

Abstract
The synthesis of compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b where the amino -NH- group of JDTic (3) was replaced with an aromatic =CH–, CH2, O, S, or SO group was accomplished and used to further characterize the SAR of the compound 3 class of κ opioid receptor antagonists. All of the compounds showed subnanomolar to low nanomolar Ke values at the κ opioid receptor. The most potent compound was 7a, where the amino -NH- group of 3 was replaced by a methylene (-CH2-) group. This compound had a Ke = 0.18 nM and was 37- and 248-fold selective for the κ relative to the μ and δ opioid receptors, respectively. Similar to compound 3, compound 7a antagonized selective κ agonist U50,488-induced diuresis after s.c. administration in rats. In contrast to 3, where κ antagonist activity lasted for three weeks, compound 7a did not show any κ antagonist activity after one week.
Introduction
Kappa opioid receptor selective antagonists are of considerable interest as potential pharmacotherapies for addiction (cocaine, opiate, alcohol, nicotine, and possibly others),1-7 depression, 1,8-10 anxiety disorders,11 obesity, 12-14 and psychosis disorders.15 Opioid antagonists with varying degrees of receptor potency and selectivity have been developed for the κ opioid receptor. The first highly selective and potent κ opioid receptor antagonist was norBNI (1).16,17 Continued studies identified 5′-GNTI (2) as a more potent and selective κ opioid antagonist.18-20 In 2001, JDTic (3), an N-substituted trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine analog, was developed as the first highly potent and selective κ opioid receptor antagonist from this class of opioid antagonists.21 More recently, the dynorphin analogues zyklophin and arodyn (4 and 5, respectively) were developed as selective κ opioid receptor antagonists.22,23
SAR studies previously conducted on 3 identified the 7′-phenolic and (S)-2′-isopropyl groups as features important for the κ selectivity and suggested that the nitrogen of the hydroxy-D-Tic group might also be uniquely required for the κ selectivity.24-26 To gain additional information as to the role that the amine nitrogen of the hydroxy-D-Tic group plays in determining the κ opioid in vitro antagonist efficacy and selectivity, the JDTic analogues 6, 7a,b, and 8a,b, 9a,b, and 10a,b, where the amino -NH- group was replaced with an aromatic =CH–, CH2, O, S, or SO group, were synthesized and evaluated for their ability to antagonize μ, δ, and κ opioid agonists in a [35S]GTPγS in vitro antagonist efficacy test. The stereochemical assignments for the position alpha to the carbonyl group for compounds 7a,b, 8a,b, and 9a,b are arbitrary. The isomer with the lowest Ke value for the κ opioid receptor in each pair was given the a designation. Compounds 10a,b are the two possible sulfoxide diastereoisomers derived from 9a.
Analogue 7a, with a Ke = 0.18 nM at the κ opioid receptor and 37- and 248-fold selectivity for the κ receptor relative to the μ and δ opioid receptors, was the most potent and selective analogue tested. The potency and duration of action of 7a to antagonize κ agonist U50,488-induced diuresis in rats was compared to that of 3.
Chemistry
Analogues 6, 7a,b, 8a,b, 9a,b, and 10a,b of 3 were synthesized by the methods depicted in Schemes 1–3. Coupling of 6-hydroxy-2-naphthoic acid (11) with 3-{1-[(2S)-amino-3-methylbutyl]-(3R,4R)-dimethyl-4-piperidinyl}phenol (12)27 using benzotriozole-1-yloxy-tris(dimethylamino)phosphonium hexafluorophorphole (BOP) in tetrahydrofuran containing triethylamine at 25 °C gave the desired analogue 6 (Scheme 1). Our initial plan for the synthesis of 7a and 7b was to start with (+)- and (-)-13. Since all attempts to resolve (±)-13 directly into its optical isomers were unsuccessful, we used the strategy outlined in Scheme 2 to prepare (+)- and (-)-13, which were then converted to 7a and 7b. This strategy was based on a report from Fox et al.28 who showed that the tetrahydro-2H-indeno[1,2d]-2-ones serve as a readily cleavable chiral auxillary suitable for large-scale normal phase separation of isomers. Thus, treatment of (3aR-cis)-3,3a,8,8a-tetrahydro-2H-indeno[1,2d]oxazol-2-one (14) with ethyl lithium provided the lithium salt, which was acetylated with the acid chloride prepared by treating (±)-1329 with thionyl chloride in toluene, to give 15a and 15b, which were separated by standard silica chromatography. Hydrolysis of 15b using lithium hydroxide in tetrahydrofuran containing 30% hydrogen peroxide yielded (+)-13.30 Coupling of (+)-13 to 1227 using BOP in tetrahydrofuran containing triethylamine at 25 °C provided 16a. Treatment of 16a with boron tribromide in methylene chloride at -78 °C afforded 7a. Subjecting 15a to the same set of reaction conditions provided (-)-13, 15b, and 7b.
Scheme 1a.

aReagents and conditions: (a) BOP, TEA, THF, 25 °C.
Scheme 3a.

aReagents: (a)  , LDA, HMPA, THF; (b) Et3SiH, TFA; (c) KOH, CH3OH, 0 °C; (d) (COCl)2, CH2Cl2; (e) 14, EtLi, THF; (f) LiOH, THF/H2O; (g) 12, BOP, TEA, THF, 25 °C; (h) BBr3, CH3Cl2, -78 °C; (i) MCPBA, CH2Cl2, 0 °C.
, LDA, HMPA, THF; (b) Et3SiH, TFA; (c) KOH, CH3OH, 0 °C; (d) (COCl)2, CH2Cl2; (e) 14, EtLi, THF; (f) LiOH, THF/H2O; (g) 12, BOP, TEA, THF, 25 °C; (h) BBr3, CH3Cl2, -78 °C; (i) MCPBA, CH2Cl2, 0 °C.
Scheme 2a.

aReagents and conditions: (a) thionyl chloride, toluene; (b) EtLi, (3aR-Cis)-3,3a,8,8a-tetrahydro-2H-indeno[1,2d]oxazol-2-one (14); (c) 30% H2O2, LiOH, 3:1 THF/H2O; (d) 12, BOP, TEA, THF 25 °C; (e) BBr3, CH2Cl2 -78 °C.
Based on the ease of separating 7a,b using chiral auxillary 14, a similar synthetic strategy was applied to the synthesis of 9a,b and 10a,b. In this case, the synthesis of the 7-methoxy-isothiochroman-3-carboxylic acid [(±)-20] had to be developed. Scheme 3 outlines the synthesis used to prepare (+)- and (-)-20 and subsequent conversion to 9a and 9b. Condensation of 7-methoxy-isothiochroman-4-one (17)31 with methyl cyanoformate using lithium diisopropylamide (LDA) as the base in tetrahydrofuran containing hexamethylphosphoramide (HMPA), gives keto ester (18).32 Reduction of 18 using triethylsilane in trifluoroacetic acid effected removal of the 4-keto group to give 19.33 Hydrolysis of 19 using potassium hydroxide in methanol provided (±)-20. The lithium salt of 14 was acylated with the acid chloride prepared by treating (±)-20 with oxalyl chloride in methylene chloride to give 21a and 21b, which were separated by normal phase silica gel chromatography. Hydrolysis of 21a using lithium hydroxide in aqueous tetrahydrofuran afforded (+)-20. It should be noted that hydrolysis of the auxillary was equally effective without the presence of peroxide in the basic media. Coupling of (+)-20 with 1227 using BOP in tetrahydrofuran containing triethylamine at 25 °C provided 22a. Treatment of 22a with boron tribromide in methylene chloride at -78 °C afforded 9a. Subjecting 21b to a set of reaction conditions analogous to those used for 21a provided (-)-20, 22b, and 9b. Oxidation of 9a using 3-chloroperoxybenzoic acid in methylene chloride provided sulfoxide diastereomers 10a and 10b that were separated using normal phase silica gel chromatography.
The synthesis of compounds 8a and 8b was more complex than that of 7a,b and 9a,b due to the requirement of phenolic protecting groups in the presence of a benzylic ether (see Scheme 4). Cyclization of 3-methoxybenzyloxyacetic acid (23) using oxalyl chloride followed by tin IV chloride in chlorobenzene yielded chromanone 24.34 Demethylation of 24 to the phenol 25 was accomplished using sodium ethanethiolate in dimethylformamide.35 The phenol group in 25 was reprotected as the methoxymethyl ether 26 using chloromethylmethyl ether and diisopropylethylamine in methylene chloride. Condensation of 26 with methyl cyanoformate using lithium diisopropylethylamine as the base in tetrahydrofuran containing hexamethylphosphoramide (HMPA) afforded the keto ester 27.32 Treatment of 27 with triethylsilane in trifluoroacetic acid afforded both removal of the 4-keto group, and the methoxylmethyl protecting group to give 28.33 Hydrolysis of 28 using lithium hydroxide in water, tetrahydrofuran, ethanol mixture provided (±)-29. Coupling of (±)-29 to 12 using BOP in tetrahydrofuran containing triethylamine at 25 °C yielded a mixture of (+)-8 that were separated using reverse phase HPLC to provide 8a,b.
Scheme 4a.

aReagents and conditions: (a) Oxalyl chloride, CH2Cl2; (b) SnCl4, chlorobenzene, 0 °C; (c) NaSEt, DMF, reflux; (d) MOMCl, i-Pr2EtN; (e) methyl cyanoformate, LDA HMPA, THF; (f) TFA, Et2SiH; (g) LiOH, THF/MeOH/H2O, (1:1:1) 0 °C; (h) 12, BOP, TEA, THF, 25 °C; (i) preparative HPLC.
Pharmacology
Compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b were first evaluated at 10 μM for intrinsic activity in the [35S]GTPγS binding assay at all three opioid receptors. As none of these compounds displayed measurable intrinsic activity at this concentration, they and the reference compound 1 were evaluated for antagonist efficacy and selectivity at the opioid receptors. These data were obtained by monitoring the ability of test compounds to inhibit stimulated [35S]GTPγS binding produced by the selective agonists DAMGO (μ), DPDPE (δ), or U69,593 (κ) using cloned human opioid receptors expressed in CHO cells.36 Agonist dose response curves were run in the presence or absence of a single concentration of test compound. The Ke values were calculated using the formula: Ke = [L]/DR-1, where [L] is the concentration of test compound and DR is the ratio of agonist EC50 value in the presence or absence of test compound, respectively. At least two different concentrations of test compound were used to calculate the Ke, and the concentrations were chosen such that the agonist EC50 exhibited at least a four-fold shift to the right and there was a clear upper asymptote to the agonist + compound concentration response curve. The Ke values along with those for the reference compound 1 are shown in Table 1.
Table 1.
