

Abstract
Niemann-Pick type C (NPC) disease is a lysosomal storage disorder characterized at the cellular level by abnormal accumulation of cholesterol and other lipids in lysosomal storage organelles. Lysosomal acid lipase (LAL) has been recently identified as a potential therapeutic target for NPC. LAL can be specifically inhibited by a variety of 3,4-disubstituted thiadiazole carbamates. An efficient synthesis of the C(3) oxygenated/C(4) aminated analogues has been developed that furnishes the products in high yields and high degrees of purity. Common intermediates can also be used for the synthesis of the C(3) carbon substituted derivatives. Herein we tested various thiadiazole carbamates, amides, esters, and ketones for inhibition of LAL. In addition, we tested a diverse selection of commercially available non-thiadiazole carbamates. Our studies show that, among the compounds examined herein, only thiadiazole carbamates are effective inhibitors of LAL. We present a mechanism for LAL inhibition by these compounds whereby LAL transiently carbamoylates the enzyme similarly to previously described inhibition of acetylcholinesterase by rivastigmine and other carbamates as well as acylation of various lipases by orlistat.
Introduction
Niemann-Pick type C disease (NPC)1 is a rare, incurable, autosomal recessive lysosomal storage disorder.2 The disease is characterized by significant accumulation of unesterified cholesterol, glycosphingolipids, and other lipids within late endosomes/lysosomes (LE/LY).3 Clinical manifestations include liver abnormalities, epilepsy, seizures, and significant neurodegeneration, ultimately resulting in fatal outcomes. While miglustat4 and cyclodextrin5 have become candidates for potential therapy, there remains a significant need to find alternative small molecules for treating NPC disease.
In normal cells, LDL-associated cholesterol esters (CE) are transported via receptor-mediated endocytosis to the LE/LY. Within this compartment, lysosomal acid lipase (LAL) hydrolyzes CE to free cholesterol and fatty acids.6 Free cholesterol is then transported from the LE/LY to various organelles, including the endoplasmic reticulum (ER) and the plasma membrane.7
LDL receptor-mediated cholesterol uptake, transport to the LE/LY, and cholesteryl ester hydrolysis by LAL are unaltered in NPC-deficient cells. However, the egress of liberated cholesterol from the LE/LY is significantly reduced, causing them to become lysosomal storage organelles (LSO's).8 Consequently, re-esterification by acyl Co-A:cholesteryl acyl transferase (ACAT) is reduced.
It is not clear what aspect of defective cholesterol transport leads to neuronal cell death. One possibility is that the accumulation of cholesterol and other lipids in the LSO's contributes to the pathology. One possible therapeutic option for NPC patients is the reduction of cholesterol within the LSO's. Treatment of NPC1- or NPC2-defective mice with β-cyclodextrins causes a significant reduction in the accumulation of cholesterol and other lipids in LSOs and significantly extends lifespan.5a, 9 These results indicate that cholesterol reduction may be a viable therapeutic option for NPC disease.
We recently presented the results of an automated filipin-based cellular assay that measures the cholesterol levels within the LSO's.10 The assay is amenable to high throughput screening, allowing libraries of compounds to be investigated for the effect of cholesterol reduction. The technique has already demonstrated some success with our findings that certain pyrrolinones increased cholesterol esterification and decreased LDL uptake in NPC-deficient cells.11 While these initial results are encouraging, the screening process simply identifies compounds that revert the NPC phenotype in terms of cholesterol accumulation. However, it does not identify the molecular target(s) affected by the compounds. An analysis of cellular mechanisms related to cellular cholesterol homeostasis shows that a decrease in cholesterol content within the LSO's can be explained by one or a combination of the following mechanisms: 1) increase in cholesterol efflux from the LSO; 2) decreasing the uptake of cholesterol; or 3) decrease in hydrolysis of cholesteryl esters by LAL.
Recently, thiadiazoles containing a carbamate moiety at C(3) (vide infra) were identified as effective compounds in reducing the LSO cholesterol content using our fluorescence-based assay.12 The cholesterol reduction was found to be a direct result of LAL inhibition. This inhibitory activity is similar to orlistat, a well-documented inhibitor of several lipases (Figure 1), which was also found as a hit in our screen.13 The IC50 values for the hit compounds against purified human LAL (phLAL) were in the mid-nanomolar range. In addition to the potent inhibition of LAL, the thiadiazoles were found to be selective for LAL, exhibiting no inhibition of human pancreatic lipase or bovine milk lipoprotein lipase at the concentrations tested. In contrast, orlistat is a potent inhibitor of many lipases.12 Furthermore, no apparent toxicity was observed for these thiadiazoles when cell viability assays were conducted at 10 μM. The combined cholesterol reduction, selective inhibition of LAL, and low toxicity of the thiadiazoles render them attractive candidates for further study as potential NPC therapeutic agents. This paper describes the next logical phase of these studies, including development of improved synthetic methods of broader scope for this new class of inhibitors, establishment of structural requirements for activity, and more detailed enzyme inhibition and cell studies.
