
| Cdc48p and its binding proteins | ||
| NPL4 | YBR170C | nme |
| DOA1 | YKL213C | nme |
| OTU1 | YFL044C | nme |
| SHP1 | YBL058W | nme |
| UBX2 | YML013W | ∼10 |
| Signal peptidase complex | ||
| SPC1 | YJR010C-A | nme |
| SPC2 | YML055W | nme |
a nme, no measurable effect.
Role of Luminal and Cytosolic Chaperones
ERAD of many misfolded proteins is aided by DnaJ domain–containing Hsp40 cochaperones that interact with the ER luminal chaperone Kar2p/BiP, improving their solubility in the ER lumen (Nishikawa et al., 2001). One way of demonstrating indirectly any role of Kar2p in RTA retrotranslocation is to examine single and double knockouts of Kar2p-interacting Hsp40s, by viability testing. The single deletion Hsp40 Δjem1 mutant was as sensitive to RTA expression as wild-type cells (Figure 2A, Table 2). However, upon extended growth (7 d), a Δscj1Δjem1 double knockout showed slightly increased survival (not shown). This suggests that these Kar2p-interacting Hsp40s in conjunction are required for optimal toxicity, confirming similar findings for the viral A/B toxin K28 (Heiligenstein et al., 2006). In the context of Δjem1 this requirement was not obvious presumably because Scj1p could largely substitute for the other Hsp40. In a strain missing the lectin chaperone Yos9p (Figure 2A), which also interacts with Kar2p (Denic et al., 2006), or Mnl1p, an ER mannosidase implicated in glycoprotein destruction (Table 2), RTA remained able to retrotranslocate, inhibiting protein synthesis and therefore cell growth to the same extent as observed in the congenic strain. We conclude that although RTA requires multiple Kar2p cochaperones for optimal retrotranslocation, it does not show an absolute requirement for proteins typically regarded as part of the ER glycoprotein surveillance machineries.
In principle, acquisition of a catalytic conformation by dislocated cytosolic RTA could be accomplished by substrate (ribosome)-mediated refolding (Argent et al., 2000), by spontaneous refolding (Rodighiero et al., 2002) or by interactions with cytosolic components such as chaperones (Spooner et al., 2008). Because chaperones can also provide routes to the proteasome (Arndt et al., 2007), knockout of these proteins and/or their cochaperones might alter the sensitivity of the yeast cells expressing toxin. We therefore examined members of the gene knockout library lacking cytosolic chaperones or their cochaperones for survival after RTA expression. No clear growth advantage was seen for cells lacking individual Hsp40, Hsp70, and Hsp90 family members or Hsp70 and Hsp90 cochaperones (Table 2). However, as for ER chaperones, redundancy of cytosolic chaperones might obscure their roles when examined in the context of single knockouts.
Cdc48p Is Required for the ERAD of Misfolded RTAΔ But Not for Native RTA
Cdc48p functions in a cofactor dependent manner in the recruitment of polyubiquitylated proteins that are destined for degradation. Because it acts between ubiquitylation and proteasomal degradation it is generally assumed that different ERAD pathways merge here (Ye et al., 2001, 2003; Bar-Nun, 2005). Because loss of CDC48 is lethal, we examined the stability of the RTA variants when expressed in conditional cdc48 mutants: the cold-sensitive cdc48-1 and the temperature-sensitive cdc48-3 (Ye et al., 2001; Jarosch et al., 2002b). Predictably, RTAΔ showed a strong requirement for Cdc48p, being stabilized at 23°C in the cold-sensitive strain and at 37°C in the temperature-sensitive strain (Figure 5A). However, no obvious Cdc48p dependence was seen in the case of native RTA (Figure 5B). This surprising finding is in contrast to the Cdc48p requirement observed when RTA is retrotranslocated from the ER of tobacco cells (Marshall et al., 2008). In plant cells, the ERAD pathway remains poorly defined although a number of plant-specific features have been recognized (Di Cola et al., 2005). We speculate that a Cdc48p requirement in plants may be needed in tissues where ribosome inactivating proteins are normally made in order to ensure a tighter coupling of extraction and degradation to prevent autointoxication of ribosomes. Tobacco leaves are known to express a RIP (termed TRIP; Sharma et al., 2004).