Comparison of Inhibition of Agonist-Stimulated [35S]GTPγS Binding in Cloned Human μ, δ, and κ Opioid Receptors for 6, 7a,b, 8a,b, 9a,b, and 10a,b to 3 and 1a,b
| compd | μ, DAMGO Ke (nM) |
δ, DPDPE Ke (nM) |
κ, U69,593 Ke (nM) |
μ/κ | δ/κ |
|---|---|---|---|---|---|
| 1 | 26.7 ± 7c | 29 ± 8c | 0.05 ± 0.02c | 520 | 580 |
| 3 | 25.1 ± 3.5c | 76.4 ± 2.7c | 0.02 ± 0.01c | 1255 | 3830 |
| 6 | 2.53 ± 0.34 | 219 ± 41 | 3.93 ± 1.2 | 0.7 | 56 |
| 7a | 6.67 ± 1.2 | 44.8 ± 7.7 | 0.18 ± 0.03 | 37 | 248 |
| 7b | 11.2 ± 2.4 | 205 ± 59 | 1.37 ± 0.36 | 8.2 | 150 |
| 8a | 7.4 ± 2.6 | 197 ± 64 | 0.71 ± 0.16 | 10 | 278 |
| 8b | 5.2 ± 1.6 | 134 ± 26 | 5.7 ± 1.6 | 0.9 | 24 |
| 9a | 5.57 ± 1.27 | 395 ± 122 | 0.99 ± 0.25 | 6 | 399 |
| 9b | 13.4 ± 2.9 | 182 ± 48 | 1.58 ± 0.07 | 8.5 | 115 |
| 10a | 7.9 ± 2.2 | 208 ± 55 | 2.7 ± 2.2 | 2.9 | 77 |
| 10b | 1.8 ± 0.3 | 61 ± 11 | 0.55 ± 0.12 | 3.3 | 111 |
Data represent means ± SE from at least three independent experiments.
The average percent stimulation and agonist EC50 values for the μ, κ, and δ [35S]GTPγS binding assays were 200% and 120 nM, 220% and 380 nM, and 50% and 6 nM, respectively. Ke values were calculated from experiments were the antagonist produced at least a 4-fold shift in the agonist ED50.
The Ke values for 3 supplied by the NIDA Opioid Treatment Discovery Program (OTDP) were 3.41, 79.3, and 0.01 nM for the μ, δ, and κ receptors, respectively (ref. 24).
Compound 7a was evaluated for its ability to antagonize U50,488-induced diuresis in male Sprague-Dawley rats (Charles River Laboratories, Raleigh, NC).
Results and Discussion
Compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b were evaluated for their μ, δ and κ agonist or antagonist activity in the [35S]GTPγS antagonist efficacy binding assay. The data are shown in Table 1 compared to 1 and 3. None of the compounds had any μ, δ, or κ intrinsic activity at 10 μM. As expected, all compounds were antagonists of the DAMGO (μ), DPDPE (δ), or U69,593 (κ) stimulated binding. Similar to 1 and 3, the compounds were more potent at the μ and κ than at the δ opioid receptor. Compounds 1 and 3 with Ke values of 0.05 and 0.02 nM, respectively, are highly potent antagonists for the κ receptor. In addition, both compounds are highly selective for the κ relative to the μ and δ opioid receptors. The compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b where the amino -NH- groups have been replaced by an aromatic =CH-, -CH2-, O, S, or SO group, showed Ke values ranging from 0.18 to 5.7 nM for κ, 1.8 to 13.4 nM for μ, and 44.8 to 395 nM for δ, and thus are potent κ and μ opioid receptor antagonists. In the cases of 7, 8, 9, and 10, one isomer was more potent than the other isomer. The differences in potencies were 7.6-, 8-, 1.6-, and 4.9-fold for 7a,b, 8a,b, 9a,b, and 10a,b, respectively. The Ke values at the κ opioid receptor for the more potent 7a, 8a, and 9a are 0.18, 0.77, and 0.99 nM, respectively. The diastereomeric sulfoxides 10a and 10b prepared from 9a had Ke values of 2.7 and 0.55 nM at the κ opioid receptor, respectively. The more potent isomers 7a, 8a, 9a, and 10b are 9-, 36-, 50-, and 28-times less potent than 3 as a κ opioid antagonist. This data shows that replacing the amino -NH- group of 3 with a -CH2- was better tolerated at the κ opioid receptor than replacement by an -O- or –S-. Compound 7a with Ke values of 6.67, 44.8, and 0.18 nM at the μ, δ, and κ opioid receptors was the most potent and selective κ opioid receptor antagonist of the compounds evaluated. The low antagonist efficacy of compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b relative to 3 strongly suggests that the –NH group of 3 is critical to its high in vitro antagonist efficacy at the κ opioid receptor.
With the exception of 8b, all of the compounds were less potent at the μ opioid receptor than the κ opioid receptor. However, with the exception of 7a, which had a 37-fold selectivity for κ over μ, the potencies of the μ and κ opioid receptor were similar. The Ke values at the μ opioid receptor ranged from 1.8 to 11.2 nM. The sulfoxide 10b with a Ke = 1.8 nM was the most potent μ opioid antagonist. All of the compounds had relative low antagonist efficacies for the δ opioid receptor. Compound 7a with a Ke = 44.8 shows the highest potency at the δ opioid receptor.
Structure activity relationship (SAR) studies of 1 and 2 and analogues led Portoghese to propose that the 17′-amino group of 1 and the 5′-guanidinyl amino moiety of 2 as the primary structural features leading to the κ opioid selectivity of these two compounds (see Metcalf and Coop37 and Aldrich38 for reviews of the SAR studies). Portoghese and co-workers explained the κ selectivity of 1 and 2 using the message-address concept developed by Schwyzer39 for peptide hormones. In this concept, molecular features common to a series of compounds recognized by a family of receptors are defined as the message. The “address” portion of this concept is a specific structural feature of the ligand that affords sub-type selectivity through interaction with a site or specific residue(s) unique to a receptor subtype. In the case of 1 and 2, Portoghese used molecular modeling studies40 coupled with site-directed mutagenesis to provide additional evidence that the 17′-amino group of 1 and the 5′-guanidinium amino group of 2 serve as the address by forming an ion pair with Glu297 at the top of the TM6 in the κ opioid receptor.19,41-44
In order to determine if 3 could be interacting with the κ opioid in a way similar to that proposed for 2 and 1, structures of 3 and 2 were constructed and compared. Low-energy conformations of 3 which provide an overlay of the primary pharmacophoric groups of 3 and 2 (as represented by the centroid of the phenolic rings of 3 and 2 and the centroid of the Tic aromatic ring of 3 and the guanidinium group of 2) were identified via conformational analysis (Figure 1). Although the geometry and dimensions of 3 conformations do not permit an atom-for-atom overlay of the 7-hydroxy-D-Tic-group hydrogen-bonding substituents of 3 with the guanidinium nitrogens of 2, both the 7-hydroxy-D-Tic secondary amine NH group and phenol oxygen are projected into the same general region and presumably could participate in some type of interactions in the κ-address locus proposed for 1 and 2 of the κ-opioid receptor (Figure 1).
Figure 1.
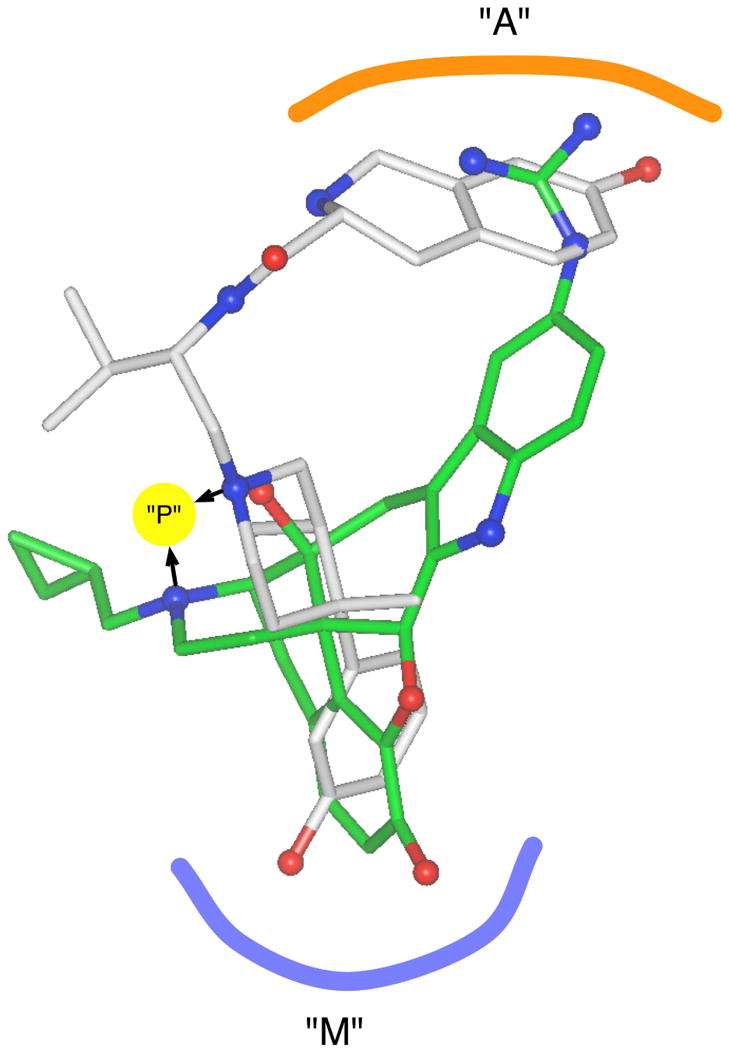
Alignment of a local-energy minimum conformation of 3 (grey) and 2 (green) with heteroatoms indicated by CPK-colored spheres (blue for nitrogen and red for oxygen). Possible shared-pharmacophore regions are labeled “A” for the opioid address region, “M” for the opioid message region and “P” for a polar interaction site adjacent to the protonated nitrogens.
Compounds 6, 7a,b, 8a,b, 9a,b, and 10a,b all have a phenol group in a location similar to the 7-hydroxy-D-Tic phenol group of 3 but lack the –NH group present in 3. Therefore, the lower κ opioid antagonist efficacy of each to these compounds seems due largely to the absence of the -NH group present in 3. Previous studies have shown that the 7-hydroxy-D-Tic phenol oxygen is essential for the high κ opioid receptor efficacy of 3.25,26 Thus, these results suggest that the highly potent κ opioid receptor efficacy of 3 is due to strong interactions at the κ opioid receptor by the 7-hydroxy-D-Tic secondary amine -NH group and the phenol oxygen as suggested by the molecular modeling studies (Figure 1). However, these results do not rule out the possibility that 3 is interacting with the κ opioid receptor in a way different from that of 1 and 2.