Figure 1.

An example of LAL inhibitor previously observed to reduce lysosomal cholesterol levels in NPC-deficient cells.
Chemistry
Previous syntheses of the 1,2,5-thiadiazole core are based upon two very different strategies.14 The first strategy begins with α-amino nitriles, which are cyclized to the corresponding 3-chloro-4-alkyl-1,2,5-thiadiazole using sulfur monochloride. This strategy has most notably been used in the synthesis of xanomeline, an alkoxythiadiazole functioning as an acetylcholine receptor agonist.15 While this is a common method for the synthesis of 1,2,5-thiadiazoles, several drawbacks exist. First, it uses the highly toxic sulfur monochloride to conduct the cyclization. Additionally, the yields are often low, and the starting α-amino nitriles must be synthesized prior to their use in this approach. The second strategy begins with commercially available 3,4-dichloro-1,2,5-thiadiazole 1. The halogens may be displaced sequentially with various nucleophiles, furnishing unsymmetrical thiadiazole products. This second strategy avoids the difficulties of the first pathway, and we consequently adapted this route for our purposes.
The synthetic route for the initial hit compounds (from this point forward, referred to as C(3) carbamates) is shown in Scheme 1. Beginning with commercially available 1, a single nucleophilic substitution with a secondary amine provided the intermediates 2–5 in high yield. Treatment with KOH afforded the hydroxy intermediates 6–9. The final step was the acylation of the alcohols with the corresponding aminocarbonyl chloride to afford the C(3) carbamates 10–17 (Table 1).
Scheme 1.

a Reagents and conditions: (a) 2° amine (4 equiv), 22 °C or Δ; (b) KOH (4 equiv), DMSO:H2O, reflux; (c) KOtBu, aminocarbonyl chloride, THF, 22 °C.
Table 1.
IC50 values and decarbamoylation rates for thiadiazoles produced via Scheme 1.
| Compound | IC50a (nM) | S.E.M. | decarbamoylation rate (min−1)b | S.E.M. |
|---|---|---|---|---|
| 10 | 496 | 34 | 0.0007 | 0.0001 |
| 11 | 473 | 62 | 0.0037 | 0.0002 |
| 12 | 152 | 50 | 0.0010 | 0.0001 |
| 13 | 68 | 21 | 0.0030 | 0.0002 |
| 14 | 424 | 53 | 0.0016 | 0.0001 |
| 15 | 492 | 69 | 0.0043 | 0.0002 |
| 16 | 300 | 106 | 0.0013 | 0.0001 |
| 17 | 189 | 47 | 0.0042 | 0.0001 |
When appropriate, apparent IC50's for selected compounds against purified human LAL were determined by fitting (MATLAB, non-linear least squares trust-region algorithm) dose-response curves (obtained with the 4MUO assay) to the rectangular hyperbola y=m/(x+b)+c, for y=50, where y = normalized enzymatic activity [%] and x = compound concentration [nM], and m, b, c are coefficients. All fits had R2 > 0.95. Data represent averages ± SEM of 3 independent experiments.
phLAL was incubated with 10 μM compound for 30 min, diluted 250× to 40 nM with substrate (0.125 mM). The reaction was monitored at 10 min intervals for 2 hours. The apparent decarbamoylation rates [min−1] were calculated in MATLAB via linear regression of the plots of the equation described previously:16 ln(Fraction Inhibited) = −k*t, Fraction Inhibited = (ActivityDMSO-ActivityCompound)/ActivityDMSO for each time point. Data represent averages ± SEM of 4 independent experiments. R2>0.96 for all fits.