Figure 5.

Cdc48p and its cofactor Npl4 are required for dislocation of misfolded RTA but not for native RTA. (A) Pulse-chase analyses at the restrictive temperatures of cold-sensitive cdc48-1 and temperature-sensitive cdc48-3 strains. Immunoprecipitates of misfolded RTAΔ (A) or RTA (B) are shown. (A and B) Bottom panels, quantitations of the radiolabeled protein remaining at each time point and expressed as a percentage of the RTA present at the end of the pulse; n = 2 or 3; bars, ±1 SD. (C) Pulse-chase analysis of misfolded RTAΔ and RTA in WT and in Δnpl4. Bottom panels, quantitations as described for A and B. (D) Viabilities of wild-type (WT) and Δnpl4 expressing RTA grown on noninducing (glucose, left) and inducing (galactose, right) medium. (E) Expression of RTA and RTAΔ was induced for 2 h in the presence and absence of cLβ-1. RTA variants were immunoprecipitated (IP) from cell extracts prepared in the presence of 10 mM N-ethyl maleimide. Proteins were identified by immunoblot (IB) for ubiquitin (left) and RTA variant (right). RbIgH, rabbit immunoglobulin heavy chain.
Npl4p is a Cdc48p cofactor that, along with its partner Ufd1p, recognizes polyubiquitylated ERAD substrates. RTAΔ was partially stabilized in a Δnpl4 strain, whereas native RTA was not (Figure 5C). Furthermore, we saw no growth advantage in Δnpl4 cells expressing native RTA (Figure 5D). Thus in yeast, polyubiquitylation may not be important for the dislocation of native RTA, a view that is consistent with our earlier observations that the ubiquitin ligase activity of Hrd1p is not required for this protein and that RTA variants lacking lysyl residues still retrotranslocate and disappear (Figure 3). It is further apparent that, although a tiny proportion of the native RTA can be trapped as monoubiquitylated g0 and g1 species in the presence of the proteasome inhibitor cLβ-l, polyubiquitylated intermediates cannot be detected (Figure 5E). In contrast there is evidence of some polyubiquitylated RTAΔ (Figure 5E), findings that are compatible with the Cdc48 complex–mediated dislocation of this misfolded substrate observed here (Figure 5, A and C) and previously (Kim et al., 2006).
The Proteasome Determines the Fate of the RTA Variants
Although efficient degradation of RTAΔ requires the Png1p–Rad23p proteasomal receptor complex (Kim et al., 2006), deletion of PNG1 or RAD23 did not confer growth advantage to cells expressing RTA (Figure 2A). This suggests that the native toxin can evade this proteasomal receptor complex, pointing perhaps to different proteasomal interactions of RTA and RTAΔ. To investigate this, we examined the RTA variants in yeast carrying defined mutations (Rubin et al., 1998) in the Walker motifs of the six AAA-ATPase Rpt subunits of the 19S proteasomal regulatory particle (RP).
Degradation of RTAΔ showed a marked requirement for Rpt2p ATPase activity, but not for the ATPase activities of the other Rpt subunits (Figure 6A). The glycosylated forms of RTAΔ disappeared selectively over time, suggesting exposure to cytosolic peptide:N-glycanase (Png1p). Because Rpt2p allows access of proteasomal substrates to the axial core of the proteasome (Kohler et al., 2001), where the proteolytic activities reside, this suggests that RTAΔ is normally degraded by the proteasome. In support of this, the disappearance of RTAΔ was significantly retarded after treatment of the drug-sensitive JN284 strain (Gaber et al., 1989) with the proteasomal inhibitor clasto-lactacystin β-lactone (Figure 6C). It was also stabilized in a pre1-1 mutant (Figure 6D) in which the core β4 catalytic subunit of the proteasome is defective (Heinemeyer et al., 1991).
