

A functional laforin–malin complex promotes the ubiquitination of AMP-activated protein kinase (AMPK), a sensor of cellular energy status. The laforin–malin complex promotes the formation of K63-linked ubiquitin chains, which are not involved in proteasome degradation but could regulate the subcellular localization of substrate proteins.
Abstract
Lafora progressive myoclonus epilepsy is a fatal neurodegenerative disorder caused by defects in the function of at least two proteins: laforin, a dual-specificity protein phosphatase, and malin, an E3-ubiquitin ligase. In this study, we report that a functional laforin–malin complex promotes the ubiquitination of AMP-activated protein kinase (AMPK), a serine/threonine protein kinase that acts as a sensor of cellular energy status. This reaction occurs when any of the three AMPK subunits (α, β, and γ) are expressed individually in the cell, and it also occurs on AMPKβ when it is part of a heterotrimeric complex. We also report that the laforin–malin complex promotes the formation of K63-linked ubiquitin chains, which are not involved in proteasome degradation. On the contrary, this modification increases the steady-state levels of at least AMPKβ subunit, possibly because it leads to the accumulation of this protein into inclusion bodies. These results suggest that the modification introduced by the laforin–malin complex could affect the subcellular distribution of AMPKβ subunits.
INTRODUCTION
Lafora progressive myoclonus epilepsy (Lafora disease [LD], OMIM 254780) is a fatal autosomal recessive neurodegenerative disorder characterized by the presence of glycogen-like intracellular inclusions named Lafora bodies (for reviews, see Delgado-Escueta, 2007; Gentry et al., 2009). LD initially manifests during adolescence with generalized tonic-clonic seizures, myoclonus, absences, drop attacks, and visual hallucinations. As the disease proceeds, a rapidly progressive dementia with apraxia, aphasia and visual loss ensues, leading patients to a vegetative state and death, usually within the first decade from onset of the first symptoms (Ganesh et al., 2006; Delgado-Escueta, 2007). Mutations causing LD have been identified in two genes, EPM2A (Minassian et al., 1998; Serratosa et al., 1999) and EPM2B (NHLRC1) (Chan et al., 2003), although there is evidence for a third locus (Chan et al., 2004). EPM2A encodes laforin, a dual-specificity phosphatase with a functional carbohydrate binding domain at the N terminus (Minassian et al., 2000; Wang et al., 2002). EPM2B encodes malin, an E3-ubiquitin ligase with a RING finger domain at the N terminus and six NCL1, HT2A, and LIN-41 domains in the C-terminal region that are involved in protein–protein interactions (Chan et al., 2003; Gentry et al., 2005; Lohi et al., 2005). We and others have recently described that laforin interacts physically with malin and that laforin recruits specific substrates to be ubiquitinated by malin (Gentry et al., 2005; Lohi et al., 2005; Solaz-Fuster et al., 2008). An alternative function of laforin on glycogen homeostasis also has been described (Worby et al., 2006; Tagliabracci et al., 2007). In this case, laforin acts as a phosphatase of complex carbohydrates, and it has been proposed that this function might be necessary for the maintenance of normal cellular glycogen (Gentry et al., 2007; Tagliabracci et al., 2008).
Our group has recently described that the activity of the laforin–malin complex is regulated by the AMP-activated protein kinase (AMPK) (Solaz-Fuster et al., 2008). AMPK is a serine/threonine protein kinase that acts as a sensor of cellular energy status. AMPK is a heterotrimer of three different subunits: α, β, and γ. AMPKα is the catalytic subunit of the AMPK complex; it contains a highly conserved kinase domain (KD) located at the N terminus of the protein, followed by an autoinhibitory domain (Pang et al., 2007) and a C-terminal domain required for interaction with the AMPKβ subunit (Xiao et al., 2007). Two isoforms of the catalytic subunit have been described, namely, α1 and α2. The AMPKγ subunit contains four tandem repeats of a structural module called a CBS motif, described initially in the enzyme cystathionine-β-synthase (Bateman, 1997), involved in AMP binding (Scott et al., 2004; Xiao et al., 2007). Three isoforms of the γ-subunit, named γ1, γ2, and γ3, have been described. Finally, the AMPKβ subunit functions as a scaffold to assemble the α and γ subunits and also may affect the subcellular localization and substrate specificity of the complex. Two isoforms of the β-subunit (β1 and β2) have been described; they differ somewhat in their N-terminal regions, but they interact with the same efficiency with AMPKα and AMPKγ subunits (Gimeno-Alcaniz and Sanz, 2003; Thornton et al., 1998). AMPK activity is regulated by allosteric activation by AMP and by the phosphorylation of Thr172 residue within the catalytic domain of the α subunit by upstream kinases (Sanz, 2008). Because we described that laforin interacts with subunits of the AMPK complex, we decided to analyze whether the laforin–malin complex could promote the ubiquitination of the AMPK subunits, in a similar way to what we described for another substrate of the laforin–malin complex, the type 1 protein phosphatase (PP1) glycogenic targeting subunit protein targeting to glycogen R5 (R5/PTG) (Solaz-Fuster et al., 2008).
Although protein ubiquitination was first described as a mechanism for targeting proteins for rapid proteasomal degradation, in recent years other functions of ubiquitination have been delineated. Ubiquitination occurs by the addition of ubiquitin monomers to a lysine side chain of a target protein by a process involving three different enzymes: E1-ubiquitin–activating, E2-ubiquitin—conjugating, and E3-ubiquitin ligase enzymes. This reaction either results in the attachment of a single ubiquitin moiety (monoubiquitination) or in the subsequent addition of further molecules of ubiquitin on the first ubiquitin (polyubiquitination). In the latter case, new ubiquitin moieties are linked to previous moieties by using internal lysines of ubiquitin. It is now becoming clear that the basis for the multiple functions of ubiquitination (proteolytic and nonproteolytic) relies in part on the type of linkages in the polyubiquitin chains. Among the seven lysines present in the ubiquitin molecule, K48- and K63-linked polyubiquitin chains are the most frequent modifications detected so far. Although K48-linked ubiquitin chains generally target proteins for proteasomal degradation, K63-linked ubiquitin chains may be involved in other functions mostly not connected with protein degradation (Ikeda and Dikic, 2008; Deshaies and Joazeiro, 2009).
In this article, we report that laforin and malin form a functional complex with E3-ubiquitin ligase activity that is able to ubiquitinate the three AMPK subunits (α, β, and γ) when they are expressed individually in the cells, and also the AMPKβ subunit when it is part of the endogenous heterotrimeric complex. The laforin–malin complex introduces K63-linked ubiquitin chains into proteins, mediating functions that are different from targeting them to the proteasome for degradation.