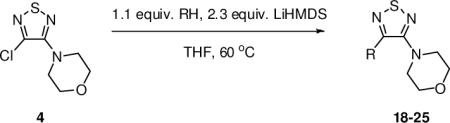
Some time dependence was previously observed in the LAL inhibition by thiadiazole carbamates.12 An increase in inhibition was observed when the enzyme was pre-incubated with the inhibitors for two hours as opposed to five minutes prior to the assay. This time dependence may have resulted from the carbamoylation of the active LAL serine residue. Previously, serine carbamoylation of acetylcholinesterase16,17 and butyrylcholinesterase18 has been shown to be an effective means of enzyme inhibition. To test this hypothesis, we sought to synthesize analogues, which were structurally similar to 10–17, but would be unable to participate in the carbamoylation pathway. Our synthetic route provided a unique opportunity to provide the appropriate analogues readily by replacing the C(3)-oxygen with a carbon. The resulting analogues, including amides, esters, and ketones, would be far less labile than the carbamates, and therefore less prone to participate in the carbamoylation of the LAL active serine residue. This strategy required that we use one of our intermediates 4 in reactions with carbon-based nucleophiles, specifically various types of enolates, to synthesize the desired analogues. This need for the use of a variety of enolates provided a good opportunity for development of synthetic methods producing a broader range of thiadiazole derivatives than were previously accessible by simple, direct means.
While it has been reported that potent nucleophiles (Grignard reagents,19 alkyl lithium reagents20) react with 1 at the sulfur atom and fragment the ring, there is lack of methods based upon enolates reacting directly with monosubstituted thiadiazoles such as 2–5. Consequently, we set out to find optimal conditions with which to conduct the substitution, using thiadiazole 4 as the model substrate with ester enolates. After a significant amount of experimentation, it was determined that using 2.3 equiv of LiHMDS and a slight excess of the corresponding ester provided the desired substituted products in moderate to high yields (Table 2). With this optimized procedure, there was no need to preform the ester enolate, but rather the ester could be added to a solution containing both the thiadiazole and the base. Generally, higher yields of the substitution products were obtained when sterically hindered esters were employed (compounds 18, 21, and 26) versus smaller esters (compounds 19 and 20). Indeed, a common byproduct was the formation of the self-Claisen product, which occurred more readily when sterically accessible esters such as ethyl acetate and methyl propionate were used. These byproducts were not observed when tert-butyl acetate and methyl isobutyrate were employed, which afforded 18 and 21, respectively.
Table 2.
The synthesis of C(3) methylene analogues.







Footnotes:
Refers to isolated, purified yields.
Used LDA (1.2 equiv) to preform the enolate.
In addition to esters, ketone enolates also performed well. Aldol byproducts were often observed under the standard conditions, but these could be avoided by quantitatively forming the lithium enolates separately with LDA, and then adding the enolate solution to a separate solution containing the thiadiazole and LiHMDS. By using this modified protocol, acetophenone and propiophenone reacted well and provided 23 and 24, respectively, in good yields. As with the sterically hindered esters, pinacolone could be employed using the standard conditions, providing ketone product 25. Aldol byproducts were not observed with pinacolone, a result consistent with the sterically hindered esters. Interestingly, isobutyrophenone failed to react under both the standard conditions and the modified protocol using the preformed enolate.
Although the corresponding amide analogues could in principle be obtained by an analogous use of amide enolates, the readily obtained ester products are easily manipulated to provide the desired amides. Two cases that we investigated were the tert-butyl acetate analogues 18 and 26. These esters were treated with TFA in CH2Cl2 to furnish the free acids, which were immediately treated with morpholine and EDC to supply the desired amides 27 and 28 (Scheme 2).
Scheme 2.

We had assumed that the enolate substitution was occurring through a direct nucleophilic attack at C(3) to displace the chloride via a SNAr mechanism. However, Merschaert recently reported that 1 cleanly reacts with LiHMDS at the sulfur atom to give a stabilized ring-opened product.21 The resulting intermediate was then able to undergo substitution reactions with various nucleophiles, including enolates. Given the similarities between our optimal conditions and those reported for the ring opening, we investigated the possibility of the reaction proceeding through an analogous ring opened intermediate.
When 4 was treated with 1.0 equiv of LiHMDS in THF at 22 °C, the starting material was consumed within minutes (Scheme 3). 13C NMR analysis of the product 29 (isolated by column chromatography) revealed a distinct shift in the carbon atoms of the thiadiazole ring which matched those of an identical compound reported by Merschaert.21 When 29 was treated with the Li enolate of tert-butyl acetate, the desired substitution product 18 was isolated in good yield, albeit in lower yield than in the one-pot protocol.
Scheme 3.