In order to gain information concerning the in vivo activity, the effect of 7a to antagonize κ agonist U50,488-induced diuresis was compared to that of 3. The results from the study are summarized in Figure 2. At week 0 (first 5 h after dosing s.c.), compound 3 showed dose-related antagonism of urine output relative to vehicle (Figure 2A). At week 1 (no further dosing with 3), 3 continued to show dose-related antagonism of U50,488-induced diuresis. In keeping with our previous findings with 3,45 the level of antagonism at week 1 was the same as or greater than at week 0.
Figure 2.

Comparison of 7a to 3 on U50,488-induced diuresis in rats. (A) Antagonism of U50,488-induced urine output in various s.c. doses (mg/kg) of 3. (B) Antagonism of U50,488-induced urine output by various s.c. doses (mg/kg) of 7a, and compound 3 and 7a were only administered once at week 0.
At week 0 (first 5 h after dosing s.c.), 7a showed dose-related antagonism of U50,488-induced diuresis but was less potent than 3, which is consistent with its nine-fold lower in vitro antagonist potency at the κ opioid receptor compared to 3 in the [35S]GTPγS in vitro antagonist efficacy test (Figure 2B). Repeated measures ANOVA (StatView v5; SAS Institute Inc., Cary, NC) with factors of Dose and Week revealed a marginally significant effect of Dose (F(3,12) = 2.74; P<0.1) but a significant Dose × Week interaction (F(1,12) = 106.8; P<0.0001). Examining the data within week indicated the ability of 7a to antagonize U50,488-mediated diuresis was dose dependent and confined to week 0. The effect of individual doses of 7a on urine output was compared to U50,488 using Dunnett's test. At week 0, ANOVA indicated there was a significant overall effect of Dose (F(3,12) = 7.53; P< 0.005) to lower urine output, with the 10 and 30 mg/kg doses causing significant antagonism of the diuretic effects of U50,488. However, at week 1 (no further dosing with 7a), antagonism of U50,488 could no longer be detected (ANOVA; F(1,12) = 0.69; NS). Thus, the activity of 7a lasted less than 1 week, which contrasts sharply with the activity of 3 as well as other κ opioid receptor antagonists such as 1, which can persist for up to 3 weeks.45 The shorter duration of action of 7a compared to 3 and 1 showed that selective, high affinity κ opioid receptor antagonists can be identified with shorter durations of activity.
In summary, replacement of the amino -NH- group of 3 with an aromatic =CH-, -CH2-, O, or S provided compounds with reduced κ opioid receptor in vitro antagonist potency and reduced κ selectivity relative to the μ and δ opioid receptors. These results confirm our earlier results showing that the amino -NH- group of 3 is highly important to its high κ opioid receptor in vitro antagonist potency as well as κ selectivity. However, molecular modeling studies show that the amino -NH- group of 3 and the amino group in GNTI are not likely to be interacting with the κ opioid receptor in the same way. Interestingly, compound 7a, where the amino -NH- of 3 has been replaced by a methylene (-CH2-) group, showed subnanomolar κ potency and reasonable selectivity for the κ relative to the μ and δ opioid receptors. Compound 7a antagonized selective κ agonist U50,488-induced diuresis with a potency that parallels its κ efficacy in the [35S]GTPγS antagonist assay. In contrast to 3, whose activity as an antagonist of U50,488-induced diuresis lasted for 3 weeks, the activity of 7a lasted less than 1 week.
Experimental Section
1H NMR spectra were determined on a Bruker 300 spectrometer using tetramethylsilane as an internal standard. Mass spectral data were obtained using a Finnegan LCQ electrospray mass spectrometer in positive ion mode at atmospheric pressure. Silica gel 60 (230–400 mesh) was used for column chromatography. All reactions were followed by thin-layer chromatography using Whatman silica gel 60 TLC plates and were visualized by UV. Optical rotations were measured on an Auto Pol III polarimeter. All solvents were reagent grade. HCl in dry diethyl ether was purchased from Aldrich Chemical Co. and used while fresh before discoloration. CMA-80 is a mixture of 80% chloroform, 18% methanol, and 2% concentrated ammonium hydroxide. MMA-80 is a mixture of 80% methylene chloride, 18% methanol, and 2% concentrated ammonium hydroxide. Purity of compounds (>95%) was established by elemental analysis except for 8a and 8b. HPLC analysis was used to establish their purity. Elemental analysis was performed by Atlantic Microlab, Inc., Norcross, GA, USA.
HPLC-grade solvents were purchased from Burdick & Jackson (Muskegon, MI, USA). Preparative HPLC was carried out utilizing a Varian Prostar HPLC system (Walnut Creek, CA, USA) equipped with Prostar 210 pumps, Prostar 701 fraction collector, and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2). Analytical HPLC was carried out utilizing a Varian Prostar HPLC system (Walnut Creek, CA, USA) equipped with Prostar 210 pumps and a Prostar 330 photodiode array detector (PDA), with data collected and analyzed using Star Chromatography Workstation software (version 6.41). For preparative HPLC, a Gemini-NX C18 (5 μM; 250 × 21.2 mm) column was used with a 19 mL/min flow rate, while for analytical HPLC, a Gemini-NX C18 (5 μm; 250 × 4.6 mm) column was used with a 1 mL/min flow rate (both from Phenomenex, Torrance, CA, USA). For analytical HPLC, a MetaTherm HPLC column temperature controller (Varian) maintained the column at 30 °C.
6-Hydroxynaphthalene-2-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide Hydrochloride (6)
6-Hydroxy-2-naphthoic acid (100 mg, 0.53 mmol) was added to a solution of 1227 (154 mg, 0.53 mmol) and BOP reagent (235 mg, 0.53 mmol) in THF (40 mL) and was allowed to stir under nitrogen for 15 min. Triethylamine (1.18 g, 0.012 mol) in THF (10 mL) was added, and the reaction mixture was allowed to stir at room temperature for 3 h. NaHCO3 (50 mL) and Et2O (50 mL) were added to the reaction mixture, and the organic layer was separated, washed with brine, dried, (Na2SO4), and concentrated under reduced pressure to afford 0.32 g of an amorphous solid. The solid was purified using medium pressure silica gel chromatography, eluting with EtOAc–CMA80 (1:1) to afford 0.095 g of a white amorphous solid. 1H NMR (CDCl3) δ 8.20 (s, 1H), 7.75 (m, 2H), 7.62 (d, J = 9.0 Hz, 1H), 7.10 (m, 4H), 6.71 (m, 2H), 6.60 (d, 6.0 Hz, 1H), 4.42 (m, 1H), 2.32–2.92 (bm, 7H), 2.25 (bt, 1H), 2.05 (m, 1H), 1.92 (bs, 1H), 1.57 (bd, 1H), 1.27 (s, 3H), 1.06 (dd, J = 4.8, 2.1 Hz, 6H), 0.55 (d, J = 6.9 Hz, 3H). The resulting solid was dissolved in CH2Cl2–MeOH (1:1) and acidified with 1M ethereal HCl. The mixture was concentrated in vacuo, then dried to yield 0.065 g (23%) of 6 as a white solid: mp 207–211 °C. Anal. (C29H37ClN2O3•2 H2O) C,H, N.
6-Hydroxy-1,2,3,4-tetrahydro-naphthalene-2(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (7a) Hydrochloride
A 1.0 M solution of BBr3 (8.2 mL. 8.2 mmol) in CH2Cl2 was added at -78 °C under N2 to 16b (0.39 g, 0.82 mmol) in CH2Cl2 (25 mL). The dark brown solution was allowed to stir at -78 °C for 0.5 h and allowed to warm to room temperature. A saturated solution of NaHCO3 (50 mL) was cautiously added, and the biphasic mixture was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography on silica using CHCl3–MeOH–NH4OH (8:1.8:0.2) as the eluent to provide 0.30 g (77%) of 7a as a colorless oil. The hydrochloride salt was prepared by adding a 1.0 M solution of HCl in Et2O to the free base in MeOH. The solution was concentrated under reduced pressure and the resulting solid recrystallized from EtOH–Et2O to provide 7a•HCl as white plates: mp 189–191 °C; [α]22D +113.7 °C (c = 0.18, MeOH). 1H NMR (CD3OH) δ 0.74 (d, J = 6.78 Hz, 3 H), 0.90 (d, J = 6.78 Hz, 3 H), 0.93 (d, J = 6.78 Hz, 3 H), 1.27 (s, 3 H), 1.55 (d, J = 12.81 Hz, 1 H), 1.68–1.89 (m, 2 H), 1.95 (m, 2 H), 2.36–2.81 (m, 12H), 4.02 (ddd, J = 9.61, 5.09, 4.90 Hz, 1 H), 6.50 (d, J = 2.26 Hz, 1 H), 6.57 (ddd, J = 15.26, 8.10, 2.26 Hz, 2 H), 6.70–6.80 (m, 2 H), 6.85 (d, J = 8.29 Hz, 1 H), 7.10 (t, J = 8.10 Hz, 1 H), 7.81 (br s, 1 H). Anal. (C29H41ClN2O3•0.75 H2O) C, H, N.
6-Hydroxy-1,2,3,4-tetrahydro-naphthalene-2(-)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (7b) Hydrochloride
A 1.0 M solution of BBr3 (8.2 mL, 8.2 mmol) in CH2Cl2 was added at -78 °C under N2 to 16a (0.70 g, 1.45 mmol) in CH2Cl2 (50 mL). The dark brown solution was allowed to stir at -78 °C for 0.5 h and allowed to warm to room temperature. A saturated solution of NaHCO3 (100 mL) was cautiously added, and the biphasic mixture was extracted with EtOAc (3 × 150 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography on using silica CHCl3–MeOH–NH4OH (8:1.8:0.2) as the eluent to provide 0.057 g (83%) of 7b as a colorless oil. The hydrochloride salt was prepared by adding a 1.0 M solution of HCl in Et2O to 7b in MeOH. The solution was concentrated under reduced pressure and recrystallized from EtOH–Et2O to provide 7b•HCl as tan cubes: mp 193–195 °C; [α]22D + 67.0 °C (c = 0.22, MeOH). 1H NMR (CD3OD) δ 0.76 (d, J = 7.32 Hz, 3 H), 0.91 (d, J = 6.84 Hz, 3 H), 0.95 (d, J = 6.84 Hz, 3 H), 1.27–1.30 (s, 3 H), 1.57 (d, J = 11.23 Hz, 1 H), 1.75–1.86 (m, 2 H), 1.95–2.03 (m, 2 H), 2.29 (td, J = 12.57, 4.15 Hz, 1 H), 2.34–2.41 (m, 1 H), 2.42–2.87 (m, 10 H), 4.02 (dt, J = 9.77, 4.88 Hz, 1 H), 6.49 (m, 1 H), 6.52 (dd, J = 8.30, 2.44 Hz, 1 H), 6.58 (dd, J = 7.81, 1.95 Hz, 1 H), 6.74 (m, 1 H), 6.77 (d, J = 7.81 Hz, 1 H), 6.82 (d, J = 8.30 Hz, 1 H), 7.10 (t, J = 8.06 Hz, 1H). Anal. (C29H41ClN2O3•1.5 H2O) C, H, N.