MATERIALS AND METHODS
Plasmids
Plasmid pCMVmyc-AMPKα2 KD was constructed by amplifying by polymerase chain reaction (PCR), using oligonucleotides α2-11 and α2-2 (Table 1) and plasmid pCMVmyc-AMPKα2 as template, a fragment containing the kinase domain (residues 1–312) of AMPKα2. The fragment was digested with EcoRI and SalI and subcloned into plasmid pCMVmyc (BD Biosciences, San Jose, CA). Similarly, a fragment containing the regulatory domain of AMPKα2 (residues 313–552) was amplified by PCR using oligonucleotides α2-RD and α2-4 (Table 1), digested with EcoRI and SalI, and subcloned into plasmid pCMVmyc. Plasmid pCMVmyc-AMPKβ1 was obtained by digesting plasmid pACT2-AMPKβ1 (Gimeno-Alcaniz and Sanz, 2003) with SfiI and BglII and subcloning the resulting fragment into pCMVmyc. To construct plasmid pCMVmyc-AMPKβ2 glycogen binding domain (GBD), oligonucleotides β2-7 and β2-6 (Table 1) were used to amplify by PCR an N-terminal fragment of AMPKβ2 (residues 1–185) containing the GBD, using pCMVmycAMPKβ2 as template; the fragment was digested with EcoRI and XhoI and subcloned into plasmid pCMVmyc. Similarly, a fragment containing the C-terminal part of AMPKβ2 (residues 186–271) was amplified by PCR using oligonucleotides β2-9 and β2-2 (Table 1), digested with EcoRI and SalI, and subcloned into plasmid pCMVmyc to obtain plasmid pCMVmyc-AMPKβ2 ASC. Finally, plasmid pcDNA4/His-AMPKβ2 was constructed by digesting plasmid pCMVmyc-AMPKβ2 with BamHI and subcloning the resulting fragment into plasmid pcDNA4/HisMaxA (Invitrogen, Carlsbad, CA).
Table 1.
Oligonucleotides used in this study (new restriction sites are underlined)
| Name | Sequence |
|---|---|
| α2-2 | 5′-GCGGGTCGACTTAGTATAAACTGTTCATCACTTCTGATTC-3′ |
| α2-4 | 5′-GCCGGTCGACTTTCAACGGGCTAAAGCAGTGATAAG-3′ |
| α2-11 | 5′-CTTCGAATTCGAATGGCTGAGAAGCAGAAGCACG-3′ |
| α2-RD | 5′-GGGCGAATTCGGAGTGGTGACCCTCAAGACCAGC-3′ |
| β2-2 | 5′-CTCGGTCGACTTCAAATGGGCTTGTATAGCAGA-3′ |
| β2-6 | 5′-CGAGCTCGAGAGGGGTGAGCTGGAAAGGTCTCT-3′ |
| β2-7 | 5′-ACTGGAATTCGAATGGGAAACACCACCAGCGAC-3′ |
| β2-9 | 5′-ACTCGAATTCCAGGGCCTTATGGTCAAGAA-3′ |
| CIDEA-1 | 5′-ACTGGAATTCGAATGGAGGCCGCCCGGGACTATGC-3′ |
| CIDEA-2 | 5′-CGAGCTCGAGAGTCCACACGTGAACCTGCCCTTGG-3′ |
Plasmid pFLAG-CIDEA was obtained in several steps. First, CIDEA cDNA was amplified by PCR from a human skeletal muscle cDNA library (Clontech, Mountain View, CA) using oligonucleotides CIDEA-1 and CIDEA-2 (Table 1). The fragment was digested with EcoRI and XhoI and subcloned into plasmid pFLAG-CMV (Sigma-Aldrich, St. Louis, MO).
All plasmids containing a fragment obtained by PCR were sequenced to confirm that the Taq polymerase had not included undesired mutations. Other plasmids used in this study were pCMV-AMPKα1, pCMVmyc-AMPKα2, pCMVmyc-AMPKβ2, and pCMVmyc-AMPKγ1 (Solaz-Fuster et al., 2006); pcDNA3-HA-malin, pCMV-HA-laforin, and pCMV-myc-R5/PTG (Solaz-Fuster et al., 2008); pCMV-His6xUbiq (from Dr. M. Rodriguez, Proteomics Unit, CIC-BioGUNE, Vizcaya, Spain); pCMV-His6xUbiq K48R and pCMV-His6xUbiq K63R (from Dr. Christine Blattner, Institute of Toxicology and Genetics, Karlsruhe Institute of Technology, Karlsruhe, Germany); pRCc/CMV-p53 (from Dr. A. Dickmanns, University of Göttingen, Göttingen, Germany); and pCMV-Mdm2 (Worby et al., 2008) (from Dr. M. Gentry, University of Kentucky, Lexington, KY).
Analysis of In Vivo Ubiquitination
To study ubiquitination in intact cells, human embryonic kidney (HEK)293 cells were transfected with plasmids pCMV-His6xUbiq (encoding a modified ubiquitin, tagged with 6 His residues); pCMVmyc plasmids encoding the protein of interest; and, when indicated, pcDNA3-HA-malin and pCMV-HA-laforin plasmids, by using the Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's instructions. After 36 h of transfection, cells were lysed in buffer A (6 M guanidinium-HCl, 0.1 M sodium phosphate, and 0.1 M Tris-HCl, pH 8.0). Four milligrams of protein of a clarified extract (CE; 12,000 × g for 15 min) was incubated in 100 μl of TALON column (Clontech) in the presence of 10 mM imidazole, for 3 h at room temperature on a rocking platform, to purify His-tagged proteins. The column was then successively washed with 2 ml each of buffer B (buffer A plus 10 mM imidazole), buffer C (buffer B but with 8 M urea instead of 6 M guanidinium-HCl), and four more times with buffer C adjusted to pH 6.0. Bound proteins (bound) were eluted with 50 μl of 2× Laemmli's sample buffer and analyzed by Western blotting using appropriated antibodies. When indicated, plasmids pCMV-His6xUbiq K48R and pCMV-His6xUbiq K63R were used in the assay instead of pCMV-His6xUbiq.
Immunoblotting
Forty micrograms of total protein from the clarified extracts prepared as described above were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting using appropriate antibodies: anti-myc (Sigma-Aldrich); anti-p53 (Exbio, Vestec Czech Republic); anti-AMPKα total (Cell Signaling Technology, Danvers, MA); anti-AMPKβ total (Cell Signaling Technology); anti-panAMPKβ, anti-AMPKγ1 (Cell Signaling Technology); anti-α-tubulin (Sigma-Aldrich); anti-ubiquitin conjugates (BIOMOL Research Laboratories, Plymouth Meeting, PA); and anti-K48 ubiquitin chain and anti-K63 ubiquitin chain conjugates (a generous gift of Genentech, South San Francisco, CA) (Newton et al., 2008).
Analysis of the Degradation Rates of Free AMPK Subunits
HEK293 cells were transfected with the appropriated combination of plasmids. Twenty-four hours after transfection, cells were treated with 355 μM cycloheximide (CHX), and, at the indicated times (from 0 to 12 h), aliquots were taken from the cultures and cell extracts (25 μg) were analyzed by SDS-PAGE and Western blotting using anti-myc antibodies. The same extracts were analyzed using anti-tubulin antibodies as a loading control. Western blots were analyzed by densitometry using an LAS-3000 electronic reader (Fujifilm, Tokyo, Japan) and the Image Gauge, version 4.0 software (Fujifilm). The levels of the corresponding myc-AMPK subunit with respect to the levels of tubulin at each time point are expressed as a percentage of the values at time 0.
Immunofluorescence and Confocal Microscopy
HEK293 cells transfected with the appropriate plasmids were grown on plates containing coverslips. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, and then the fixation was stopped with 10 mM glycine for 5 min. Cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min. Samples were washed three times with PBS and blocked with 10% fetal bovine serum, 5% nonfat dried milk, and 0.1% Triton X-100 in PBS for 5 h at 4°C. Samples were then incubated overnight at 4°C with a 1/200 dilution of anti-AMPKβ total (Cell Signaling Technology) with or without anti-laforin (1/2000) in blocking solution. Samples were washed five times with PBS during 1 h at room temperature and incubated again with a 1/500 dilution of appropriated fluorescent-labeled secondary antibodies (anti-rabbit Alexa-Fluor 488 and anti-mouse Texas Red; Invitrogen). Nuclei were stained with a 1/500 dilution of a Tropro3 fluorescent probe in PBS for 10 min. Finally, samples were washed again with PBS and mounted on slices using Fluoromount G. Images were acquired for 2 μm slices with a TCS/SP2 confocal microscope (Leica, Wetzlar, Germany) using a 63× oil immersion objective. Images were treated with the ImageJ 1.43c software (Wayne Rasband, National Institutes of Health, Bethesda, MD). More than 100 green cells (therefore containing AMPKβ) were analyzed for each condition and used to determine the presence or absence or large intracellular inclusions. Because in cotransfection experiments some of the green cells may not contain the rest of the plasmids, the quantification of cells containing large inclusions could be underestimated because in this case the cells were considered as punctuated pattern of AMPKβ.