a Reagents and conditions: (a) 1.0 equiv LiHMDS, THF, 22 °C, 3 min; (b) 1.3 equiv LiHMDS, tBuOAc, THF, 60 °C, 77%; (c) 2.3 equiv LiHMDS, 1.1 equiv tBuOAc, THF, 60 °C, 80%.
Based on the above data, it is not likely that a direct nucleophilic substitution occurs at C(3) of the thiadiazole ring. Rather, the substitution more likely occurs through a LiHMDS ring-opened intermediate generated in situ, which undergoes facile reaction with enolate nucleophiles and regenerates the thiadiazole ring.
Biological Activity
Our previous studies have identified several LAL inhibitors as potential therapeutics for NPC disease.12 Among these LAL inhibitors were three thiadiazole carbamates: compounds 13, 14, 15 (named as 3a2, 3a7 and 3a6, respectively, in the previous publication), which showed significant potency and specificity towards LAL. In this follow up study, we have examined the structure-activity relationship (SAR) of these thiadiazoles and their derivatives in terms of their ability to inhibit LAL in vitro and in cultured cells.
Enzyme kinetics analysis
In order to determine the type of inhibition of LAL by thiadiazole carbamates, we performed a Lineweaver-Burk analysis for 13 using purified human LAL (phLAL). As shown in Figure 2, compound 13 acts as an apparent competitive inhibitor. Since other compounds examined herein are close structural analogues of 13 it would be reasonable to assume that their mode of inhibition is also competitive.
Figure 2. Lineweaver-Burk analysis of LAL inhibition.

Compound 13 was pre-incubated for 30 min at 37 °C with phLAL, the reaction was started by addition of 4MUO at various concentrations and monitored for 45 min. Compound concentration indicated refers to the concentration in the final reaction mixture. Linear regression fits to the data were computed in MATLAB. R2>0.95 for all fits. Data are from two independent experiments (6<n<8 for treated samples, where n is total number of wells per condition used for quantification).
In vitro activity assay and IC50 determinations
We have examined alterations of the compound structure that involve changes to both the ring system at C4 and the specific groups located at C(3) of the thiadiazole ring. Only the carbamate analogues (10–17) were found to be effective inhibitors. Their apparent IC50's, as determined by an in vitro LAL activity assay, lie in the range of approximately 70 to 500 nM, as summarized in Table 1. Only one ester analogue (20) showed some limited efficacy in inhibiting LAL, with an apparent IC50=13 ± 2 μM. None of the ketone analogues and neither of the two amides tested showed any inhibition of LAL activity at 10 μM, as shown in Supplemental Figure 1. Thus, it appears that the carbamate moiety is required for inhibition of LAL by thiadiazoles. In an attempt to test whether any carbamate would inhibit LAL, we acquired a series of commercially available carbamates (see the Supporting Information for the structures) of various degrees of complexity (methyl carbamate; guaiacol glyceryl ether carbamate; 1-methyl-4-piperidinyl- N-(4-fluorophenyl) carbamate; benzyl N-(6-aminohexyl) carbamate hydrochloride; eserine; neostigmine methyl sulfate). None of these carbamates inhibited LAL at 10 μM, as shown in Supplemental Figure 2.
It appears that piperidine is the preferred substituent at the C4 position and morpholine at the C(3) carbamate (13). Also an increase of the C4 substituent ring size from piperidine to azepane was better tolerated than a decrease in ring size to pyrrolidine or a change to morpholine (compare changes in IC50's between 13 and 16, 11 or 15). On the other hand, the C(3) substituent ring seemed to be less sensitive to modifications in this in vitro assay as both piperidine and morpholine substituents seemed equally tolerated (compare compounds 14 and 15).
Time-dependence of inhibition
We previously demonstrated that 13, 14, and 15 showed some time-dependence of inhibition based upon pre-incubation of compounds with phLAL prior to reaction.12 As shown in Figure 3, we observed similar pre-incubation time-dependence for all of the compounds tested, except for 10, which showed significantly higher time-dependence than other compounds. This slow time-dependence is in agreement with the hypothesis that LAL inhibition is associated with serine carbamoylation.
Figure 3.

Time-dependence of LAL inhibition by thiadiazole carbamates. phLAL was pre-incubated with the various compounds at 833 nM for the indicated times at 37 °C. The reaction was started by addition of 125 μM 4MUO and monitored for 30 min at 37 °C. The final compound concentration in the assay was 500 nM. Data represent averages ± SEM of 2 independent experiments normalized to control (DMSO) average value for each experiment (n=12, where n is total number of wells per condition used for quantification).