Figure 6.

Native RTA avoids proteasomal degradation. Top panels, pulse-chase of RTAΔ (A) and RTA (B) in WT (SUB62) and rpt1-6 mutant strains. Bottom panels, overlapping error bars have been omitted for clarity. (C and D) Pulse-chase analysis of RTAΔ and RTA from JN284 cells treated with DMSO (vehicle) or DMSO/lactone cLβ-1 (C) and pulse-chase analysis of CPY*, RTAΔ, and RTA in a pre1-1 proteasomal core mutant strain and corresponding WT (cl3-ABYS-86) (D). Bottom panels in C as in A and B.
By contrast, native RTA dislocates in a process involving the 19S proteasome cap protein Rpt4p (Figure 6B), which can extract some ERAD substrates from the ER (Lipson et al., 2008). However, it shows no obvious requirement for the other Rpt subunits, Ubr1p (the N-recognin E3 ligase associated with Rpt6p; Xia et al., 2008; Figure 1A) or the proteasome core itself (Figure 6, C and D). Native RTA was not selectively deglycosylated (Figure 6B), consistent with our observation that a PNG1 deletion had no discernable effect on the native protein (Figure 2A). Finally, RTA degradation is independent of ubiquitylation because RTA and RTA(0K) exhibit identical kinetics of turnover in pulse-chase analyses (Figure 3D). It remains unclear what protease or pathway removes the bulk of this protein in the cytosol. Nevertheless, it is evident that at least a fraction of dislocated RTA can recover ribosome-inactivating ability because growth is severely affected upon toxin expression in the ER (Figure 2A).
DISCUSSION
Through the use of a relatively small number of luminal ERAD substrates a map of ERAD-L components has emerged (Carvalho et al., 2006). Glycoprotein substrate recognition by these pathways is currently being unraveled by defining critical glycan structures in positions of the substrate where overall protein folding and stability can be monitored (Spear and Ng, 2005; Xie et al., 2009). However, it remains unclear how a common substrate that presents ERAD-detectable motifs in more than one location is handled. We have therefore studied the fate of a newly made native and misfolded toxin in order to highlight similarities and differences in their handling by ERAD constituents. Such a study also probes the requirements for the uncoupling that must occur before reactivation of native toxin in the cytosol. Our data reveal that the yeast ERAD system can discriminate between two subtly different forms of this substrate, showing that although the steps in preparation for retrotranslocation are generally similar, the downstream cytosolic stages are strikingly different (Figure 7).
Figure 7.

Interactions of RTA with components of the ERAD pathway. RTA variants (1), require ongoing transport to and from the Golgi (2), before recognition by the Hrd1p–Hrd3p–Der1p membrane-bound dislocation complex (3). Their fates then differ. Although a proportion of RTA requires Rpt4p functions for cytosolic activation (4) (the rest being degraded in a nonproteasomal manner), RTAΔ is directed to the proteasome core by the Cdc48p complex, the proteasomal receptor Rad23p–Png1p complex and the proteasomal cap component Rpt2p (5).
RTA inhibits yeast cell growth dramatically (Allen et al., 2007) through the catalyzed depurination of the sarcin-ricin domain of 26S rRNA that results in ribosome inactivation and the inhibition of protein synthesis (Endo et al., 1987). By monitoring survival of otherwise sensitive yeast cells, and/or by analyzing acquired biochemical stability of the toxin substrate, we tested a range of strains lacking or carrying defects in many known ERAD components. RTA was also expressed in a form that is unable to acquire its native structure, either before or after dislocation to the cytosol (Simpson et al., 1999). The fate of this RTAΔ was monitored by following its kinetics of degradation in selected strains of yeast.