Statistical Analyses
Values are given as means ± SD of at least three independent experiments. Differences between groups were analyzed by two-tailed Student's t tests. The significance has been considered at *p < 0.05 and **p < 0.01, as indicated in each case.
RESULTS
The Laforin–Malin Complex Promotes the Ubiquitination of AMPK Subunits
We have recently described that the activity of the laforin–malin complex is modulated by the AMPK complex and that AMPK interacted physically with laforin (Solaz-Fuster et al., 2008). Because we also described that the laforin–malin complex interacted with the PP1 glycogenic-targeting subunit R5/PTG and promoted its ubiquitination (Solaz-Fuster et al., 2008), we decided to analyze whether the laforin–malin complex was also able to promote the ubiquitination of the AMPK subunits. With this aim, we set up an assay for the analysis of ubiquitinated proteins based on the expression in mammalian HEK293 cells of a 6xHis-tagged version of ubiquitin (Kaiser and Tagwerker, 2005). Cell extracts were made in the presence of the chaotropic agent guanidinium chloride, to inhibit deubiquitinating enzymes, and ubiquitinated proteins were purified by metal-affinity chromatography. This purified fraction (bound material) was analyzed by SDS-PAGE and Western blotting, using specific antibodies against the protein of interest (Figure 1A). We then coexpressed in these HEK293 cells individual AMPK subunits and laforin, malin, or a combination of laforin and malin. As shown in Figure 1B, we observed an efficient ubiquitination of the three individual AMPK subunits tested, i.e., AMPKα2, -β2, and -γ1 but only when we coexpressed these subunits with a combination of laforin and malin. For AMPKα2 and AMPKγ1, we also detected a minor modification when only laforin was overexpressed, perhaps because it can force endogenous malin to carry out the corresponding ubiquitination. These results confirmed our previous observation that both laforin and malin are needed to form a functional complex, in which laforin recruits putative substrates to be modified by the E3-ubiquitin ligase activity of malin (Solaz-Fuster et al., 2008).
Figure 1.

The laforin–malin complex is able to promote ubiquitination of individual AMPK subunits. (A) Diagram of the protocol used to determine the presence of ubiquitinated proteins, based on the use of a 6xHis-tagged version of ubiquitin (Kaiser and Tagwerker, 2005). (B) HEK293 cells were transfected with plasmid pCMV-His6xUbiq and the indicated combination of plasmids (pLaforin, pCMV-HA-laforin; and pMalin, pcDNA3-HA-malin). Cell extracts were then obtained as described in Materials and Methods and the clarified extract (CE; 40 μg), and the material bound to the metal-affinity chromatography column (bound; 40 μl) was analyzed by SDS-PAGE and Western blotting using anti-myc antibodies. Molecular mass markers are indicated on the left of each panel. For AMPKα2 and AMPKγ1, we also detected a minor modification when only laforin was overexpressed, perhaps because it can force endogenous malin to carry out the corresponding ubiquitination. (C) HEK293 cells were transfected with plasmid pCMV-His6xUbiq and the indicated combination of plasmids (Mdm2, pCMV-Mdm2; Laf/Mal, pCMV-HA-laforin/pcDNA3-HA-malin; and empty, pCMV-HA). Cell extracts were analyzed as described above using anti-myc antibodies. (D) HEK293 cells were transfected with plasmid pCMV-His6xUbiq and plasmids pCMV-p53 and pCMV-Mdm2 or pCMV-HA (empty). Cell extracts were analyzed as described above using anti-p53 antibodies.
To check the possibility that the ubiquitination of the AMPK subunits was nonspecific and could be carried out by the overexpression of any E3-ubiquitin ligase, we repeated the experiment expressing Mdm2, another RING E3-ubiquitin ligase involved in the ubiquitination of p53 (Brady et al., 2005). As shown in Figure 1C, compared with the laforin–malin complex, Mdm2 did not carry out an efficient ubiquitination of AMPK subunits, whereas it was active in promoting the ubiquitination of its known substrate p53 (Figure 1D). Together, all these results indicate that the laforin–malin complex can promote the selective ubiquitination of the three individual AMPK subunits tested.
We extended our analysis to other isoforms of AMPK subunits, such as AMPKα1 and AMPKβ1. The laforin–malin complex could also promote the ubiquitination of these subunits (Supplemental Figure S1). We next attempted to map the regions in AMPKα2 involved in the laforin–malin-dependent ubiquitination. We constructed two truncated forms of AMPKα2, one form containing an N-terminal fragment (residues 1–312), comprising the catalytic domain (KD); and another form containing a C-terminal fragment (residues 313–552), comprising the regulatory domain (RD) involved in binding to the AMPKβ and -γ subunits. Previous structural analysis indicated that both forms are able to form a stable fold (Sanz, 2008). However, when we expressed these truncated forms, we did not observe any efficient laforin–malin-dependent ubiquitination (Supplemental Figure S1). These results suggest that both the KD and RD domains of AMPKα2 are needed to form a motif competent to be ubiquitinated by the laforin–malin complex. We also attempted a mapping analysis of the AMPKβ2 subunit. We constructed two truncated forms, one form containing the N-terminal region (residues 1–185), including the GBD; and another form containing the C-terminal region (residues 186–271), containing the so called “association with Snf1 complex domain” (ASC), involved in binding to both AMPKα and AMPKγ subunits. Previous structural analysis indicates that both forms are able to form a stable fold (Sanz, 2008). However, when we expressed these truncated forms, we only detected traces of modified products in the case of the ASC construct, and these were much less abundant than using full-length AMPKβ2 (Supplemental Figure S1). These results suggest that both segments of the AMPKβ2 subunit are required to form a motif competent to be ubiquitinated by the laforin–malin complex.
Next, we checked whether laforin and malin also could ubiquitinate recombinant AMPK subunits when they were forming part of a stable trimeric complex. With this aim, we coexpressed AMPKα2, -β2, and -γ1 in cells also expressing laforin and malin. As shown in Figure 2A, we did not detect any modification of the AMPKα2 and AMPKγ1 subunits, but the AMPKβ2 subunit was still ubiquitinated by the laforin–malin complex, although to a much lesser extent than when it was expressed individually (Figure 2A). We also analyzed the laforin–malin-dependent ubiquitination of the endogenous AMPK complex. When both laforin and malin were expressed, only ubiquitination of the endogenous AMPKβ subunit was detected (Figure 2B). These results suggest, first, that AMPKα and AMPKγ subunits become poor substrates for the laforin–malin-dependent ubiquitination when they are part of a stable heterotrimeric complex; and, second, that AMPKβ subunits are substrates for laforin–malin-dependent ubiquitination even when they are part of a heterotrimeric complex.
Figure 2.