7-Hydroxy-3,4-dihydro-1H-isochromene-3-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (8a,b)
7-Hydroxy-3,4-dihydro-1H-isochromene-3-carboxylic acid [(±)-29, 66 mg, 0.34 mmol] was added to a solution of 1227 (98 mg, 0.34 mmol) and BOP reagent (150 mg, 0.34 mmol) in THF (20 mL) and was allowed to stir under nitrogen for 15 min. Triethylamine (69 mg, 0.68 mmol) in THF (10 mL) was added, and the reaction was allowed to stir at room temperature for 3 h. To the reaction mixture were added NaHCO3 (30 mL) and Et2O (30 mL), and the organic layer was separated, washed with brine, dried, (Na2SO4), and concentrated under reduced pressure to afford an oil. The oil was purified by medium pressure silica gel chromatography, eluting with CHCl3–CMA80 (1:1) to afford 0.129 g of a pale yellow oil as a mixture of 8a and 8b. The diastereomers were purified using preparative HPLC. A sample of 41 mg of 8a,b in DMSO (200 μL) was purified via reversed phase HPLC using an isocratic MeOH–H2O (0.1% DEA) solvent system at 19 mL/min for 35 min, and 19-mL fractions were collected. Fractions were combined to obtain two pools. The first pool contained 21.6 mg of 100% pure compound 8a (the first compound to elute), and the second pool contained 16 mg of 8b, the second peak determined to be 87% pure. The second peak was repurified to afford 9.2 mg of 8b at 98.7% purity. The purity of isolates was determined via analytical HPLC using an isocratic solvent system at MeOH–H2O (70:30) (0.1% DEA) for 15 min. Chromatograms were observed at 220 nm, and the samples were dissolved in MeOH.
7-Hydroxy-3,4-dihydro-1H-isochromene-3-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (8a, peak 1)
[α]22D +108.6 °C, (c 0.11, MeOH). 1H NMR (CDCl3) δ 0.71 (d, J = 6Hz, 3H), 0.93 (m, 6H), 1.28 (m, 4H), 1.56 (d, J = 12 Hz, 1H), 1.87 (m, 1H), 1.95 (m, 1H), 2.26 (ddd, 1H), 2.41–2.55 (m, 4H), 2.64–2.80 (m, 2H), 2.90–2.96 (dd, J = 3, 18 Hz, 1H), 3.99 (m, 1H), 4.14 (dd, J = 6, 12 Hz, 1H), 4.84 (d, 1H), 4.87 (d, J = 12 Hz, 1H), 6.45 (s, 1H), 6.58 (m, 2H), 6.71 (m, 2H), 6.91 (d, J = 9 Hz, 1H), 7.07 (dd, J = 9, 9 Hz, 1H). HRMS m/z 467.2908 (M+H)+, predicted 467.2910.
7-Hydroxy-3,4-dihydro-1H-isochromene-3-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (8b, peak 2)
[α]22D −16.3 °C (c 0.08, MeOH). 1H NMR (CDCl3) δ 0.71 (d, J = 6Hz, 3H), 0.93 (m, 6H), 1.28 (m, 4H), 1.56 (d, J = 12 Hz, 1H), 1.87 (m, 1H), 1.95 (m, 1H), 2.26 (ddd, 1H), 2.41–2.55 (m, 4H), 2.64–2.80 (m, 2H), 2.90–2.96 (dd, J = 3, 18 Hz, 1H), 3.99 (m, 1H), 4.14 (dd, J = 6, 12 Hz, 1H), 4.84 (d, 1H), 4.87 (d, J = 12 Hz, 1H), 6.45 (s, 1H), 6.58 (m, 2H), 6.71 (m, 2H), 6.91 (d, J = 9 Hz, 1H), 7.07 (dd, J = 9, 9 Hz, 1H). HRMS m/z 467.2905 (M+H)+, predicted 467.2910.
7-Hydroxyisothiochroman-3(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (9a)
A 1.0 M solution of BBr3 (9.1 mL, 9.1 mmol) in CH2Cl2 was added at -78 °C under N2 to 22a (0.45 g, 0.91 mmol) in CH2Cl2 (100 mL). The dark brown solution was allowed to stir at -78 °C for 0.5 h and allowed to warm to 0 °C for 2 h. A saturated solution of NaHCO3 (100 mL) was cautiously added, and the biphasic mixture was extracted with EtOAc (3 × 150 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography on silica gel using CHCl3–MeOH–NH4OH (8:1.8:0.2) as an eluent to provide a 0.43 g (98%) of 9a tan semisolid. The solid was recrystallized from acetone–petroleum ether to afford 9a as white needles: mp 133–135 °C; [α]22D +108.7 °C, (c 0.20, MeOH). 1H NMR (CD3OD) δ 0.69–0.74 (m, 9H), 0.89–0.98 (m, 1H), 1.28 (s, 3H), 1.52–1.59 (d, J = 12.9 Hz, 1H), 1.64–1.68 (m, 1H), 1.94–1.96 (m, 1H), 2.17–2.47 (m, 4H), 2.58–2.62 (d, J = 11.3 Hz, 1H), 2.72–2.75 (d, J = 11.3 Hz, 1H), 2.89–2.96 (dd, J = 5.3, 15 Hz, 1H), 3.11–3.18 (dd, J = 7.54, 14.3 Hz, 1H), 3.62–3.77 (m, 3H), 3.83–3.90 (m, 1H), 6.54–6.65 (m, 3H), 6.71–6.76 (m, 2H), 6.91–6.94 (d, J = 8.2 Hz, 1H), 7.06–7.11 (t, J = 7.9 Hz, 1H). Anal. (C28H38N2O3S•0.25 H2O) C, H, N.
7-Hydroxyisothiochroman-3(-)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide Hydrochloride (9b)
A 1.0 M solution of BBr3 (9.0 mL, 9.0 mmol) in CH2Cl2 was added at -78 °C under N2 to 22b (0.44 g, 0.90 mmol) in CH2Cl2 (100 mL). The dark brown solution was allowed to stir at -78 °C for 0.5 h and allowed to warm to 0 °C for 2 h. A saturated solution of NaHCO3 (100 mL) was cautiously added, and the biphasic mixture was extracted with EtOAc (3 × 150 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography on silica gel using CHCl3–MeOH–NH4OH (8:1.8:0.2) as an eluent to provide 0.40 g (93%) of 9b as a tan semisolid. 1H NMR (CD3OD) δ 0.71–0.74 (d, J = 6.8 Hz, 3H), 0.83–0.85 (d, J = 6.8 Hz, 3H), 0.88–0.90 (d, J = 6.8 Hz, 3H), 1.06–1.13 (m, 1H), 1.26 (s, 3H), 1.50–1.54 (d, J = 12.4 Hz, 1H), 1.79–1.94 (m, 2H), 2.15–2.39 (m, 4H), 2.48 (brs, 1H), 2.71–2.75 (d, J = 11Hz, 1H), 2.97–3.10 (m, 2H), 3.58–3.78 (m, 3H), 3.85–3.91 (m, 1H), 6.57–6.60 (d, J = 7.9 Hz, 1H), 6.63 (m, 2H), 6.73 (m, 2H), 6.95–6.98 (d, J = 8.2 Hz, 1H), 7.06–7.12 (t, J = 7.9 Hz, 1H). The hydrochloride salt was prepared by adding a 1.0 M solution of HCl in Et2O to 9b in MeOH. The solution was concentrated under reduced pressure and recrystallized from EtOH–Et2O to provide 9b•HCl as white cubes: mp 224–227 °C (191–194 °C softens), [α]22D +62.1 °C, (c 0.38, MeOH). Anal. C, H, N.
7-Hydroxy-2-oxo-2-isothiochroman-3(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (10a,b) Resorcylate
3-Chloroperoxybenzoic acid (0.94 g, 0.41 mmol) was added to an ice-cold solution of 9a (0.20 g, 0.41 mmol) in CH2Cl2 (20 mL). The solution was allowed to stir at 0 °C for 30 min and was quenched by the addition of saturated sodium bicarbonate. The slurry was extracted with CH2Cl2 (3 × 30 mL), dried (MgSO4), and concentrated under reduced pressure to afford a mixture of diastereomers as a pale yellow oil. The diastereomers were separated by silica gel chromatography using CHCl3–MeOH–NH4OH (8:1.8:0.2) as the eluent to provide 10a (first to elute, 28 mg, 14 %) and 10b (second to elute, 21 mg, 10%) as pale yellow oils. Additional product was collected as a mixture of diastereomers.
7-Hydroxy-2-oxo-2-isothiochroman-3(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (10a) Resorcylate
The first spot to elute off the column was converted to the resorcylate salt by dissolving 3,5-dihydroxybenzoic acid (1.1 eq) in acetone and adding the amine in acetone. The solution was concentrated, and the solid was recrystallized from EtOAc and hexane to provide 17 mg of 10a as a white solid: mp 170–173 °C; [α]22D +4.7 °C, (c 0.11, MeOH). 1H NMR (CD3OD) δ 0.80 (d, J = 6.8 Hz, 3H), 1.01 (m, 6H), 1.27–1.40 (m, 4H), 1.72 (d, 1H), 1.85 (m, 1H), 2.13 (m, 1H), 2.41 (m, 1H), 2.79–2.86 (m, 3H), 3.0–3.08 (m, 3H), 3.18–3.23 (m, 1H), 3.62 (dd, J = 6, 9Hz, 1H), 4.10–4.16 (m, 3H), 6.38 (dd, 1H), 6.59–6.79 (m, 5H), 6.91 (dd, 1H), 6.96–6.99 (d, J = 9 Hz, 1H), 7.10–7.15 (m, 2H). Anal. (C35H44N2O8S•1.5 H2O) C, H, N.
7-Hydroxy-2-oxo-2-isothiochroman-3(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (10b) Resorcylate
The second spot to elute off the column was converted to the resorcylate salt by dissolving 3,5-dihydroxybenzoic acid (1.1 eq) in acetone and adding the amine in acetone. The solution was concentrated, and the solid was recrystallized from EtOAc and hexane to provide 12 mg of 10b resorcylate as a white solid: mp 186–189 °C; [α]22D +102.0 °C, (c 0.15, MeOH). 1H NMR (CD3OD) δ 0.82 (dd, J = 6, 3Hz, 3H), 0.97 (m, 6H), 1.26 (dd, J = 4, 12Hz 1H), 1.36 (s, 3H), 1.72–1.76 (d, 8 Hz, 1H), 1.90 (m, 1H), 2.18 (m, 1H), 2.46 (m, 1H), 2.91–3.32 (m, 6H), 3.51 (m, 1H), 3.72–3.87 (m, 1H), 4.0–4.33 (m, 3H), 6.37 (dd, J = 6, 6Hz, 1H), 6.60–6.78 (m, 5H), 6.91 (d, J = 3Hz, 2H), 7.01–7.15 (m, 2H). Anal. (C35H44N2O8S•1.25 H2O) C, H, N.