For dislocation to the cytosol, both forms of RTA need ongoing traffic to the Golgi and they show a specific requirement for Erp2p, a protein of the p24 family that has a poorly understood role in bidirectional transport (cycling) between the ER and Golgi. Other Δerp strains, including the p24Δ8 that lacks all eight p24 genes (Springer et al., 2000), exhibit severe growth inhibition when expressing ER-targeted RTA. The p24 proteins are proposed to act as negative regulators of COPII vesicle entry for different substrates (Springer et al., 2000), where individual p24-COPII complexes preclude entry of specific cargoes into transport vesicles unless they display high affinity for budding components. It follows, however, that in the absence of all the members of this family, RTA would have unfettered access to budding vesicles and may recycle by default. A similar dependency on COPII-associated proteins has been reported for the Erv29p-dependent ER–Golgi transport of CPY* (Haynes et al., 2002), suggesting that it is the cycling of the substrate that is important. As with other examples of ERAD substrates that cycle at the ER–Golgi interface (Caldwell et al., 2001; Vashist et al., 2001; Haynes et al., 2002; Vashist and Ng, 2004), RTA lacks a C-terminal HDEL sequence so the mechanism of retrieval remains unknown.
Both RTAs show a common requirement for the central Hrd1p–Hrd3p–Der1p ubiquitylation complex, supporting the view that the structurally native RTA can pose as an ERAD-L substrate. The requirement for Hrd1p reflects its structural role and is not related to canonical ubiquitylation because it is observed even with a catalytically inactive Hrd1p and when the native toxin lacks all internal lysyl residues. This lends further support to the idea that this Hrd1–Hrd3–Der1 complex may not only serve as a focal point for the recruitment of luminal and cytosolic ERAD components, but that it may constitute the core of a membrane-spanning retrotranslocon (Gauss et al., 2006). The lesser requirement for the membrane anchor Cue1p suggests that a poorly ubiquitylated substrate like RTA might also dislocate through mobile, nonanchored Hrd1–Hrd3p complexes. RTAΔ becomes more heavily glycosylated than RTA, presumably because in the more structurally defective toxin, the two N-glycan sequons have had prolonged exposure to oligosaccharyl transferase. For the weakly glycosylated native RTA, we observed no dependence on calnexin or on the intermediaries that extricate glycosylated substrates for passage to the E3 ligase complex (e.g., the mannosidase-like protein of the ER, Mnl1p). Furthermore, there is no clear role for the Yos9p lectin component of the Hrd1p complex either. It is known that the OS-9 mammalian ortholog of Yos9p can bind to both glycosylated and nonglycosylated ERAD substrates to deliver them to SEL1L (Hrd3p; Christianson et al., 2008; Alcock and Swanton, 2009). OS-9 thereby acts as a kind of gatekeeper to the HRD1 complex in mammalian cells. In the absence of a role for Yos9p in the ERAD of RTA in yeast cells, it remains unclear precisely how this substrate is delivered to the central Hrd1p–Hrd3p complex. Furthermore, the ubiquitin ligase that monoubiquitylates RTA has not been identified and any role for Sec61p is also unresolved at this time.
After convergence at the Hrd1p–Hrd3p–Der1p complex, RTA then requires the 19S proteasome cap subunit Rpt4p that has been associated with extracting CPY* from the ER membrane (Lipson et al., 2008). However, this phase of the toxin ERAD process shows no obvious requirement for other Rpt subunits including Rpt2p (the base subunit of the cap that can open the proteolytic chamber; Kohler et al., 2001) and does not involve Cdc48p, Ubr1p (an N-recognin E3 ligase associated with Rpt6p; Xia et al., 2008), or rather curiously, the proteasome core itself. The effect of rpt4 on the ERAD of native RTA is probably not related to a general unfolded protein response (UPR) effect (Lipson et al., 2008) because this defective RP subunit has a clear consequence on the ERAD of native RTA, but it has no effect on the ERAD of RTAΔ. It is not unreasonable therefore to speculate that in yeast, native RTA is dislocated without a polyubiquitin tag in a process facilitated by a specific base subunit of the proteasome cap.