Laforin–malin-dependent ubiquitination of the heterotrimeric AMPK complex. (A) Analysis of overexpressed AMPK complex. HEK293 cells were transfected with plasmid pCMV-His6xUbiq and the indicated combination of plasmids (αβγ, pCMVmyc-AMPKα2 + pCMVmyc-AMPKβ2 + pCMVmyc-AMPKγ1; α2, pCMVmyc-AMPKα2; β2, pCMVmyc-AMPKβ2; γ1, pCMVmyc-AMPKγ1; and pLaf/pMal, pCMV-HA-laforin/pcDNA3-HA-malin). Cell extracts were analyzed as described in Figure 1 by using anti-AMPKα total, anti-AMPKβ total, or anti-AMPKγ1 antibodies. (B) Analysis of endogenous AMPK complex. HEK293 cells were transfected with plasmid pCMV-His6xUbiq and plasmids pCMV-HA-laforin (pLaf) and pcDNA3-HA-malin (pMal), when indicated. Cell extracts were analyzed as described in A but using anti-AMPKα total, anti-panAMPKβ, and anti-AMPKγ1 antibodies to determine the components of the endogenous AMPK complex (*, nonspecific band). Molecular mass markers are indicated on the left of each panel.
To exclude the possibility that the observed modification could occur just by having high concentrations of the laforin–malin complex and substrate proteins, we treated cells with the proteasomal inhibitor MG132, which induces the accumulation of polyubiquitinated proteins. As shown in Figure 3A, inhibition of proteasome activity with MG132 resulted in the accumulation of polyubiquitinated forms of all three individual AMPK subunits tested, i.e., AMPKα2, -β2, and -γ1. Therefore, these individual subunits could be modified by endogenous E3-ubiquitin ligases. We also studied whether the AMPK subunits forming part of the endogenous heterotrimeric complex were modified by ubiquitination. As shown in Figure 3B, treatment with MG132 promoted the accumulation of ubiquitinated AMPKβ subunits but had no effect on AMPKα and AMPKγ subunits. These results suggest that when AMPKα and AMPKγ subunits form part of a heterotrimeric AMPK complex, they are less sensitive to ubiquitination by endogenous E3-ubiquitin ligases. However, the AMPKβ subunits remain sensitive to this type of modification, whether present as free subunits or within heterotrimeric complexes. Because the ubiquitination profile of AMPK subunits by endogenous E3-ubiquitin ligases was similar to the one obtained by the expression of laforin and malin, these results confirmed a positive role of the laforin–malin complex in the ubiquitination of AMPK subunits.
Figure 3.

AMPK subunits accumulate as ubiquitinated proteins upon proteasome inhibition. (A) Analysis of the expression of individual AMPK subunits. HEK293 cells were transfected with plasmid pCMV-His6xUbiq (when indicated) and either pCMVmyc-AMPKα2, pCMVmyc-AMPKβ2, or pCMVmyc-AMPKγ1. Eighteen hours after transfection, cells were treated with or without 50 μM MG132 and incubated 18 h further. Cell extracts were then analyzed as described in Figure 1 by using anti-myc antibodies. (B) Analysis of the subunits of the endogenous AMPK complex. HEK293 cells were transfected only with plasmid pCMV-His6xUbiq (when indicated) and analyzed as described in A but using anti-AMPKα total, anti-panAMPKβ, and anti-AMPKγ1 antibodies to determine the components of the endogenous AMPK complex (*, nonspecific band). Molecular mass markers are indicated on the left of each panel.
Effect of the Laforin–Malin-dependent Ubiquitination on Protein Stability
We next studied the effect of the laforin–malin-dependent ubiquitination on protein stability. With this aim, we expressed individual subunits of AMPK with or without laforin and malin. Cells were treated with cycloheximide to inhibit new protein synthesis, and the degradation rates of the corresponding proteins were measured. In agreement with published results (Crute et al., 1998), the half-lives of the individual AMPK subunits (AMPKα2, -β2, and -γ1) were ≈4–6 h (Figure 4, A–C). Surprisingly, we found that coexpression of laforin and malin did not accelerate the degradation of any of the AMPK subunits tested (Figure 4, A–C). We also analyzed the half-life of the subunits when they were coexpressed (AMPKα2, -β2, and -γ1) and found that laforin and malin did not change the half-lives of these proteins either (data not shown). These results suggest that the laforin–malin-dependent ubiquitination of AMPK subunits does not promote their rapid degradation.
Figure 4.

Degradation rates of individual AMPK subunits in the presence or absence of the laforin–malin complex. HEK293 cells were transfected with plasmid pCMVmyc-AMPKα2 (A), pCMVmyc-AMKβ2 (B), or pCMVmyc-AMPKγ1 (C) without or with a combination of plasmids pCMV-HA-laforin and pcDNA3-HA-malin. Twenty-four hours after transfection, cells were treated with 355 μM CHX, and at the indicated times, aliquots were taken from the cultures and cell extracts (25 μg) were analyzed by SDS-PAGE and Western blotting using anti-myc antibodies. The same blots were analyzed using anti-tubulin antibodies as loading controls. A representative blot is shown in A, B, and C. M, molecular mass standards lane. The right of each panel shows a plot of the levels of the corresponding myc-AMPK subunit with respect to the levels of tubulin of each time point, expressed as a percentage of the value at time 0. Plots are the mean of three different experiments (bars are SD). (D) The laforin–malin complex increases the steady-state levels of AMPKβ subunits. HEK293 cells were cotransfected with plasmid pCMV-GFP, and pCMVmyc-AMPKβ1 or pCMVmyc-AMPKβ2 and a combination or not of plasmids pCMV-HA-laforin and pcDNA3-HA-malin (L/M; empty: pCMV-HA). After 24 h of transfection, cell extracts (30 μg) were analyzed by SDS-PAGE and Western blotting using anti-AMPKβ (top) and anti-GFP (bottom) antibodies. (E) HEK293 cells cotransfected with plasmid pCMVmyc-AMPKβ2 and a combination or not of plasmids pCMV-HA-laforin and pcDNA3-HA-malin (pLaforin + pMalin). After 18 h of transfection, cells were treated for 5 h with either 20 mM ammonium chloride and 100 μM leupeptin (to inhibit lysosomes) or with 5 μM lactacystin (to inhibit proteasomes) or left untreated (Untr). Cell extracts (30 μg) were then analyzed by SDS-PAGE and Western blotting using anti-myc (top) and anti-tubulin (bottom) antibodies. (F) CIDEA and the laforin–malin complex have opposite effects on AMPKβ2 accumulation. HEK293 cells were transfected with plasmid pCMVmyc-AMPKβ2 and with plasmids pFLAG-CIDEA or pCMV-HA-laforin and pcDNA3-HA-malin (Laf/Mal) or with an empty plasmid (pCMV-HA). Then, cell extracts (30 μg) were analyzed by SDS-PAGE and Western blotting using anti-myc (top) and anti-tubulin (bottom) antibodies.
During the course of this study, we noticed that the steady-state levels of AMPKβ subunits were higher in cells coexpressing the laforin–malin complex (Figure 4B). We studied this accumulation in more detail and found, first, that it was not related to enhanced general transcription/translation, because the levels of a coexpressed protein such as green fluorescent protein (GFP), whose expression was driven by the same promoter as the one found in the AMPKβ construct, were unchanged (Figure 4D). Second, in regular cells the steady-state levels of AMPKβ2 subunit increased upon inhibition of either the lysosomal (treatment with ammonium chloride and leupeptin) or proteasomal (treatment with lactacystin) pathways (Figure 4E). However, in the presence of the laforin–malin complex, treatment of the cells with lysosome or proteasome inhibitors did not increase the steady-state levels of the AMPKβ2 subunit (Figure 4E) (similar results were obtained when we analyzed the steady-state levels of the AMPKβ1 subunit; data not shown). These results indicated that the laforin–malin complex promoted the accumulation of AMPKβ subunits in forms that were resistant to their rapid proteolytic turnover.