Compound efficacy in cells
In order to examine compound efficacy in inhibiting LAL in cells, we measured enzyme activity in lysates of cells that were treated for 4 days with thiadiazole carbamates at various concentrations (Figure 4A). We observed that the compounds segregated into two distinct groups based on the level of residual inhibition, i.e., 10, 12, 14, and 16 showed much higher level of inhibition of LAL upon cell lysis than 11, 13, 15, and 17. The two groups of compounds vary primarily in the nature of the C(3) substituent: a morpholine (10, 12, 14, 16) or piperidine (11, 13, 15, 17), which is expected to carbamoylate the catalytic serine of LAL. Since the volume of a cell monolayer is much smaller than that of our reaction mixture upon cell lysis, we postulated that the differences in inhibition observed was a consequence of the differences in the apparent decarbamoylation rate of LAL modified by these compounds upon the dilution of the enzyme with reaction buffer. We also observed that the compounds that had a piperidine C(3) substituent inhibited LAL significantly better in cells upon withdrawal of the compounds from the growth medium for 1 day after 3 days of treatment (Figure 4B). The compounds that had both the C(3) and the C4 substituent non-oxygenated rings (12,16 and 10) exhibited maximal potency in this withdrawal assay.
Figure 4.

Compound efficacy in cells. Enzymatic assay for inhibition of LAL in cell lysates of cells treated with compound continuously for ~4 days (A) or treated for ~3 days and allowed to recover in growth media for ~1 additional day (B). Data represent averages ± SEM of 3 independent experiments normalized to control (DMSO) average value for each experiment (n=12, where n is total number of wells per condition used for quantification). Quantification of thiadiazole effects after ~3 days of treatment on cholesterol accumulation as measured by filipin LSO assay in NPC1 mutant cells (C) or neutral lipid accumulation in apparently normal human fibroblasts as measured by quantification of LipidTOX green staining (D). Data are presented as percentage of the average value of the DMSO controls (C) or normalized to the 10 μM 12 average value (D) in each experiment. Error bars, where visible, are SEM. (n=8 for treated samples, where n is total number of wells per condition used for quantification).
LAL decarbamoylation rate determination
In order to test whether indeed the difference in inhibition between the morpholine and piperidine thiadiazole carbamates observed was the result of different decarbamoylation rates of the modified enzyme for the two groups, we examined enzyme reactivation (decarbamoylation) in vitro under our standard reaction conditions (37 °C, 200 mM sodium acetate pH 5.5, 0.02% Tween, 1.6 % Triton X-100, final) using phLAL. We pre-incubated phLAL with compounds at high concentration (10 μM) to carbamoylate most of the enzyme and then extensively diluted the enzyme (250X) with substrate and monitored the reaction at 10 min intervals for 2 hours at 37 °C. The decarbamoylation rates listed in Table 1 were calculated as described previously.16 Indeed, we observed that the C(3) thiadiazole morpholine carbamates 11, 13, 15, and 17 decarbamoylated several fold faster than their piperidine analogues (10, 12, 14, 16).
In order to further test LAL inhibition in intact cells, we measured the decrease in cholesterol accumulation in NPC1 mutant cells (Figure 4C) and the increase in neutral lipid accumulation in normal cells (Figure 4D). It appears that 12 is the most potent thiadiazole carbamate eliciting these effects, while 15 was least potent in all four assays, with the rest of compounds exhibiting various degrees of efficacy.
Discussion and Conclusions
In summary, we have devised an efficient route for the synthesis of unsymmetrical 3,4-disubstituted-1,2,5-thiadiazoles beginning with the commercially available 3,4-dichloro-1,2,5-thiadiazole 1. The route produces C(3) oxygen-bonded carbamates in high yields, is quite general, and is amenable to large scale work. Additionally, we have found that C(3) carbon substituted thiadiazoles can be obtained by a simple one-pot substitution with either ester or ketone enolates without isolation of the intermediates as reported previously. The enolate reaction takes place rapidly and provides good to excellent yields of the substituted products. Given the importance of the 1,2,5-thiadiazole core as a scaffold for drug development,14,22 the new enolate-based transformation reported in this paper opens up a wide range of new possibilities for the use of this heterocycle in medicinal chemistry.