2(+)-6-Methoxy-1,2,3,4-tetrahydronaphthalene-2-carboxylic Acid [(+)-13]
A 30% solution of hydrogen peroxide (6.96 mmol, 0.24 mL) in H2O was added at 0 °C to a solution of 15a (0.42 g, 1.16 mmol) in 3:1 THF–H2O (25 mL). Lithium hydroxide hydrate (0.098 g, 2.32 mmol) was added to the solution in portions. The suspension was allowed to stir for 0.5 h at 0 °C and for 2 h at room temperature. A 1.5 N solution of Na2SO3 (15 mL) was added in a dropwise manner, and the biphasic solution was basified (pH ≈ 9) with saturated sodium bicarbonate solution. The solution was extracted (2 × 50 mL) with EtOAc, made acidic to pH 3 with HCl (10 M solution), and extracted (3 × 100 mL) with CH2Cl2. The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a white solid. The solid was recrystallized from EtOAc–petroleum ether to provide 0.22 g (92%) of (+)-13 as white needles: mp 129–130 °C; [α]22D +57.27 °C, (c 0.22, CHCl3). 1H-NMR (CDCl3) δ 1.87–1.90 (m, 1H), 2.20–2.25 (m, 1H), 2.74–2.98 (m, 5H), 3.77 (s, 3H), 6.63 (s, 1H), 6.68–6.72 (dd, J = 2.7, 8.4 Hz, 1H), 7.0–7.03 (d, J = 8.4 Hz, 1H).
2(-)-6-Methoxy-1,2,3,4-tetrahydronaphthalene-2-carboxylic Acid [(-)-13]
A 30% solution of hydrogen peroxide (3.3 mmol, 0.11 mL) in H2O was added at 0 °C to a solution of 15b (0.20 g, 0.55 mmol) in THF–H2O (3:1) (15 mL). Lithium hydroxide hydrate (0.046 g, 1.10 mmol) was added to the solution in portions. The suspension was allowed to stir for 0.5 h at 0 °C and for 2 h at room temperature. A 1.5 N solution of Na2SO3 (10 mL) was added in a dropwise manner, and the biphasic solution was basified (pH ≈ 10) with saturated sodium bicarbonate solution. The solution was extracted (2 × 50 mL) with EtOAc, made acidic to pH 3 with HCl (10 M solution), and extracted (3 × 100 mL) with CH2Cl2. The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a white solid. The solid was recrystallized from EtOAc–petroleum ether to provide 0.102 g (90%) of (-)-13 as white needles: mp 121–122 °C; [α]22D -56.9 °C (c 0.25, CHCl3). 1H NMR (CDCl3) δ 1.87–1.90 (m, 1H), 2.20–2.25 (m, 1H), 2.74–2.98 (m, 5H), 3.77 (s, 3H), 6.63 (s, 1H), 6.68–6.72 (dd, J = 2.7, 8.4 Hz, 1H), 7.0–7.03 (d, J = 8.4 Hz, 1H).
(3aR-cis)-3-(6-Methoxy-1,2,3,4-tetrahydronaphthalene-2(+ and -)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (15a,b)
A 2.0 M solution of thionyl chloride (7.25 mL, 14.3 mmol) in CH2Cl2 was added to a solution of (±)-1329 (0.29 g, 1.43 mmol) in toluene (20 mL). The solution was heated at reflux for 8 h, cooled to room temperature, and concentrated under reduced pressure to provide 6-methoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl chloride as a tan solid. In a separate flask, a 0.50 M solution of ethyl lithium (3.0 mL, 1.50 mL) in benzene–cyclohexane (90:10) was added to a solution of (3aR-cis)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (0.25 g, 1.43 mmol) in THF (20 mL) at 0 °C under N2. The suspension was allowed to stir at 0 °C for 0.5 h and was cooled to -78 °C. A solution of 6-methoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl chloride (0.29 g, 1.43 mmol) in THF (10 mL) was then added in a dropwise manner to the -78 °C slurry. The resulting slurry was allowed to warm to room temperature over 2 h, and water (100 mL) was added. The suspension was extracted with CH2Cl2 (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a mixture of 15a and 15b as a tan solid. The two compounds were separated using silica gel medium pressure column chromatography petroleum ether–Et2O (70:30) to provide each of the diastereomers in approximately 50% theoretical yield. The yield improves with additional chromatography. The less polar spot was later identified as the (+) isomer while the more polar was (-).
(3aR-cis)-3-(6-Methoxy-1,2,3,4-tetrahydronaphthalene-2(+)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (15a)
The solid was recrystallized from EtOAc–petroleum ether to provide 0.12 g (46%) of 15a as a white solid: mp 168–169 °C. 1H-NMR (CDCl3) δ 1.80–1.85 (m, 1H), 2.10–2.21 (m, 1H), 2.71–3.13 (m, 4H), 3.38 (d, J = 3.3 Hz, 2H), 3.76 (s, 3H), 3.84 (m, 1H), 5.27 (m, 1H), 5.96–5.99 (d, J = 9 Hz, 1H), 6.62 (s, 1H), 6.70–6.71 (dd, J = 2.4, 8.1 Hz, 1H), 6.99–7.04 (dd, J = 3.6, 8.4 Hz, 1H), 7.24–7.32 (m, 3H), 7.57–7.60 (d, J = 7.5 Hz, 1H).
(3aR-cis)-3-(6-Methoxy-1,2,3,4-tetrahydronaphthalene-2(-)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (15b)
The solid was recrystallized from EtOAc-petroleum ether to provide 0.13 g (50%) of 15b as a white solid: mp 162–164 °C. 1H-NMR (CDCl3) δ 1.85–1.98 (m, 1H), 2.12–2.18 (m, 1H), 2.84–2.95 (m, 4H), 3.40–3.41 (d, J = 3.3 Hz, 2H), 3.77 (s, 3H), 3.85–3.95 (m, 1H), 5.28–5.33 (m, 1H), 5.97–5.99 (d, J = 6.9 Hz, 1H), 6.57–6.69 (m, 2H), 6.95–6.98 (d, J = 8.4 Hz, 1H), 7.26–7.42 (m, 3H), 7.60–7.62 (d, J = 7.5 Hz, 1H).
6-Methoxy-1,2,3,4-tetrahydro-naphthalene-2(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (16a)
2(+)-6-Methoxy-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [(+)-13, 0.22 g, 1.07 mmol] was added under N2 to a solution of BOP (0.47 g, 1.07 mmol), TEA (0.23 g, 2.35 mmol), and 1227 (0.31 g, 1.07 mmol) in anhydrous THF (50 mL). The solution was allowed to stir at room temperature for 6 h, and a saturated NaHCO3 solution (100 mL) was added. The biphasic mixture was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide an oil. The oil was purified using medium pressure column chromatography on silica gel using CHCl3–MeOH–NH4OH (9:0.8:0.2) at the eluent to provide 0.39 g (77%) of 16a as a colorless oil. 1H NMR (CDCl3) δ ppm 0.72 (d, J = 6.78 Hz, 3 H), 0.82–0.96 (m, 6 H), 1.26 (s, 3 H), 1.55 (d, J = 12.43 Hz, 1 H), 1.78–2.07 (m, 4 H), 2.18–2.88 (m, 12 H), 3.73 (s, 3 H), 4.00–4.16 (m, 1 H), 6.05 (d, J = 7.54 Hz, 1 H), 6.57 (d, J = 2.64 Hz, 1 H), 6.62–6.77 (m, 3 H), 6.84 (m, 1 H), 6.93 (d, J = 8.67 Hz, 1 H), 7.11 (t, J = 7.91 Hz, 1 H).
6-Methoxy-1,2,3,4-tetrahydro-naphthalene-2(-)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (16b)
2(-)-6-Methoxy-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [(-)-13, 0.31 g, 1.48 mmol] was added under N2 to a solution of BOP (0.65 g, 1.48 mmol), TEA (0.33 g, 3.26 mmol), and 12 (0.43 g, 1.48 mmol) in anhydrous THF (65 mL). The solution was allowed to stir at room temperature for 6 h, and a saturated NaHCO3 solution (100 mL) was added. The biphasic mixture was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide an oil. The oil was purified using medium pressure column chromatography on silica gel using CHCl3–MeOH–NH4OH (9:0.8:0.2) as the eluent to provide 0.70 g (98%) of 16b as a colorless oil. 1H NMR (CDCl3) δ ppm 0.66–0.78 (d, J = 6.9 Hz, 3 H), 0.83–0.97 (m, 6 H), 1.25 (s, 3 H), 1.53 (d, J = 12.43 Hz, 1 H), 1.78–2.10 (m, 4 H), 2.20–2.97 (m, 12 H), 3.73 (s, 3 H), 4.03 (m, 1 H), 6.03 (d, J = 7.54 Hz, 1 H), 6.57 (d, J = 2.26 Hz, 1 H), 6.61–6.75 (m, 3 H), 6.82 (m, 1H), 6.90 (d, J = 8.29 Hz, 1 H), 7.10 (t, J = 7.72 Hz, 1 H).
7-Methoxy-isothiochroman-4-one-3-carboxylic Acid Methyl Ester (18)
A 2.0 M solution of LDA in heptane–THF–ethylbenzene (1.61 mL, 3.21 mmol) was added in a dropwise manner to a solution of 7-methoxyisothiochroman-4-one (17)31 (0.50 g, 2.57 mmol) in THF (50 mL) at -78 °C under N2. After 30 min at -78 °C, HMPA (0.46 g, 2.57 mmol) and methyl cyanoformate (0.27 g, 3.21 mmol) were added, and the yellow solution was allowed to stir at -78 °C for 30 min. The solution was then allowed to warm to room temperature, and a saturated solution of NH4Cl (100 mL) was added. The slurry was extracted with EtOAc (3 × 75 mL), and the organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a bright yellow oil. The oil was purified on silica gel medium pressure chromatography using petroleum ether–EtOAc (9:1) as the eluent to provide 0.51 g (78%) of 18 as a bright yellow oil. 1H-NMR (CDCl3) δ 3.73 (s, 2H), 3.83 (s, 3H), 3.85 (s, 3H), 6.67 (d, J = 3 Hz, 1H), 6.45 (dd, J = 3, 8.7 Hz, 1H), 7.80 (d, J = 8.7 Hz, 1H), 12.52 (s, 1H).