The action of the laforin–malin complex was therefore contrary to what has been recently described for the action of cell death-inducing DFFA-like effector A (CIDEA) that induced the rapid degradation of AMPKβ subunits (Qi et al., 2008). To confirm these two types of action on the same substrate (AMPKβ2), we analyzed the steady-state levels of AMPKβ2 in cells expressing CIDEA and found, in agreement with a previous report (Qi et al., 2008), that they were much lower than in control cells (Figure 4F). By contrast, the expression of laforin and malin resulted in higher steady-state levels of AMPKβ2 (Figure 4F). These results indicated that the modifications induced by CIDEA and by the laforin–malin complex resulted in opposite effects: whereas CIDEA decreased the stability of AMPKβ2, the laforin–malin complex promoted the long-term accumulation of the protein.
The Laforin–Malin Complex Introduces K63-Ubiquitin Chains into Its Corresponding Substrates
To understand why the laforin–malin-dependent ubiquitination of AMPK subunits did not promote their rapid degradation, we analyzed the topology of ubiquitin chains introduced by this E3-ubiquitin ligase complex. With this aim, we expressed in HEK293 cells forms of ubiquitin that contained either K48R or K63R mutations, to prevent the assembly of the corresponding ubiquitin chains. Because in the ubiquitination of AMPKγ1 the background level was higher than in AMPKα2 and AMPKβ2 (Figure 1C), we focused our attention on the modification that suffered only the last two AMPK subunits. As shown in Figure 5, the laforin–malin complex produced ubiquitinated forms of AMPKα2 and AMPKβ2 in the presence of K48R-ubiquitin (Figure 5, A and B) that were even more abundant than in wild-type ubiquitin, perhaps because of a better efficiency of the laforin–malin-dependent ubiquitination under these conditions. However, AMPKα2 and AMPKβ2 did not result significantly ubiquitinated in the presence of K63R-ubiquitin (Figure 5, A and B), indicating that the laforin–malin complex introduced K63-ubiquitin chains into the AMPK subunits tested. Crude extracts contained similar amounts of total ubiquitinated proteins in all the cases which indicated that the three modified ubiquitins were functional. In addition, control experiments using the E3-ubiquitin ligase Mdm2 and its substrate p53 demonstrated that the K48R- and K63R-ubiquitins yielded modified forms of p53 (Figure 5C). We extended this type of analysis to the ubiquitination of the PP1 glycogenic targeting subunit R5/PTG, another substrate of the laforin–malin complex (Solaz-Fuster et al., 2008), and we found that the complex also promoted the ubiquitination of R5/PTG by the acquisition of K63-linked ubiquitin chains (Figure 5D).
Figure 5.

The laforin–malin complex promotes the formation of K63-linked ubiquitin chains into its corresponding substrates. HEK293 cells were transfected with plasmids pCMVmyc-AMPKα2 (A), pCMVmyc-AMPKβ2 (B), or pCMVmyc-R5/PTG (D), and pCMV-HA-laforin, pcDNA3-HA-malin and either pCMV-His6xUbiq (WT), pCMV-His6xUbiq K48R (K48R), or pCMV-His6xUbiq K63R (K63R). Cell extracts were analyzed as described in Figure 1 using anti-myc antibodies. (C) HEK293 cells were transfected with plasmids pCMV-p53, pCMV-Mdm2, and either pCMV-His6xUbiq (WT), pCMV-His6xUbiq K48R (K48R), or pCMV-His6xUbiq K63R (K63R). Cell extracts were analyzed as described in Figure 1 by using anti-p53 antibodies. Molecular mass markers are indicated on the left of each panel. Total ubiquitinated proteins were analyzed in the crude extracts (40 μg) of each panel by using anti-ubiquitin conjugates antibodies. (E) His-AMPKβ2 is also modified with K63-ubiquitin linkages by the laforin–malin complex. HEK293 cells were transfected with plasmid pcDNA4/His-AMPKβ2 (6xHis-AMPKβ2) with or without plasmids pCMV-HA-laforin and pcDNA3-HA-malin (pLaf/pMal). Cell extracts were prepared as described in Figure 1, and the bound material was analyzed using antibodies against conjugated ubiquitin (anti-ubiq), K48-linked poly-ubiquitin (anti-K48), or K63-linked poly-ubiquitin (anti-K63) chains. CEs (40 μg) also were analyzed using anti-AMPKβ antibodies. (F) Ubiquitination of endogenous AMPKβ subunits. HEK293 cells were transfected with plasmids pCMV-HA-laforin, pcDNA3-HA-malin, and either pCMV-Ubiqx6His (WT), pCMV-Ubiqx6His K48R (K48R), or pCMV-Ubiqx6His K63R (K63R). Cell extracts were analyzed as described in A: the bound material and CE (40 μg) were analyzed using anti-pan AMPKβ antibodies.
To confirm the topology of ubiquitin linkages introduced by the laforin–malin complex we used an alternative strategy based on the use of antibodies that recognize specifically K48- and K63-ubiquitin chains (Newton et al., 2008). We expressed a 6xHis-tagged-AMPKβ2 fusion protein in HEK293 cells with or without laforin and malin, and we purified the fusion protein by metal-affinity chromatography. The purified fractions were then analyzed by Western blotting using an antibody that recognizes total ubiquitin conjugates and antibodies that specifically recognize K48- and K63-polyubiquitin conjugates. As expected, coexpression of laforin and malin induced greater degree of ubiquitination of 6xHis-AMPKβ2 (assessed using a general antiubiquitin conjugates antibody) (Figure 5E). This increase in laforin–malin-mediated ubiquitination was also clearly observed when we used the antibody that recognized K63-ubiquitin chains (anti-K63) but only to a very low extent when we used the antibody that recognized K48-ubiquitin chains (anti-K48).
We also analyzed the topology of ubiquitin chains introduced by the laforin–malin complex on endogenous AMPKβ subunits. We found that it also promoted the acquisition of K63-linked ubiquitin chains (Figure 5F).
We extended our analysis to the ubiquitination that suffered AMPKβ2 subunit by endogenous E3-ubiquitin ligases (Figure 6). We observed that treatment of cells expressing AMPKβ2 subunit with MG132 resulted in an accumulation of polyubiquitinated forms, which were more evident if the cells were expressing K48R-ubiquitins. However, in the presence of K63R-ubiquitins, no polyubiquitinated forms of AMPKβ2 were detected even in the presence of MG132. Crude extracts contained similar amounts of total ubiquitinated proteins in MG132 treated or untreated cells, respectively. These results indicate that endogenous E3-ubiquitin ligases also promote the acquisition of K63-linked ubiquitin chains into AMPKβ2 subunits.
Figure 6.