The specific thiadiazole analogues synthesized exhibited a strong SAR in terms of LAL inhibition in vitro and in cultured cells. Of the compounds tested herein, only the thiadiazole carbamates were effective inhibitors. Only one ester (20) exhibited some low inhibitory activity in vitro, and none of the amides or ketones studied showed activity at the concentrations tested. Moreover, none of the commercially acquired non-thiadiazole carbamates were effective in inhibiting LAL at the concentrations tested (Suppl. Fig. 2). These findings point to the dual requirement of both the carbamate and the 3,4-disubstituted thiadiazole moieties for successful inhibition of LAL. We also observed that thiadiazole carbamates were competitive inhibitors of LAL (Figure 2) and exhibited time-dependence in their ability to inhibit the enzyme (Figure 3). Thus, our data point to LAL carbamoylation as the mechanism of inhibition by thiadiazole carbamates. Carbamate inhibitors of enzymes such as acetylcholinesterase have been previously characterized.16,17 These inhibitors have been shown to transiently carbamoylate the catalytic serine of acetylcholinesterase. A similar mode of inhibition of LAL can be envisaged for the thiadiazole carbamates examined in this study. The proposed mechanism of the modification of LAL by thiadiazole carbamates is presented in Scheme 4.
Scheme 4.

Kinetic outline of transient LAL modification by 3,4-disubstituted thiadiazole carbamates.
In this mechanism, the apparent efficacy of the compound would be determined by the three reaction rates: the reversible rates of the binding of the enzyme to the compound (k1, k−1), the enzyme carbamoylation rate (k2) and the rate of decarbamoylation of the modified LAL (k3). The rates k1 and k2 combine into the apparent, observable, inhibition rate ki. Previous work has demonstrated that the compound IC50 is related to ki/k3, under steady state conditions.23
In order to explore the SAR of the thiadiazole carbamates' inhibition of LAL, we examined their ability to inhibit LAL in vitro using purified enzyme and in cultured cells. Based on the IC50 data summarized in Table 1, the best compound for in vitro inhibition of phLAL was 13, followed by 12. However, in the cellular context we observed that 12 was much more potent in inhibiting LAL than 13. This was shown by the significant changes in the dose-dependent decrease in cholesterol accumulation in NPC-mutant cells as well as the increased neutral lipid accumulation in normal fibroblasts treated with these compounds. We hypothesize that the difference in the time-scales of the two different sets of experiments (i.e., in vitro vs. in cultured cells) was important in determining the apparent compound efficacy. The in vitro experiments were usually conducted for a few hours or less (e.g., 90 min for IC50 determination), whereas the tissue culture experiments were conducted for several days (e. g., 3 days for filipin/LipidTOX experiments). As outlined in Scheme 4, the modified (and thus inhibited) enzyme will recover based upon the magnitude of the decarbamoylation rate (k3). We observed strong preference for morpholine (11, 13, 15, 17) as opposed to piperidine modification (10, 12, 14, 16) as shown in Table 1 in regards to the ability of the enzyme to spontaneously recover after carbamoylation. This preference appeared to affect compound efficacy in cells since this recovery rate would significantly affect the more prolonged cell experiments, as opposed to the relatively short in vitro ones. Compound 12 is a bispiperidine thiadiazole carbamate. The C(3) piperidine carbamate moiety, after its attachment to the enzyme, would decarbamoylate at a significantly slower rate than its morpholine analogue (13) and thus would be a major contributing factor to this compound's increased potency in cells. The difference in the relative rates of decarbamoylation is consistent with the difference in pKa values of piperidine and morpholine, the latter being significantly lower.24
In conclusion, we have explored the SAR of thiadiazole derivatives for LAL inhibition in vitro and in cultured cells leading to further optimization of compound efficacy in a cellular context and to significant elucidation of the molecular mechanism of action for this series of compounds. Further structural studies would be needed to elucidate the precise mechanism of LAL inhibition by these novel inhibitors as well as in vivo studies for the determination of their pharmacokinetics and pharmacodynamics.