7-Methoxy-isothiochroman-3-carboxylic Acid Methyl Ester (19)
Triethylsilane (8.08 mmol, 0.94 g) was added to a solution of 18 (0.51 g, 2.02 mmol) in trifluoroacetic acid (15 mL) at room temperature under N2. The reaction mixture was allowed to stir at room temperature for 2 h and was concentrated under reduced pressure. The resulting oil was dissolved in EtOAc (100 mL) and washed with a saturated NaHCO3 solution (3 × 75 mL). The organic extracts were combined, dried (MgSO4), and concentrated to provide an oil. The oil was purified by silica gel medium pressure chromatography using petroleum ether–EtOAc (9:1) as the eluent to provide 0.34 g, (70%) of 19 as a pale yellow oil. 1H NMR (CDCl3) δ 3.14 (m, 2H), 3.58–3.63 (d, J = 15 Hz, 1H), 3.73–3.86 (m, 8H), 6.70 (d, J = 3 Hz, 1H), 6.75 (dd, J = 3, 9 Hz, 1H), 7.10 (d, J = 9 Hz, 1H).
7-Methoxy-isothiochroman-3-carboxylic Acid [(±)-20]
Potassium hydroxide (0.80 g, 14.3 mmol) was added to a solution of 19 (0.34 g, 1.43 mmol) in MeOH (50 mL). The solution was heated at 60 °C for 2 h, cooled to room temperature, and diluted with H2O (100 mL). The solution was made acidic with 6 N HCl and extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated to give 0.28 g (88%) of (±)-20 as a pale yellow solid. The solid was used in the next step without further purification.
3(+)-7-Methoxy-isothiochroman-3-carboxylic Acid [(+)-20]
Lithium hydroxide hydrate (0.093 g, 2.2 mmol) was added at 0 °C to a solution of 21a (0.42 g, 1.10 mmol) in THF–H2O (3:1) (25 mL). The suspension was allowed to stir for 0.5 h at 0 °C. The reaction was made basic (pH ≈ 9) with saturated sodium bicarbonate solution, and the solution was extracted with Et2O (1 × 100 mL), made acidic to pH 3 with HCl (6 N solution), and extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide 0.25 g (100%) of (+)-20 as a white solid. The solid was recrystallized from toluene–petroleum ether to provide (+)-20 as tan cubes: mp 117–118 °C; [α]22D +98.3 °C (c 0.24, MeOH). 1H NMR (CD3OD) δ 2.97–3.01 (dd, J = 9.3, 15 Hz, 1H), 3.10–3.17 (dd, J = 5.1, 15.3, 1H), 3.63–3.88 (m, 6H), 6.75–6.77 (m, 2H), 7.08–7.11 (d, J = 8.2 Hz, 1H).
3(-)-7-Methoxy-isothiochroman-3-carboxylic Acid [(-)-20]
Lithium hydroxide hydrate (0.055 g, 1.32 mmol) was added at 0 °C to a solution of 21b (0.25 g, 0.66 mmol) in THF–H2O (3:1) (15 mL). The suspension was allowed to stir for 0.5 h at 0 °C. The reaction was made basic (pH ≈ 9) with saturated sodium bicarbonate solution, and the solution was extracted with Et2O (1 × 100 mL), made acidic to pH 3 with 6 N HCl, and extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide 0.14 g (92%) of (-)-20 as a white solid (0.136 g, 92%). The solid was recrystallized from toluene–petroleum ether to provide (-)-20 as pale yellow needles: mp 121–122 °C; [α]22D -100.8 °C (c 0.26, MeOH). 1H NMR (CD3OD) δ 2.97–3.01 (dd, J = 9.3, 15 Hz, 1H), 3.10–3.17 (dd, J = 5.1, 15.3, 1H), 3.63–3.88 (m, 6H), 6.75–6.77 (m, 2H), 7.08–7.11 (d, J = 8.2 Hz, 1H).
(3aR-cis)-3-(7-Methoxy-isothiochroman-3(+ and -)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (21a,b)
A 2.0 M solution of oxalyl chloride (3.57 mL, 7.14 mmol) in CH2Cl2 was added under N2 to a solution of (±)-20 (0.80 g, 3.57 mmol) and a drop of DMF in CH2Cl2 (100 mL). The solution was allowed to stir at room temperature for 3 h and was concentrated under reduced pressure to provide 7-methoxy-isothiochroman-3-carbonyl chloride as a tan oil. 7-Methoxy-isothiochroman-3-carbonyl chloride was used in the next step without further purification. In a separate flask, a 0.50 M solution of ethyl lithium (8.6 mL, 4.28 mmol) in benzene–cyclohexane 90:10 was added to a solution of (3aR-cis)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (14, 0.75 g, 4.28 mmol) in THF (100 mL) at 0 °C under N2. The suspension was allowed to stir at 0 °C for 0.5 h and was then cooled to -78 °C. A solution of 7-methoxyisothiochroman-3-carbonyl chloride (0.86 g, 3.57 mmol) in THF (10 mL) was added in a dropwise manner to the -78 °C slurry. The resulting slurry was allowed to warm to room temperature over 2 h, and water (150 mL) was then added. The suspension was extracted with CH2Cl2 (3 × 150 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide a mixture of 21a and 21b as a tan solid. The mixture was separated by silica gel medium pressure column chromatography using petroleum ether–Et2O (60:40) as the eluent to provide each of the diastereomers in 62% [21a, (+)-isomer] and 37% [21b, (-)-isomer] theoretical yield. The yield improves with additional chromatography. The higher Rf spot on TLC was identified as the (+)-isomer while the more polar spot was the (-)-isomer.
(3aR)-cis)-3-(7-Methoxy-isothiochroman-3(+)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (21a)
The solid was recrystallized from EtOAc–petroleum ether to provide 0.42 g (62%) of 21a as a white solid: mp 146–147 °C. 1H-NMR (CDCl3) δ 3.09–3.16 (dd, J = 6, 15.6 Hz, 1H), 3.20–3.28 (dd, J = 7.2, 15.3 Hz, 1H), 3.38–3.39 (d, J = 3.6 Hz, 2H), 3.59–3.64 (d, J = 15 Hz, 1H), 3.79 (s, 3H), 3.85–3.90 (d, J = 15 Hz, 1H), 4.97–5.01 (m, 1H), 5.30–5.35 (m, 1H), 5.92–5.95 (d, J = 7 Hz, 1H), 6.71–6.72 (d, J = 2.7 Hz, 1H), 6.75–6.79 (dd, J = 2.4, 8.4 Hz, 1H), 7.06–7.09 (d, J = 8.4 Hz, 1H), 7.23–7.38 (m, 3H), 7.58–7.61 (d, J = 7.5 Hz, 1H).
(3aR-cis)-3-(7-Methoxy-isothiochroman-3(-)-carbonyl)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-one (21b)
The solid was recrystallized from EtOAc–petroleum ether to provide 0.25 g (37%) of 21b as a white solid: mp 176–178 °C. 1H-NMR (CDCl3) δ 3.14–3.19 (dd, J = 6, 12.6 Hz, 1H), 3.24–3.29 (dd, J = 7.5, 15.3 Hz, 1H), 3.39–3.40 (d, J = 3.6 Hz, 2H), 3.48–3.55 (d, J = 15 Hz, 1H), 3.79 (s, 3H), 3.83–3.88 (d, J = 15 Hz, 1H), 4.91–4.95 (m, 1H), 5.29–5.33 (m, 1H), 5.96–5.99 (d, J = 7 Hz, 1H), 6.71–6.72 (d, J = 2.7 Hz, 1H), 6.76–6.80 (dd, J = 2.4, 8.4 Hz, 1H), 7.10–7.12 (d, J = 8.4 Hz, 1H), 7.26–7.38 (m, 3H), 7.58–7.61 (d, J = 7.5 Hz, 1H).
7-Methoxy-isothiochroman-3(+)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R)-(4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (22a)
3(+)-7-Methoxyisothiochroman-3-carboxylic acid [(+)-20, (0.25 g, 1.12 mmol)] was added under N2 to a solution of BOP (0.50 g, 1.12 mmol), TEA (0.23 g, 2.24 mmol), and 1227 (0.33 g, 1.12 mmol) in anhydrous THF (50 mL). The solution was allowed to stir at room temperature for 6 h, and a saturated NaHCO3 solution (100 mL) was added. The biphasic mixture was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide an oil. The oil was purified using medium pressure column chromatography using CHCl3–MeOH–NH4OH (9:0.8:0.2) to provide 0.5 g (81%) of 22a as a pale yellow semisolid. 1H NMR (CDCl3) δ 0.49–0.55 (m, 6H), 0.67–0.69 (d, J = 6 Hz, 3H), 1.24 (s, 3H), 1.48–1.52 (d, J = 12 Hz, 1H), 1.62–1.70 (m, 1H), 1.86–1.88 (m, 1H), 2.14–2.52 (m, 6H), 2.61–2.71 (m, 2H), 2.89–2.95 (dd, J = 5.1, 14.4 Hz, 1H), 3.31–3.38 (dd, J = 5.4, 14.4 Hz, 1H), 3.57–3.62 (d, J = 13.8 Hz, 1H), 3.65–3.69 (d, J = 13.8 Hz, 1H), 3.74 (s, 3H), 3.84–3.87 (m, 1H), 6.70–6.72 (m, 3H), 6.84–6.89 (m, 2H), 7.03–7.12 (m, 2H).
7-Methoxy-isothiochroman-3(-)-carboxylic acid-{1-[4-(3-hydroxyphenyl)-(3R,4R)-trans-dimethyl-piperidinylmethyl]-(2S)-methylpropyl}amide (22b)
3(-)-7-Methoxy-isothiochroman-3-carboxylic acid [(-)-20, 0.24 g, 1.07 mmol] was added under N2 to a solution of BOP (0.47 g, 1.07 mmol), TEA (0.21 g, 2.14 mmol), and 1227 (0.31 g, 1.07 mmol) in anhydrous THF (50 mL). The solution was allowed to stir at room temperature for 6 h, and a saturated NaHCO3 solution (100 mL) was added. The biphasic mixture was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide an oil. The oil was purified by silica gel medium pressure column chromatography using CHCl3–MeOH–NH4OH (9:0.8:0.2) as the eluent to provide 0.44 g (84%) of 22b as a pale yellow semisolid. 1H NMR (CDCl3) δ 0.65–0.68 (d, J = 6.9 Hz, 3H), 0.77–0.79 (d, J = 4.2 Hz, 3H), 0.84–0.86 (d, J = 6.6 Hz, 3H), 1.27 (s, 3H), 1.47–1.51 (d, J = 12.3 Hz, 1H), 1.80–2.70 (m, 11H), 3.03–3.09 (dd, J = 5.4, 14.7 Hz, 1H), 3.17–3.24 (dd, J = 6.3, 14.4 Hz, 1H), 3.60–3.65 (d, J = 14.1 Hz, 1H), 3.67–3.72 (d, J = 14.1 Hz, 1H), 3.77 (s, 3H), 3.83–3.87 (m, 1H), 6.59–6.83 (m, 5H), 7.05–7.16 (m, 2H).