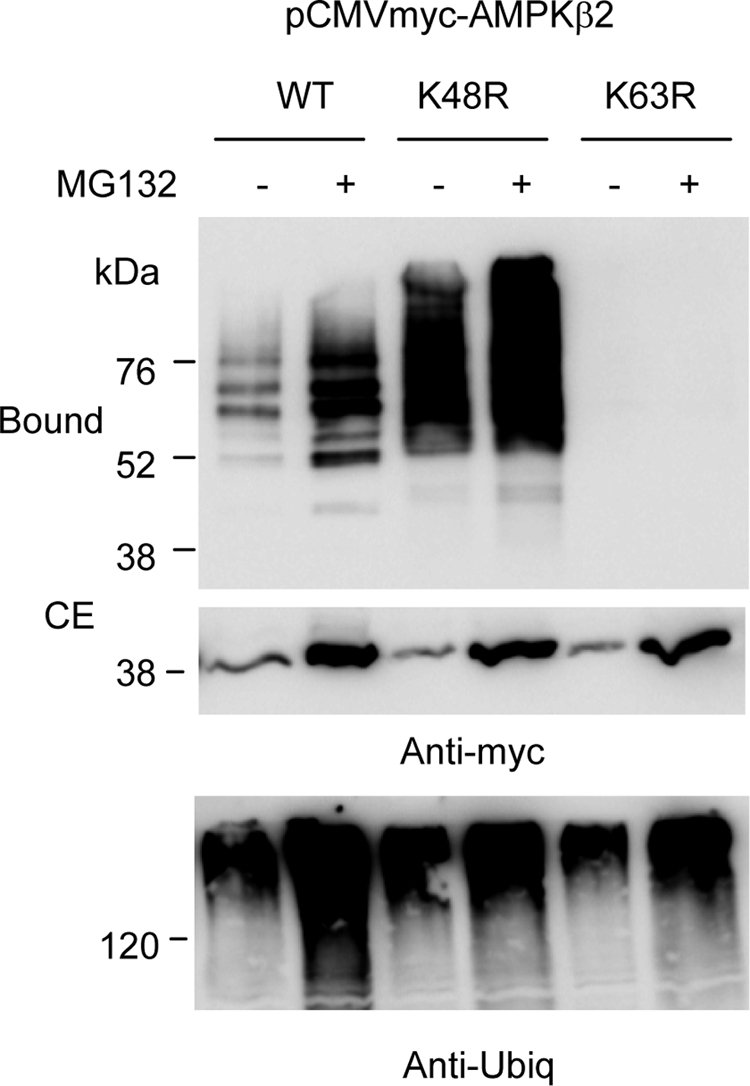
Endogenous E3-ubiquitin ligases also promote the K63-linked ubiquitination of AMPKβ2. HEK293 cells were transfected with plasmid pCMVmyc-AMPKβ2 and either pCMV-His6xUbiq (WT), pCMV-His6xUbiq K48R (K48R), or pCMV-His6xUbiq K63R (K63R). Eighteen hours after transfection, cells were treated with or without 50 μM MG132 and incubated 18 h further. Cell extracts were then analyzed as described in Figure 1 by using anti-myc antibodies. Total ubiquitinated proteins were analyzed in the crude extracts (40 μg) of each panel by using anti-ubiquitin conjugates antibodies.
Effect of the Laforin–Malin-dependent Ubiquitination on AMPK Function and Localization
We analyzed next the effect of the laforin–malin-dependent ubiquitination on the function of the AMPK complex. The activity of the whole AMPK complex was measured by its capacity to phosphorylate acetyl-CoA carboxylase (ACC), one of its specific substrates. However, we did not detect any difference in either HEK293 or HeLa (data not shown) cells when we coexpressed or not laforin and malin: the basal phosphorylation status of ACC was similar in all the cases and its phosphorylation was induced to similar levels upon treatment with the AMPK activator phenformin (Supplemental Figure S2). We also assayed whether the deficiency in laforin could affect the expression or activation of the endogenous AMPK complex. With this aim we used a mouse model of Lafora disease, deficient in the expression of laforin (Epm2a−/−) (Ganesh et al., 2002). Our results indicate that in the absence of laforin, the levels of endogenous AMPKα and AMPKβ subunits and the regulation of AMPKα-Thr172 phosphorylation in liver and brain of Epm2a−/− mice are similar to control animals (Supplemental Figure S3).
We also analyzed the subcellular distribution of AMPK subunits upon treatment with the laforin–malin complex, as a possible way to regulate AMPK function (Figure 7). The expression of AMPKβ2 subunit resulted in a punctuated distribution of the protein throughout the cell, although it was less abundant within the nucleus (Figure 7A). However, the coexpression of laforin and malin produced some distinct inclusion bodies (sometimes perinuclear), which contained laforin and AMPKβ2 (Figure 7B; 38% of cells transfected with AMPKβ2, laforin, and malin contained these inclusions, Figure 7E). Localization analysis of AMPKβ1 subunit gave similar results (data not shown). These inclusion bodies contained ubiquitin as they were stained with anti-ubiquitin antibodies (Figure 7C). However, the presence of these aggregates diminished if we transfected the cells with a K63R-ubiquitin–expressing plasmid (Figure 7D; only 14% of cells expressing AMPKβ2 contained the inclusions, Figure 7E). All these results suggested that the laforin–malin complex promoted the formation of inclusion bodies containing at least AMPKβ2, laforin, and ubiquitin, by introducing K63-linked ubiquitin chains into the corresponding substrates.
Figure 7.

The laforin–malin complex promotes the aggregation of coexpressed AMPKβ subunits. (A–D) HEK293 cells were transfected with plasmid pCMVmyc-AMPKβ2 with (B and C) or without (A) plasmids pCMV-HA-laforin and pcDNA3-HA-malin (Laf+Mal). When indicated, cells were also transfected with plasmid pCMV-His6xUbiq K63R (D). The subcellular localization of AMPKβ2 subunit was carried out as described in Materials and Methods by using anti-AMPKβ total as primary and anti-rabbit Alexa-Fluor 488 as secondary antibodies. The same samples were treated with Topro3 to stain the nucleus and with anti-laforin or anti-ubiquitin as primary and anti-mouse Texas Red as secondary antibodies to determine the localization of laforin and ubiquitin conjugates. The three images were subjected to a merge analysis. (E) Quantification of cells expressing AMPKβ2 and showing either a punctuated distribution or large inclusion bodies. One hundred cells expressing AMPKβ2 from each of the above conditions were used to estimate the proportion of cells with or without inclusions. Bars indicate SD; statistical significance was considered at *p < 0.05 and **p < 0.01.
DISCUSSION
We report in this study that a functional complex formed by laforin, a dual-specificity protein phosphatase, and malin, an E3-ubiquitin ligase, related to Lafora disease, promotes the ubiquitination of AMPK subunits. This reaction occurs when any of the three AMPK subunits (α, β, and γ) are expressed individually in cells, but it also occurs on AMPKβ subunit when it is part of a heterotrimeric AMPK complex. The different ability of AMPKα and AMPKγ to be ubiquitinated by the laforin–malin complex depending on whether they are forming part or not of a trimeric complex could be due to different folding. It has been described that when AMPK subunits are forming part of a heterotrimeric complex they have a longer half-life than when they are expressed individually (12–13 h instead of 5–6 h) (Crute et al., 1998). Perhaps subunits forming part of a complex have specific domains less exposed to modifier enzymes because they participate in the interaction with other subunits. The reaction needs both components of the laforin–malin complex, where laforin has been previously shown to function by recruiting putative substrates to be modified by the E3-ubiquitin ligase activity of malin. These results complement previous findings of our group that indicated that laforin was able to interact physically with different subunits of the AMPK complex (Solaz-Fuster et al., 2008). Because a similar ubiquitination profile of AMPK subunits was detected when we treated cells with the proteasome inhibitor MG132, we suggest that laforin and malin may form part of a collection of endogenous E3-ubiquitin ligases that are responsible for the ubiquitination of AMPK subunits.