In the disposal of RTAΔ, an essentially similar substrate save for a short internal deletion that renders it incapable of attaining correct native structure, there is a contrary and very noticeable involvement of Cdc48p, Npl4p, the Png1p–Rad23p proteasomal receptor complex, Rpt2p, and the proteasome core. Such a profile suggests that RTAΔ behaves as a classical ERAD substrate, most likely extracted by the Cdc48 complex as a polyubiquitinated protein that is then deglycosylated in the cytosol by peptide:N-glycanase and degraded in the proteasome after gating interactions with Rpt2p.
The site or number of lesions that lead to recognition by ERAD components clearly influences downstream pathways. RTA presents as an ERAD-L substrate, most likely through structural alterations that involve interaction of a C-terminal hydrophobic patch in this protein with ER lipids (Mayerhofer et al., 2009). In a normal mammalian cellular intoxication this patch is occluded by RTB (Simpson et al., 1995) until revealed by reduction of the holotoxin in the ER (Spooner et al., 2004). However, after dislocation, native RTA is handled very differently to RTAΔ, which contains this same hydrophobic interface together with an internal lesion that affects its folding. We propose that engagement or otherwise with Cdc48p determines subsequent presentation of RTA to the proteasome, whereupon ATPase subunits of the proteasome RP discriminate between the two toxin forms. The ability of native RTA to evade the proteasome core and gain activity shows that the fate of a protein dislocated by Hrd1p-dependent processes is not necessarily destruction. We conclude that interactions with Cdc48p and the manner of toxin presentation to the proteasome RP permit an unexpected divergence in fate on the cytosolic face of the ER membrane that is distinct from the Doa10p scrutiny of cytosolic proteins (Metzger et al., 2008).
ACKNOWLEDGMENTS
We are grateful to Randy Schekman (University of California, Berkeley) for supplying strain RSY188, Mike Lewis (Medical Research Council Laboratory of Molecular Biology, Cambridge) for supplying strains RSY263 and RSY281, and Professor Shoshana Bar-Nun (Tel Aviv University) for supplying rpt strains and for critical reading of this manuscript. S.L. and S.C.H.A. were supported by UK Department of Health and Home Office grants (to L.M.R., G.L., and J.M.L.); R.A.S. was supported by Wellcome Trust Programme Grant 080566/Z/06/Z (to L.M.R. and J.M.L.); and T.S. was supported by Deutsche Forschungsgemeinschaft Grant GRK845 (to M.J.S.). This work was also supported by National Institutes of Health Grant 5U01AI65869-02 (to J.M.L. and L.M.R.); and a UK BBSRC studentship (to C.P.G.).
Abbreviations used:
- BFA
brefeldin A
- cLβ-1
clasto-lactacystin β lactone
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- RP
proteasome regulatory particle
- RTA
ricin A chain
- RTB
ricin B chain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-08-0743) on June 2, 2010.