7-Methoxy-1H-isochromen-4(3H)-one (24)
Oxalyl chloride (92.5 mL, 2.0 M in CH2Cl2, 185 mmol) was added dropwise to a solution of (3-methoxybenzyloxy)acetic acid (23)34 (23.2 g, 0.12 mol) in CH2Cl2 (850 mL) at 0 °C under N2, and a few drops of DMF were added to initiate the reaction. The reaction mixture was stirred for 2 h at room temperature and was concentrated to give a brown oil. Chlorobenzene (500 mL) was then added, and the mixture was allowed to stir using a mechanical stirrer. The mixture was cooled to 0 °C in an ice bath, SnCl4 (29 mL, 246.8 mmol) was added slowly, and the reaction mixture was allowed to stir at 0 °C under N2 for 1 h. The reaction mixture was quenched with saturated NaHCO3 (150 mL) and H2O (150 mL) and then extracted with CH2Cl2 (3 × 250 mL). The combined organics were dried over NaSO4, and the solvent was removed under reduced pressure to afford crude compound which was purified by silica gel medium pressure column chromatography using 10–40% EtOAc in hexane to give 10.9 g (52%) of 24 as a white solid. 1H NMR (CDCl3) δ 8.02 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 7.9 Hz, 1H), 6.67 (s, 1H), 4.85 (s, 2H), 4.33 (s, 2H), 3.88 (s, 1H). 13C NMR (CDCl3, 75MHz) δ 193.1, 164.6, 144.6, 129.4, 123.6, 114.5, 109.1, 73.7, 68.4, 56.0. LCMS (APCI) m/z (M+H)+ 179.2.
7-Hydroxy-1H-isochromen-4(3H)-one (25)
7-Methoxy-1H-isochromen-4(3H)-one (24, 11.0 g, 0.062 mol) was dissolved in DMF (320 mL), and EtSNa (7.8 g, 0.093 mol) was added. The reaction mixture was heated at reflux for 3 h, H2O (450 mL) was added, and the mixture was extracted with EtOAc (300 mL). The aqueous layer was cooled, acidified to pH ≈ 4 with 1 M HCl, and then extracted with EtOAc (3 × 300 mL). The combined organics were washed with bleach (200 mL) and dried over sodium sulfate, and the solvent was removed under reduced pressure to afford crude product which was purified by silica gel medium pressure column chromatography using 10–50% EtOAc in hexane as the eluent to give 7.2 g (71%) of 25 as a colorless solid. 1H NMR (CD3OD) δ 7.85–7.80 (m, 1H), 6.77 (d, J = 6.4 Hz, 1 H), 6.6 (s, 1H), 4.76 (s, 3H), 4.22 (s, 2H). 13C NMR (CD3OD, 75MHz) δ 195.5, 165.2, 147.0, 130.3, 123.3, 116.8, 111.7, 74.4, 69.2. LCMS (ESI) m/z 165.3(M+H)+; 163.4 (M-H)+.
7-(Methoxymethoxy)-1H-isochromen-4(3H)-one (26)
7-Hydroxy-1H-isochromen-4(3H)-one (25, 7.0 g, 0.43 mol) was dissolved in CH2Cl2 (350 mL), and the solution was cooled to 0 °C under N2. i-Pr2EtN (8.3g, 0.064 mol) was added, and then MOMCl (5.15 g, 0.064 mol) was added slowly. The reaction mixture was stirred for 8 h at room temperature, and saturated NaHCO3 (200 mL) was added. The suspension was extracted with EtOAc (3 × 250 mL), the combined organics were dried over sodium sulfate, and the solvent was removed under reduced pressure. The solid was purified by silica gel medium pressure column chromatography using 10–40% EtOAc in hexane as the eluent to give 7.8 g (88%) of 26 as a colorless solid. 1H NMR (CDCl3) δ 8.02 (d, J = 8.6 Hz, 1H), 7.03 (d, J = 8.6 Hz, 1H), 6.84 (s, 1H), 5.24 (s, 2H), 4.85 (s, 2H), 4.33 (s, 2H), 3.49 (s, 3H). 13C NMR (CDCl3, 75MHz) δ 192.8, 161.8, 144.2, 128.9, 124.0, 116.1, 110.7, 94.1, 73.4, 68.1, 56.4. LCMS (APCI) m/z 209.3(M+H)+.
Methyl 7-(methoxymethoxy)-4-oxo-3,4-dihydro-1H-isochromen-3-carboxylate (27)
LDA (31 mL, 2.0 M in THF–heptane–ethylbenzene, 62.0 mmol) was then added to a solution of 26 (6.6 g, 0.032 mol) in THF (400 mL) at -78°C. The mixture was stirred at -78 °C for 30 min, HMPA (5.74 g, 5.6 mL, 0.032mol) was added followed by methylcyanoformate (5.4 g, 0.064 mol). The reaction mixture was stirred at -78 C for 2 h and warmed to room temperature. The reaction mixture was quenched with saturated NH4Cl (250 mL) and then extracted with EtOAc (3 × 300 mL). The combined organics were dried over sodium sulfate, and the solvent was removed under reduced pressure to afford crude compound which was purified by silica gel medium pressure column chromatography using 10–30% EtOAc in hexane as eluent to give 4.35 g (52%) of 27 as a colorless solid. NMR shows a mixture of ketone and enol tautomers. 1H NMR (CDCl3) δ 8.02 (d, J = 8.7 Hz, 0.7 H), 7.61 (d, J = 8.5 Hz, 0.2H), 7.03 (d, J = 8.7 Hz, 1H), 6.81 (s, 1H), 5.24 (s, 1H), 5.23 (s, 2H), 4.97–4.85 (m, 2H), 3.91 (s, 0.7H), 3.84 (s, 2.5H), 3.48 (s, 3H). 13C NMR (CDCl3, 75MHz) δ 187.1, 166.9, 162.3, 143.5, 129.6, 124.4, 116.5, 110.5, 94.2, 80.5, 65.9, 56.4, 52.8. LCMS (ESI) m/z 267.1(M+H)+.
Methyl 7-hydroxy-3,4-dihyro-1H-isochromene-3-carboxylate (28)
TFA (80 mL) was added to 27 (4.3 g, 0.016 mol) at 0 °C, and Et3SiH (7.52 g, 0.065 mol) was added. The reaction mixture was stirred at room temperature for 2 h and then was concentrated under reduced pressure. Saturated NaHCO3 (100 mL) was added, and the mixture was extracted with EtOAc (3 × 150 mL). The combined organics were dried (Na2SO4), and the solvent was removed under reduced pressure to afford crude compound which was purified by silica gel medium pressure column chromatography using 10–40% EtOAc in hexane as eluent to give 2.22 g (66%) of 28 as a colorless solid. 1H NMR (CDCl3) δ 6.99 (d, J = 8.0 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.48 (s, 1H), 5.76 (s, 1H), 4.91 (d, J = 15.0 Hz, 1H), 4.78 (d, J = 15.0Hz, 1H), 4.36 (t, J = 7.0 Hz, 1H), 3.82 (s, 3H), 2.99 (d, J = 6.9 Hz, 2H). 13C NMR (CDCl3, 75MHz) δ 171.9, 154.4, 134.8, 129.9, 123.3, 114.4, 110.7, 73.6, 67.8, 52.4, 30.1. LCMS (ESI) m/z 209.3 (M+H)+; 231.5 (M+Na)+.
7-Hydroxy-3,4-dihydro-1H-isochromene-3-carboxylic Acid (29)
Lithium hydroxide hydrate (45 mg, 1.08 mmol) was added to a solution of 28 (0.100 g, 0.48 mmol) in a mixture of THF–H2O–MeOH (1:1:1) (25 mL) at 0 °C. The solution was allowed to stir for 1 h and warmed to room temperature. A 1 N solution of HCl was added until the mixture was acidic and was then extracted with CHCl3 (3 × 50 mL). The organic extracts were combined, dried (MgSO4), and concentrated under reduced pressure to provide 0.75 g (82%) of (±)-29 as a white solid. This solid was used in the next step without further purification. 1H NMR (CD3OD) δ 2.96, (m, 3H), 4.32 (dd, J = 3, 9 Hz, 1H), 4.74 (m, 2H), 6.46 (s, 1H), 6.63 (d, J = 9 Hz, 1H), 6.95 (d, J = 9 Hz, 1H).
Compounds 3 and 2 Alignment
Three-dimensional structures of 3 and 2 were built using the Sybyl fragment library and optimized using the MMFF94 force-field and charges. A systematic conformational search followed by a multi-fit flexible structural alignment was performed using 2 as the rigid template (excepting the guanidinium group of 2 which was allowed to freely rotate during the multi-fit alignment). Six 2-3 atom pairs and a spring constant of 20 kcal/mol were used to align 3 to the 2 template.The atom pairs were the 3 and 2 phenolic ring centroid, 3 atoms 3, 4, 5, 3-methyl, 4-methyl paired with 2 atoms 14, 13, 15, 10, and 5 (morphine numbering). The centroid of the Tic aromatic ring of 3 was paired with the centroid of the guanidinium group of 2. After alignment, a final energy minimization was performed using the MMFF94 force-field to allow the structure of 3 to relax to a local minimum energy conformation. This process was repeated for each of the 10 lowest energy conformations of 3 obtained from an initial systematic conformational search in order to identify the lowest energy conformation of 3 that could provide a close alignment to the 2 pharmacophore template.
Evaluation of 7a to Block U50,488-Induced Diuresis
Adult male Sprague-Dawley rates (Charles River Laboratories, Raleigh, NC, USA) were used for these studies. The test compound and U50,488 doses were prepared fresh in distilled deionized water (vehicle) and administered (1 mL/kg body weight) via subcutaneous injection. Six groups of four rats were used to evaluate each test compound: vehicle control; agonist control (10 mg/kg); test compound 7a at 3, 10, or 30 mg/kg followed by agonist (10 mg/kg); and compound 7a followed by vehicle (30 mg/kg). Each rat was weighed prior to dosing. One rat from each group was dosed in succession and the pattern repeated to distribute any effects of time of day across all groups. After dosing, each rat was placed into a metabolic chamber and urine output was collected hourly for 5 h. Urine output for each collection period was calculated as (urine + collection tube weight)-collection tube tare weight. The effect of test compound on total urine output was assessed by analysis of variance with repeated measures (subject within Group) and factors of Group and Time and their interaction, or one-way ANOVA, where appropriate. A univariate ANOVA was run only if a significant effect was observed following the multivariate ANOVA. Significance was assumed at p<0.05 for the individual factors and p<0.1 for their interaction.