We studied in more detail the topology of the linkages present in the polyubiquitin chains of the modified AMPK subunits. We used two different strategies: the use of mutated forms of ubiquitin, i.e., K48R and K63R, and the use of antibodies that recognize specific type of polyubiquitin chains. We found that the laforin–malin complex promotes the formation of K63-linked ubiquitin chains in AMPK subunits. The same topology was found in the ubiquitination of AMPKβ promoted by endogenous E3-ubiquitin ligases (Figure 6). These results indicated that only E3-ubiquitin ligases with the specificity of promoting K63-ubiquitination are able to modify AMPKβ subunits. In addition, a similar topology of ubiquitin linkages was introduced by the laforin–malin complex into R5/PTG, another substrate of the complex (Solaz-Fuster et al., 2008). All these results indicate that the laforin–malin complex has the specificity of introducing K63-linked ubiquitin chains into its corresponding substrates. We do not know yet the E2-conjugating enzyme that participates in this reaction. So far, it has been described that malin can promote an efficient ubiquitination when is associated with the E2 UbcH2, UbcH5, and UbcH6 (Gentry et al., 2005; Solaz-Fuster et al., 2008), although all these results were obtained in vitro. Recently, it has been proposed that the U-box cochaperone carboxyl terminus of the Hsc70-interacting protein (CHIP) stabilizes malin (Rao et al., 2010). Because CHIP may interact with the E2 UbcH13/Uev1a, a heterodimeric E2 enzyme that exclusively forms K63-linked ubiquitin chains (Windheim et al., 2008; Xu et al., 2008), we suggest that laforin and malin could form a macrocomplex with CHIP and UbcH13/Uev1a that would be responsible for the K63-linked ubiquitination of specific substrates. In this way, malin would resemble parkin, an E3-ubiquitin ligase related to Parkinson disease, because it was described that parkin mediated the K63-linked ubiquitination of misfolded DJ-1 (Olzmann et al., 2007), synphilin-1 (Lim et al., 2005), and α-synuclein (Liu et al., 2007), promoting in this way their aggregation, and that the action of parkin was possibly mediated by its interaction with CHIP and UbcH13/Uev1a [(Lim et al., 2006), (Yoshida, 2007), (Kostova et al., 2007)]. Alternatively, laforin and malin could interact directly with UbcH13/Uev1a.
Although our results suggest that the laforin–malin complex introduces K63-linked ubiquitin chains into its corresponding substrates, we do not discard the possibility that under certain circumstances, it could also promote the formation of K48-linked ubiquitins. This possibility also has been recently described for parkin that in addition to modifying synphilin-1 by K63-linked ubiquitin chains, also mediates the proteasomal degradation of this protein by introducing K48-linked ubiquitin chains, but only at an unusually high parkin-to-synphilin-1 expression ratio or when primed with K48-linked ubiquitination (Lim et al., 2005). Perhaps this is the reason why the overexpression of laforin and malin could promote the proteasomal degradation of R5/PTG (Solaz-Fuster et al., 2008). In this sense, the results presented in Figure 5, A and B, might underestimate a possible ubiquitination of AMPK subunits by K48-linked ubiquitins that would be absent in the K63R lanes because of their rapid proteasomal degradation. However, the importance of this possible modification must be very low because no differences in the degradation rates of AMPK subunits were observed in the presence or absence of the laforin–malin complex.
Consistent with the idea that K63-linked polyubiquitin chains are not involved in protein degradation but in other intracellular signaling functions (Ikeda and Dikic, 2008; Deshaies and Joazeiro, 2009), we found that the laforin–malin-dependent ubiquitination of AMPK subunits did not accelerate their rapid proteolytic degradation (Figure 4). On the contrary, the laforin–malin-dependent ubiquitination of at least AMPKβ subunits resulted in an increase of the steady-state levels of these proteins. Recently, it was reported that CIDEA promotes the ubiquitination and subsequent degradation of AMPKβ subunits (Qi et al., 2008). Because CIDEA is not itself an E3-ubiquitin ligase, the authors suggested that, by binding to AMPKβ, CIDEA could recruit specific E3-ubiquitin ligases that would ubiquitinate AMPKβ, targeting it for proteasomal degradation. The authors did not study the type of polyubiquitin chains that CIDEA promoted on AMPKβ, but we suspect that they must be different from the K63-linkages that the laforin–malin complex is able to produce. As a result, whereas CIDEA-promoted ubiquitination of AMPKβ enhanced the degradation of the subunit, the laforin–malin-promoted ubiquitination of AMPKβ resulted in its accumulation. In CIDEA-induced ubiquitination, the authors mapped the domain that was involved in this modification at the C terminus of AMPKβ (residues 231–248) (Qi et al., 2008). We constructed similar truncated forms of AMPKβ2 as those reported in the study of CIDEA (Qi et al., 2008). However, this modification was abolished when either the N-terminal (residues 1–185) or the C-terminal (residues 186–271) part of the protein was used in the assay, compared with the full-length AMPKβ subunit. These results suggest that the ubiquitination induced by CIDEA and by the laforin–malin complex must depend on different lysine residues of the AMPKβ subunit.
The laforin–malin-dependent K63-linked polyubiquitination of AMPK subunits has no apparent effect on AMPK activity. However, we observed that cells coexpressing laforin, malin, and AMPKβ subunits produced inclusion bodies that were positive for at least ubiquitin, laforin, and AMPKβ (Figure 7). The overexpression of laforin and malin was the trigger for the formation of these protein inclusions, in agreement with their aggregation prone capabilities (Mittal et al., 2007; Liu et al., 2009; Rao et al., 2010), because when laforin and malin were not coexpressed no inclusion bodies were formed. The fact that the presence of these aggregates diminished if the cells coexpressed K63R-ubiquitin suggests that K63-linked ubiquitination of AMPKβ subunits could be relevant to form the inclusion bodies. This would be in agreement with previous results that indicated that K63-linked ubiquitination favors the formation of protein aggregates (Lim et al., 2005; Tan et al., 2008). Therefore, two pools of AMPKβ might coexist in the cells, one pool forming part of these intracellular inclusions, and another pool that would be soluble. The pool of AMPKβ subunits present in these inclusion bodies would remain less accessible to rapid proteolytic degradation and this would be the reason of the observed increase in the steady-state levels of these proteins. The rest of AMPKβ would remain in the soluble fraction displaying its regular function (i.e., ACC phosphorylation). This increase is however absent in endogenous AMPKβ subunits upon treatment with laforin and malin. Perhaps the levels of modified AMPKβ subunits required a minimal concentration to initiate the formation of these intracellular inclusions and this minimum could not be reached with the endogenous AMPKβ subunits.
In summary, our results indicate that the laforin–malin complex promote the K63-linked ubiquitination of several substrate proteins and reinforces the idea that Lafora disease should be considered as a disorder of protein clearance (Delgado-Escueta, 2007). In the absence of a functional laforin–malin complex unmodified substrates would accumulate, which could eventually affect the unfolded protein response pathway, the proteasome activity (already described in Vernia et al., 2009) and possibly autophagy.
Supplementary Material
ACKNOWLEDGMENTS
We want to thank Drs. A. Dickmanns, M. Gentry, Christine Blattner, and M. Rodriguez for providing the described plasmids. We also thank Genentech for providing the K48- and K63-ubiquitin chain-specific antibodies. This work was supported by the Spanish Ministry of Education and Science grant SAF2008-01907 (to P. S.). M.C.T. and D.G.H. were supported by programme grant 080982 from the Wellcome Trust. P. S. and D.G.H. also were supported by EXGENESIS, an Integrated Project (LSHM-CT-2004-005272) of the European Commission. E. K. was supported by grant BFU2008-00186/BMC from the Spanish Ministry of Education and Science.