REFERENCES
- Alcock F., Swanton E. Mammalian OS-9 is upregulated in response to endoplasmic reticulum stress and facilitates ubiquitination of misfolded glycoproteins. J. Mol. Biol. 2009;385:1032–1042. doi: 10.1016/j.jmb.2008.11.045. [DOI] [PubMed] [Google Scholar]
- Allen S., Naim H. Y., Bulleid N. J. Intracellular folding of tissue-type plasminogen activator. Effects of disulfide bond formation on N-linked glycosylation and secretion. J. Biol. Chem. 1995;270:4797–4804. doi: 10.1074/jbc.270.9.4797. [DOI] [PubMed] [Google Scholar]
- Allen S. C., Moore K. A., Marsden C. J., Fulop V., Moffat K. G., Lord J. M., Ladds G., Roberts L. M. The isolation and characterization of temperature-dependent ricin A chain molecules in Saccharomyces cerevisiae. FEBS J. 2007;274:5586–5599. doi: 10.1111/j.1742-4658.2007.06080.x. [DOI] [PubMed] [Google Scholar]
- Argent R. H., Parrott A. M., Day P. J., Roberts L. M., Stockley P. G., Lord J. M., Radford S. E. Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 2000;275:9263–9269. doi: 10.1074/jbc.275.13.9263. [DOI] [PubMed] [Google Scholar]
- Arndt V., Rogon C., Hohfeld J. To be, or not to be—molecular chaperones in protein degradation. Cell Mol. Life Sci. 2007;64:2525–2541. doi: 10.1007/s00018-007-7188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nun S. The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr. Top. Microbiol. Immunol. 2005;300:95–125. doi: 10.1007/3-540-28007-3_5. [DOI] [PubMed] [Google Scholar]
- Barlowe C., Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- Bays N. W., Wilhovsky S. K., Goradia A., Hodgkiss-Harlow K., Hampton R. Y. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol. Biol. Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazirgan O. A., Hampton R. Y. Cue1p is an activator of Ubc7p E2 activity in vitro and in vivo. J. Biol. Chem. 2008;283:12797–12810. doi: 10.1074/jbc.M801122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellisola G., Fracasso G., Ippoliti R., Menestrina G., Rosen A., Solda S., Udali S., Tomazzolli R., Tridente G., Colombatti M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004;67:1721–1731. doi: 10.1016/j.bcp.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Biederer T., Volkwein C., Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Bordallo J., Wolf D. H. A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae. FEBS Lett. 1999;448:244–248. doi: 10.1016/s0014-5793(99)00362-2. [DOI] [PubMed] [Google Scholar]
- Caldwell S. R., Hill K. J., Cooper A. A. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 2001;276:23296–23303. doi: 10.1074/jbc.M102962200. [DOI] [PubMed] [Google Scholar]
- Carroll S. M., Hampton R. Y. Usa1p is required for optimal function and regulation of the Hrd1p ER-associated degradation (ERAD) ubiquitin ligase. J. Biol. Chem. 2010;285:5146–5156. doi: 10.1074/jbc.M109.067876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P., Goder V., Rapoport T. A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chaddock J. A., Roberts L. M. Mutagenesis and kinetic analysis of the active site Glu177 of ricin A-chain. Protein Eng. 1993;6:425–431. doi: 10.1093/protein/6.4.425. [DOI] [PubMed] [Google Scholar]
- Christianson J. C., Shaler T. A., Tyler R. E., Kopito R. R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks E. D., Cook J. P., Day P. J., Smith D. C., Roberts L. M., Lord J. M. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry. 2002;41:3405–3413. doi: 10.1021/bi011580v. [DOI] [PubMed] [Google Scholar]
- Denic V., Quan E. M., Weissman J. S. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Di Cola A., Frigerio L., Lord J. M., Roberts L. M., Ceriotti A. Endoplasmic reticulum-associated degradation of ricin A chain has unique and plant-specific features. Plant Physiol. 2005;137:287–296. doi: 10.1104/pp.104.055434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden R. ER-to-Golgi transport: COP I and COP II function [Review] Mol. Membr. Biol. 2003;20:197–207. doi: 10.1080/0968768031000122548. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y., Shapira I., Rabinovich E., Bar-Nun S. Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplasmic reticulum-bound p97/Cdc48p and proteasome. J. Biol. Chem. 2004;279:3980–3989. doi: 10.1074/jbc.M309938200. [DOI] [PubMed] [Google Scholar]
- Endo Y., Mitsui K., Motizuki M., Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987;262:5908–5912. [PubMed] [Google Scholar]