Supplementary Material

Acknowledgments
This research was supported by the National Institute on Drug Abuse Grant DA09045.
a Abbreviations
- GPCRs
G-protein-coupled receptors
- cDNAs
complementary deoxyribonucleic acid
- SAR
structure activity relationship
- [35S]GTPγS
sulfur-35 guanosine-5′-O-(3-thio)triphosphate
- DAMGO
[D-Ala2,MePhe4,Gly-ol5]enkephalin
- DPDPE
[D-Pen2,D-Pen5]enkephalin
- U69,593
(5α,7α,8β)-(-)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide
- CHO
Chinese hamster ovary
- GDP
guanosine diphosphate
- BOP
benzotriazole-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HMPA
hexamethylphosphoramide
- LOA
lithium diisopropylamide
- Tic
tetrahydroisoquinolinecarboxylic acid
- SAR
structure-activity relationship
Footnotes
Supporting Information Available: Elemental analysis data for compounds 6, 7a,b, 9a,b, and 10a,b and HPLC traces for 8a and 8b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- 2.Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll FI, Harris LS, Aceto MD. Effects of JDTic, a selective kappa-opioid receptor antagonist, on the development and expression of physical dependence on morphine using a rat continuous-infusion model. Eur J Pharmacol. 2005;524:89–94. doi: 10.1016/j.ejphar.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker BM, Koob GF. Pharmacological evidence for a motivational role of κ-opioid systems in ethanol dependence. Neuropsychopharmacology. 2007:1–10. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson KJ, Carroll FI, Negus SS, Damaj MI. Effect of the selective kappa-opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology (Berl) 2010:285–294. doi: 10.1007/s00213-010-1803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Shi YG, Woods JH, Watson SJ, Ko MC. Central kappa-opioid receptor-mediated antidepressant-like effects of nor-Binaltorphimine: behavioral and BDNF mRNA expression studies. Eur J Pharmacol. 2007;570:89–96. doi: 10.1016/j.ejphar.2007.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 11.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-Like Effects of κ-Opioid Receptor Antagonists in Models of Unlearned Learned Fear in Rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 12.Jarosz PA. The effect of kappa opioid receptor antagonism on energy expenditure in the obese Zucker rat. Biol Res Nurs. 2007;8:294–299. doi: 10.1177/1099800406298774. [DOI] [PubMed] [Google Scholar]
- 13.Calcagnetti DJ, Calcagnetti RL, Fanselow MS. Centrally administered opioid antagonists, nor-binaltorphimine, 16-methyl cyprenorphine and MR2266, suppress intake of a sweet solution. Pharmacol Biochem Behav. 1990;35:69–73. doi: 10.1016/0091-3057(90)90206-w. [DOI] [PubMed] [Google Scholar]
- 14.Jewett DC, Grace MK, Jones RM, Billington CJ, Portoghese PS, Levine AS. The kappa-opioid antagonist GNTI reduces U50,488-, DAMGO-, and deprivation-induced feeding, but not butorphanol- and neuropeptide Y-induced feeding in rats. Brain Res. 2001;909:75–80. doi: 10.1016/s0006-8993(01)02624-5. [DOI] [PubMed] [Google Scholar]
- 15.Bortolato M, Aru GN, Frau R, Orru M, Fa M, Manunta M, Puddu M, Mereu G, Gessa GL. Kappa opioid receptor activation disrupts prepulse inhibition of the acoustic startle in rats. Biol Psychiatry. 2005;57:1550–1558. doi: 10.1016/j.biopsych.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 16.Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor-binaltorphimine, potent and selective κ-opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 17.Portoghese PS, Lipkowski AW, Takemori AE. Bimorphinans as highly selective, potent kappa opioid receptor antagonists. J Med Chem. 1987;30:238–239. doi: 10.1021/jm00385a002. [DOI] [PubMed] [Google Scholar]
- 18.Stevens WC, Jr, Jones RM, Subramanian G, Metzger TG, Ferguson DM, Portoghese PS. Potent selective indolomorphinan antagonists of the kappa-opioid receptor. J Med Chem. 2000;43:2759–2769. doi: 10.1021/jm0000665. [DOI] [PubMed] [Google Scholar]
- 19.Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Mutational evidence for a common kappa antagonist binding pocket in the wild-type kappa and mutant mu[K303E] opioid receptors. J Med Chem. 1998;41:4911–4914. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- 20.Jones RM, Portoghese PS. 5′-Guanidinonaltrindole, a highly selective and potent kappa-opioid receptor antagonist. Eur J Pharmacol. 2000;396:49–52. doi: 10.1016/s0014-2999(00)00208-9. [DOI] [PubMed] [Google Scholar]
- 21.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J Med Chem. 2001;44:2687–2690. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 22.Patkar KA, Yan X, Murray TF, Aldrich JV. [Nalpha-benzylTyr1,cyclo(D-Asp5,Dap8)]-dynorphin A-(1-11)NH2 cyclized in the “address” domain is a novel kappa-opioid receptor antagonist. J Med Chem. 2005;48:4500–4503. doi: 10.1021/jm050105i. [DOI] [PubMed] [Google Scholar]
- 23.Bennett MA, Murray TF, Aldrich JV. Identification of arodyn, a novel acetylated dynorphin A-(1-11) analogue, as a kappa opioid receptor antagonist. J Med Chem. 2002;45:5617–5619. doi: 10.1021/jm025575g. [DOI] [PubMed] [Google Scholar]
- 24.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 25.Thomas JB, Fix SE, Rothman RB, Mascarella SW, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Importance of phenolic address groups in opioid kappa receptor selective antagonists. J Med Chem. 2004;47:1070–1073. doi: 10.1021/jm030467v. [DOI] [PubMed] [Google Scholar]
- 26.Cai TB, Zou Z, Thomas JB, Brieaddy L, Navarro HA, Carroll FI. Synthesis and in vitro opioid receptor functional antagonism of analogues of the selective kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) J Med Chem. 2008;51:1849–1860. doi: 10.1021/jm701344b. [DOI] [PubMed] [Google Scholar]
- 27.Thomas JB, Fall MJ, Cooper JB, Rothman RB, Mascarella SW, Xu H, Partilla JS, Dersch CM, McCullough KB, Cantrell BE, Zimmerman DM, Carroll FI. Identification of an opioid κ receptor subtype-selective N-substituent for (+)-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine. J Med Chem. 1998;41:5188–5197. doi: 10.1021/jm980511k. [DOI] [PubMed] [Google Scholar]
- 28.Liao LA, Zhang F, Yan N, Golen JA, Fox JM. An efficient and general method for resolving cyclopropene carboxylic acids. Tetrahedron. 2004;60:1803–1816. [Google Scholar]
- 29.Chatterjee A, Roy D, Chatterjee SK. An Efficient General-Method for the Synthesis of Some Intermediates for Heterocyclic Steroids. Synthesis-Stuttgart. 1981:449–451. [Google Scholar]
- 30.Hamada Y, Hayashi K, Shioiri T. Efficient Stereoselective Synthesis of Dolastatin-10, an Antineoplastic Peptide from a Sea Hare. Tetrahedron Lett. 1991;32:931–934. [Google Scholar]
- 31.Venugopal VK, Rao N, Rahman MF, Bhalerao UT, Thyagarajan G. A Study of Electronic Effects Involving Electron Deficient Nitrogen. 4. Schmidt and Beckmann Rearrangements of Isoquinolinones and Isothiachromanone. Indian Journal of Chemistry Section B-Organic Chemistry Including Medicinal Chemistry. 1981;20:156–157. [Google Scholar]
- 32.Mander LN, Sethi SP. Regioselective Synthesis of Beta-Ketoesters from Lithium Enolates and Methyl Cyanoformate. Tetrahedron Lett. 1983;24:5425–5428. [Google Scholar]
- 33.West CT, Donnelly SJ, Kooistra DA, Doyle MP. Silane Reductions in Acidic Media .2. Reductions of Aryl Aldehydes and Ketones by Trialkylsilanes in Trifluoroacetic Acid - Selective Method for Converting Carbonyl Group to Methylene. J Org Chem. 1973;38:2675–2681. [Google Scholar]
- 34.Ramadas SR, Chaudhuri AP, Suryaprakash GK. Heterocyclic Steroids .5. Studies on Total Synthesis of Racemic 3-Methoxy-7-Oxaestra-1,3,5(10), 8-Tetraen-17(E)-Ol. Steroids. 1976;28:197–209. doi: 10.1016/0039-128x(76)90109-4. [DOI] [PubMed] [Google Scholar]
- 35.Feutrill GI, Mirringt Rn. Demethylation of Aryl Methyl Ethers with Thioethoxide Ion in Dimethyl Formamide. Tetrahedron Lett. 1970:1327–1328. [Google Scholar]
- 36.Carroll FI, Chaudhari S, Thomas JB, Mascarella SW, Gigstad KM, Deschamps J, Navarro HA. N-Substituted cis-4a-(3-hydroxyphenyl)-8a-methyloctahydroisoquinolines are opioid receptor pure antagonists. J Med Chem. 2005;48:8182–8193. doi: 10.1021/jm058261c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldrich JV, Vigil-Cruz SC. Narcotic Analgesics. In: Abraham DJ, editor. Burger's Medicinal Chemistry and Drug Discovery. 6th. Vol. 6. John Wiley & Sons; New York, NY: 2003. pp. 329–481. Chapter 7. [Google Scholar]
- 39.Schwyzer R. ACTH: A short introductory review. Ann N Y Acad Sci. 1977;297:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 40.Metzger TG, Paterlini MG, Portoghese PS, Ferguson DM. Application of the message-address concept to the docking of naltrexone and selective naltrexone-derived opioid antagonists into opioid receptor models. Neurochem Res. 1996;21:1287–1294. doi: 10.1007/BF02532369. [DOI] [PubMed] [Google Scholar]
- 41.Hjorth SA, Thirstrup K, Grandy DK, Schwartz TW. Analysis of selective binding epitopes for the kappa-opioid receptor antagonist nor-binaltorphimine. Mol Pharmacol. 1995;47:1089–1094. [PubMed] [Google Scholar]
- 42.Larson DL, Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Binding of norbinaltorphimine (norBNI) congeners to wild-type and mutant mu and kappa opioid receptors: molecular recognition loci for the pharmacophore and address components of kappa antagonists. J Med Chem. 2000;43:1573–1576. doi: 10.1021/jm000059g. [DOI] [PubMed] [Google Scholar]
- 43.Metzger TG, Paterlini MG, Ferguson DM, Portoghese PS. Investigation of the selectivity of oxymorphone- and naltrexone-derived ligands via site-directed mutagenesis of opioid receptors: exploring the “address” recognition locus. J Med Chem. 2001;44:857–862. doi: 10.1021/jm000381r. [DOI] [PubMed] [Google Scholar]
- 44.Portoghese PS. From models to molecules: Opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 45.Carroll FI, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: A novel κ-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.