Abbreviations used:
- AMPK
AMP-activated protein kinase
- CHIP
carboxy-terminus Hsc70-interacting protein
- CHX
cycloheximide
- LD
Lafora disease
- PP1
type 1 protein phosphatase
- R5/PTG
protein targeting to glycogen R5.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-03-0227) on June 9, 2010.
REFERENCES
- Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- Brady M., Vlatkovic N., Boyd M. T. Regulation of p53 and MDM2 activity by MTBP. Mol. Cell. Biol. 2005;25:545–553. doi: 10.1128/MCB.25.2.545-553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crute B. E., Seefeld K., Gamble J., Kemp B. E., Witters L. A. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- Chan E. M., Omer S., Ahmed M., Bridges L. R., Bennett C., Scherer S. W., Minassian B. A. Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology. 2004;63:565–567. doi: 10.1212/01.wnl.0000133215.65836.03. [DOI] [PubMed] [Google Scholar]
- Chan E. M., et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- Delgado-Escueta A. V. Advances in lafora progressive myoclonus epilepsy. Curr. Neurol. Neurosci. Rep. 2007;7:428–433. doi: 10.1007/s11910-007-0066-7. [DOI] [PubMed] [Google Scholar]
- Deshaies R. J., Joazeiro C. A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Ganesh S., et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002;11:1251–1262. doi: 10.1093/hmg/11.11.1251. [DOI] [PubMed] [Google Scholar]
- Ganesh S., Puri R., Singh S., Mittal S., Dubey D. Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 2006;51:1–8. doi: 10.1007/s10038-005-0321-1. [DOI] [PubMed] [Google Scholar]
- Gentry M. S., Dixon J. E., Worby C. A. Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem. Sci. 2009;34:628–639. doi: 10.1016/j.tibs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M. S., Dowen R. H., 3rd, Worby C. A., Mattoo S., Ecker J. R., Dixon J. E. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J. Cell Biol. 2007;178:477–488. doi: 10.1083/jcb.200704094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M. S., Worby C. A., Dixon J. E. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno-Alcaniz J. V., Sanz P. Glucose and type 2A protein phosphatase regulate the interaction between catalytic and regulatory subunits of AMP-activated protein kinase. J. Mol. Biol. 2003;333:201–209. doi: 10.1016/j.jmb.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Ikeda F., Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser P., Tagwerker C. Is this protein ubiquitinated? Methods Enzymol. 2005;399:243–248. doi: 10.1016/S0076-6879(05)99016-2. [DOI] [PubMed] [Google Scholar]
- Kostova Z., Tsai Y. C., Weissman A. M. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin. Cell Dev. Biol. 2007;18:770–779. doi: 10.1016/j.semcdb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. L., et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1, implications for Lewy body formation. J. Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. L., Dawson V. L., Dawson T. M. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol. Aging. 2006;27:524–529. doi: 10.1016/j.neurobiolaging.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Liu C., Fei E., Jia N., Wang H., Tao R., Iwata A., Nukina N., Zhou J., Wang G. Assembly of lysine 63-linked ubiquitin conjugates by phosphorylated alpha-synuclein implies Lewy body biogenesis. J. Biol. Chem. 2007;282:14558–14566. doi: 10.1074/jbc.M700422200. [DOI] [PubMed] [Google Scholar]
- Liu Y., Wang Y., Wu C., Liu Y., Zheng P. Deletions and missense mutations of EPM2A exacerbate unfolded protein response and apoptosis of neuronal cells induced by endoplasm reticulum stress. Hum. Mol. Genet. 2009;18:2622–2631. doi: 10.1093/hmg/ddp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohi H., Ianzano L., Zhao X. C., Chan E. M., Turnbull J., Scherer S. W., Ackerley C. A., Minassian B. A. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 2005;14:2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- Minassian B. A., Ianzano L., Meloche M., Andermann E., Rouleau G. A., Delgado-Escueta A. V., Scherer S. W. Mutation spectrum and predicted function of laforin in Lafora's progressive myoclonus epilepsy. Neurology. 2000;55:341–346. doi: 10.1212/wnl.55.3.341. [DOI] [PubMed] [Google Scholar]
- Minassian B. A., et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- Mittal S., Dubey D., Yamakawa K., Ganesh S. Lafora disease proteins malin and laforin are recruited to aggresomes in response to proteasomal impairment. Hum. Mol. Genet. 2007;16:753–762. doi: 10.1093/hmg/ddm006. [DOI] [PubMed] [Google Scholar]
- Newton K., et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Olzmann J. A., Li L., Chudaev M. V., Chen J., Perez F. A., Palmiter R. D., Chin L. S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T., Xiong B., Li J. Y., Qiu B. Y., Jin G. Z., Shen J. K., Li J. Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J. Biol. Chem. 2007;282:495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- Qi J., Gong J., Zhao T., Zhao J., Lam P., Ye J., Li J. Z., Wu J., Zhou H. M., Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. N., Sharma J., Maity R., Jana N. R. Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 2010;285:1404–1413. doi: 10.1074/jbc.M109.006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz P. AMP-activated protein kinase: structure and regulation. Curr. Protein Peptide Sci. 2008;9:478–492. doi: 10.2174/138920308785915254. [DOI] [PubMed] [Google Scholar]
- Scott J. W., Hawley S. A., Green K. A., Anis M., Stewart G., Scullion G. A., Norman D. G., Hardie D. G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serratosa J. M., et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum. Mol. Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- Solaz-Fuster M. C., Gimeno-Alcaniz J. V., Casado M., Sanz P. TRIP6 transcriptional co-activator is a novel substrate of AMP-activated protein kinase. Cell Signal. 2006;18:1702–1712. doi: 10.1016/j.cellsig.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Solaz-Fuster M. C., et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 2008;17:667–678. doi: 10.1093/hmg/ddm339. [DOI] [PubMed] [Google Scholar]
- Tagliabracci V. S., Girard J. M., Segvich D., Meyer C., Turnbull J., Zhao X., Minassian B. A., Depaoli-Roach A. A., Roach P. J. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J. Biol. Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci V. S., et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. USA. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J. M., et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Thornton C., Snowden M. A., Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J. Biol. Chem. 1998;273:12443–12450. doi: 10.1074/jbc.273.20.12443. [DOI] [PubMed] [Google Scholar]
- Vernia S., Rubio T., Heredia M., Rodriguez de Cordoba S., Sanz P. Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PloS One. 2009;4:e5907. doi: 10.1371/journal.pone.0005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Stuckey J. A., Wishart M. J., Dixon J. E. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J. Biol. Chem. 2002;277:2377–2380. doi: 10.1074/jbc.C100686200. [DOI] [PubMed] [Google Scholar]
- Windheim M., Peggie M., Cohen P. Two different classes of E2 ubiquitin-conjugating enzymes are required for the mono-ubiquitination of proteins and elongation by polyubiquitin chains with a specific topology. Biochem. J. 2008;409:723–729. doi: 10.1042/BJ20071338. [DOI] [PubMed] [Google Scholar]
- Worby C. A., Gentry M. S., Dixon J. E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby C. A., Gentry M. S., Dixon J. E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG) J. Biol. Chem. 2008;283:4069–4076. doi: 10.1074/jbc.M708712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B., et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- Xu Z., Kohli E., Devlin K. I., Bold M., Nix J. C., Misra S. Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct. Biol. 2008;8:26. doi: 10.1186/1472-6807-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.