Experimental Section
Unless otherwise stated, all synthetic reactions were conducted under an inert atmosphere of either nitrogen or argon. Reactions were monitored using thin layer chromatography (TLC) performed on EMD plates. THF was distilled from sodium/benzophenone prior to use. Methylene chloride (DCM) was distilled from CaH2 prior to use. 3,4-Dichloro-1,2,5-thiadiazole was purchased from Alfa Aesar and used as received. MEM and LipidTOX green were from Invitrogen (Carlsbad, CA). All other reagents were purchased from Aldrich and were used as received. Bicinchoninic acid assay kit, Triton X-100 and Tween-20 were from Thermo Scientific (Rockford, IL). Purified recombinant human LAL (phLAL) was a gift of Drs. Greg Grabowski and Hong Du (Cincinnati Children's Hospital Medical Center, OH) and was prepared as described.25 1H and 13C spectra were obtained using a Varian INOVA instrument operating at 600 MHz and 150 MHz, respectively. All spectral data are reported in ppm (δ) relative to the residual solvent peaks present. For 1H NMR, the ppm are relative to 7.27 (CDCl3) and 2.5 (DMSO-d6). For 13C NMR, the ppm are relative to 77.23 (CDCl3) and 39.43 (DMSO-d6). Coupling constants (J) are reported in Hz. High resolution mass spectrometry (HRMS) was performed on a JEOL GCMate system. Flash chromatography was performed using EMD silica gel (200–400 mesh). Infrared spectra were recorded on a Perkin Elmer Paragon 1000 FT-IR Spectrometer. All tested compounds were assessed to be ≥ 95% pure by analytical HPLC.
Typical procedure for the synthesis of 4-amino-3-chloro-1,2,5-thiadiazoles. 3-Pyrrolidinyl-4-chloro-1,2,5-thiadiazole (2)
3,4-Dichloro-1,2,5-thiadiazole (0.61 mL, 6.45 mmol) was added dropwise to pyrrolidine (2.2 mL, 25.8 mmol) at 22 °C. The solution immediately began to warm and turn orange in color. After 5 min, TLC (DCM) showed complete consumption of the starting thiadiazole. The reaction mixture was poured into ice water and acidified to pH 2.0 by addition of concentrated aq HCl. The mixture was extracted with DCM (2 × 10 mL) and the combined organic extracts were dried with a2SO4, filtered, and evaporated. The product was purified by column chromatography (DCM as eluent) to yield 1.07 g (87%) of the desired product. 1H NMR (600 MHz, CDCl3) δ 3.72 (t, J = 7.2 Hz, 4H), 1.98 (t, J = 7.2 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 156.4, 131.2, 49.9, 25.9; HRMS (ESI) calcd for C6H8ClN3S [M]+ 189.0127, found 189.0122; IR (neat): 2962, 2899, 2872, 1502 cm−1.
Typical procedure for the synthesis of 3-hydroxy-4-amino-1,2,5-thiadiazoles. 3-Hydroxy-4-pyrrolidinyl-1,2,5-thiadiazole (6)
2 (600 mg, 3.16 mmol) was suspended in a H2O/DMSO mixture (4:1, 5 mL) containing KOH (710 mg, 12.65 mmol). The mixture was stirred under reflux for 4 h. After this time, TLC (DCM) showed all starting thiadiazole to be consumed. The reaction was cooled to 0 °C. The solution was acidified to pH 2.0 by addition of concentrated HCl. A white precipitate formed, which was filtered and washed with water. After drying, the product was recrystallized from MeOH to yield 514 mg (95%) of a white solid. mp > 200 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.4 (bs, 1H), 3.54 (t, J = 7.2 Hz, 4H), 1.84 (t, J = 7.2 Hz, 4H); 13C NMR (150 MHz, DMSO-d6) δ 152.4, 148.7, 48.3, 24.8; HRMS (ESI) calcd for C6H9N3OS [M + Na]+ 194.0359, found 194.0342; IR (KBr): 3227, 2961, 2859, 1501 cm−1.
Typical procedure for the synthesis of 3-carbamoyl-4-amino-1,2,5-thiadiazoles. 4-(Pyrrolidin-1-yl)-1,2,5-thiadiazol-3-yl piperidine-1-carboxylate (10)
6 (70 mg, 0.406 mmol) was suspended in THF (1 mL) at 22 °C. KOtBu (46 mg, 0.406 mmol) was added as a solid, and the mixture was stirred for 10 min. Piperidinecarbonyl chloride (0.04 mL, 0.339 mmol) was then added via syringe. The reaction was stirred for 12 h, during which time it became cloudy and turbid. Water (10 mL) was added and the mixture was extracted with DCM (2 × 10 mL). The combined organic extracts were dried with Na2SO4, filtered, and concentrated. The residue was dissolved in MeOH (1 mL) and cooled to 0 °C. Water was added, and scratching with a spatula induced a white solid to form. The solid was filtered, washed with water, and dried to yield 69 mg (72%) of the title compound. mp = 57–60 °C. 1H NMR (600 MHz, CDCl3) δ 3.60-3.56 (m, 6H), 3.53-3.51 (m, 2H), 1.95 (t, J = 6.6 Hz, 4H), 1.68-1.60 (m, 6H); 13C NMR (150 MHz, CDCl3) δ 151.9, 151.5, 144.4, 48.5, 46.0, 45.7, 26.2, 25.7, 25.6, 24.3; HRMS (ESI) calcd for C12H18N4O2S [M + Na]+ 305.1043, found 305.1039; IR (film): cm−1.