- Gaber R. F., Copple D. M., Kennedy B. K., Vidal M., Bard M. The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell-cycle-sparking sterol. Mol. Cell. Biol. 1989;9:3447–3456. doi: 10.1128/mcb.9.8.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R., Sommer T., Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain M., Udvardy A., Mann C. S. cerevisiae 26S protease mutants arrest cell division in G2/metaphase. Nature. 1993;366:358–362. doi: 10.1038/366358a0. [DOI] [PubMed] [Google Scholar]
- Gietz R. D., Woods R. A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gillece P., Luz J. M., Lennarz W. J., de La Cruz F. J., Romisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T. R., Scott P. A., Emr S. D. Brefeldin A reversibly blocks early but not late protein transport steps in the yeast secretory pathway. EMBO J. 1993;12:869–877. doi: 10.1002/j.1460-2075.1993.tb05727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. M., Caldwell S., Cooper A. A. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 2002;158:91–101. doi: 10.1083/jcb.200201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B., Read R. J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- Heiligenstein S., Eisfeld K., Sendzik T., Jimenez-Becker N., Breinig F., Schmitt M. J. Retrotranslocation of a viral A/B toxin from the yeast endoplasmic reticulum is independent of ubiquitination and ERAD. EMBO J. 2006;25:4717–4727. doi: 10.1038/sj.emboj.7601350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer W., Kleinschmidt J. A., Saidowsky J., Escher C., Wolf D. H. Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J. 1991;10:555–562. doi: 10.1002/j.1460-2075.1991.tb07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt R., Wolf D. H. Der1p, a protein required for degradation of malfolded soluble proteins of the endoplasmic reticulum: topology and Der1-like proteins. FEMS Yeast Res. 2004;4:721–729. doi: 10.1016/j.femsyr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Jarosch E., Geiss-Friedlander R., Meusser B., Walter J., Sommer T. Protein dislocation from the endoplasmic reticulum—pulling out the suspect. Traffic. 2002a;3:530–536. doi: 10.1034/j.1600-0854.2002.30803.x. [DOI] [PubMed] [Google Scholar]
- Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D. H., Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 2002b;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Kaiser C. A., Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S., Ng D. T. SnapShot: ER-associated protein degradation pathways. Cell. 2007;129:1230. doi: 10.1016/j.cell.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Kim I., Ahn J., Liu C., Tanabe K., Apodaca J., Suzuki T., Rao H. The Png1-Rad23 complex regulates glycoprotein turnover. J. Cell Biol. 2006;172:211–219. doi: 10.1083/jcb.200507149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Mlsna D., Monzingo A. F., Ready M. P., Frankel A., Robertus J. D. Structure of a ricin mutant showing rescue of activity by a noncatalytic residue. Biochemistry. 1992;31:3294–3296. doi: 10.1021/bi00127a035. [DOI] [PubMed] [Google Scholar]
- Knop M., Finger A., Braun T., Hellmuth K., Wolf D. H. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kohler A., Cascio P., Leggett D. S., Woo K. M., Goldberg A. L., Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol. Cell. 2001;7:1143–1152. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- Lipson C., Alalouf G., Bajorek M., Rabinovich E., Atir-Lande A., Glickman M., Bar-Nun S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J. Biol. Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- Lord J. M., Roberts L. M., Lencer W. I. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- Marshall R. S., Jolliffe N. A., Ceriotti A., Snowden C. J., Lord J. M., Frigerio L., Roberts L. M. The role of CDC48 in the retro-translocation of non-ubiquitinated toxin substrates in plant cells. J. Biol. Chem. 2008;283:15869–15877. doi: 10.1074/jbc.M709316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerhofer P. U., Cook J. P., Wahlman J., Pinheiro T. T., Moore K. A., Lord J. M., Johnson A. E., Roberts L. M. Ricin A-chain insertion into ER membranes is triggered by a temperature increase to 37uC. J. Biol. Chem. 2009;284:10232–10242. doi: 10.1074/jbc.M808387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B., Kostova Z., Schaefer A., Wolf D. H. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004;5:692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger M. B., Maurer M. J., Dancy B. M., Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J. Biol. Chem. 2008;283:32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B., Hirsch C., Jarosch E., Sommer T. ERAD: the long road to destruction. Nat. Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K., Brodsky J. L. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S. I., Fewell S. W., Kato Y., Brodsky J. L., Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E., Kerem A., Frohlich K. U., Diamant N., Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodighiero C., Tsai B., Rapoport T. A., Lencer W. I. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouiller I., DeLaBarre B., May A. P., Weis W. I., Brunger A. T., Milligan R. A., Wilson-Kubalek E. M. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat. Struct. Biol. 2002;9:950–957. doi: 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- Rubin D. M., Glickman M. H., Larsen C. N., Dhruvakumar S., Finley D. Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 1998;17:4909–4919. doi: 10.1093/emboj/17.17.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N., Park S. W., Vepachedu R., Barbieri L., Ciani M., Stirpe F., Savary B. J., Vivanco J. M. Isolation and characterization of an RIP (ribosome-inactivating protein)-like protein from tobacco with dual enzymatic activity. Plant Physiol. 2004;134:171–181. doi: 10.1104/pp.103.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J. C., Lord J. M., Roberts L. M. Point mutations in the hydrophobic C-terminal region of ricin A chain indicate that Pro250 plays a key role in membrane translocation. Eur. J. Biochem. FEBS. 1995;232:458–463. doi: 10.1111/j.1432-1033.1995.tb20831.x. [DOI] [PubMed] [Google Scholar]
- Simpson J. C., Roberts L. M., Romisch K., Davey J., Wolf D. H., Lord J. M. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999;459:80–84. doi: 10.1016/s0014-5793(99)01222-3. [DOI] [PubMed] [Google Scholar]
- Spear E. D., Ng D. T. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner R. A., Hart P. J., Cook J. P., Pietroni P., Rogon C., Hohfeld J., Roberts L. M., Lord J. M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2008;105:17408–17413. doi: 10.1073/pnas.0809013105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner R. A., Smith D. C., Easton A. J., Roberts L. M., Lord J. M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006;3:26–35. doi: 10.1186/1743-422X-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner R. A., Watson P. D., Marsden C. J., Smith D. C., Moore K. A., Cook J. P., Lord J. M., Roberts L. M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004;383:285–293. doi: 10.1042/BJ20040742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer S., Chen E., Duden R., Marzioch M., Rowley A., Hamamoto S., Merchant S., Schekman R. The p24 proteins are not essential for vesicular transport in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2000;97:4034–4039. doi: 10.1073/pnas.070044097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C., Hitt R., Park S. H., Deak P. M., Kostova Z., Wolf D. H. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- Vashist S., Kim W., Belden W. J., Spear E. D., Barlowe C., Ng D. T. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 2001;155:355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S., Ng D. T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J. P., Lee J. N., Kirsch D. R., Rose M. D., Sztul E. S. Brefeldin A causes a defect in secretion in Saccharomyces cerevisiae. J. Biol. Chem. 1993;268:3040–3043. [PubMed] [Google Scholar]
- Wesche J., Rapak A., Olsnes S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999;274:34443–34449. doi: 10.1074/jbc.274.48.34443. [DOI] [PubMed] [Google Scholar]
- Wilson J. D., Liu Y., Bentivoglio C. M., Barlowe C. Sel1p/Ubx2p participates in a distinct Cdc48p-dependent endoplasmic reticulum-associated degradation pathway. Traffic. 2006;7:1213–1223. doi: 10.1111/j.1600-0854.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- Xia Z., Webster A., Du F., Piatkov K., Ghislain M., Varshavsky A. Substrate-binding sites of UBR1, the ubiquitin ligase of the N-end rule pathway. J. Biol. Chem. 2008;283:24011–24028. doi: 10.1074/jbc.M802583200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W., Kanehara K., Sayeed A., Ng D. T. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum-associated degradation system. Mol. Biol. Cell. 2009;20:3317–3329. doi: 10.1091/mbc.E09-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Meyer H. H., Rapoport T. A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y., Meyer H. H., Rapoport T. A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]