Typical procedure for the reaction between 4-amino-3-chloro-1,2,5-thiadiazoles and enolates. tert-Butyl 2-(4-morpholino-1,2,5-thiadiazol-3-yl)acetate (18)
Monochloride 4 (200 mg, 0.97 mmol) was charged into a flame-dried flask and evacuated for 10 min. The flask was brought into a glovebox and LiHMDS (374 mg, 2.24 mmol) was added. The flask was capped, removed from the glovebox, and placed under argon. THF (3 mL) was added via syringe, and the solution immediately darkened in color. tert-Butyl acetate (0.14 mL, 1.07 mmol) was then added via a syringe. The mixture was heated to 60 °C and monitored by TLC (DCM as eluent). Upon completion, the mixture was cooled to 22 °C and quenched with 1.0 N HCl. The mixture was extracted with Et2O and the combined organic extracts were dried with MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (2% EtOAc in DCM) to furnish 222 mg (80%) of the title compound as an oil. 1H NMR (600 MHz, CDCl3) δ 3.82 (m, 4H), 3.78 (s, 2H), 3.26 (m, 4H), 1.44 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 168.1, 163.1, 148.3, 82.2, 66.6, 50.4, 37.9, 28.1; HRMS (ESI) calcd for C12H19N3O3S [M + Na]+ 308.1039, found 308.1002; IR (neat): 2975, 2928, 2853, 1729, 1501, 1440 cm−1.
1-Morpholino-2-(4-morpholino-1,2,5-thiadiazol-3-yl)ethanone (26)
18 (100 mg, 0.35 mmol) was dissolved in a 1:1 solution of DCM:TFA (2 mL) at 22 °C. The solution was stirred for 3 h. After this time, TLC analysis (2% EtOAc in DCM) showed complete consumption of the ester. The solution was evaporated to dryness, and the residue was redissolved in DCM (2 mL). The flask was cooled to 0 °C and morpholine (37 mg, 0.42 mmol) was added via syringe, followed by EDC (202 mg, 1.05 mmol). The reaction mixture was stirred for 12 h and gradually warmed to 22 °C. After this time, TLC analysis (hexane:EtOAc 2:1) showed complete consumption of the starting material and a new more polar spot. The mixture was diluted with DCM (10 mL) and washed with 1.0 N HCl (2 × 10 mL). The organic layer was dried with Na2SO4, filtered, and concentrated to yield a yellow solid (82 mg, 78%). mp = 66–68 °C. 1H NMR (600 MHz, CDCl3) δ 3.88 (s, 2H), 3.84-3.82 (m, 4H), 3.68-3.65 (m, 4H), 3.63-3.62 (m, 2H), 3.53-3.56 (m, 2H), 3.33-3.32 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 166.8, 163.5, 148.9, 66.9, 66.79, 66.77, 50.7, 47.0, 42.6, 36.2; HRMS (ESI) calculated for C12H18N4O3S [M + Na]+ 321.0992, found 321.1001; IR (film): 2958, 2919, 2850, 1640, 1501, 1437, 1112 cm−1.
Cell lines
Human fibroblast cell lines GM03123 (NPC1) and GM05659 (apparently normal) were obtained from the Coriell Institute (Camden, NJ). Fibroblasts were grown in Minimal Essential Medium (MEM) with 2.2 g/L sodium bicarbonate, supplemented with 10 % (v/v) FBS (growth medium).
LAL activity assay
LAL activity was determined mainly as described previously.12 For IC50 (compound concentration required for 50 % inhibition of activity) determinations, three-fold serial dilutions of the compounds were prepared from DMSO stocks and diluted in the assay buffer (200 mM sodium acetate, pH 5.5, 0.02 % (w/v) Tween-20). Purified enzyme (phLAL, 1 mU/ml final concentration, 105 U/mg, 1 U of enzyme releases 1.0 μmol of 4-methylumbelliferone per minute at pH 5.5 at 37 °C) was then added. The enzyme/compound mixture was incubated at 37 °C for 30 min. The reaction was started by addition of 125 μM (final) 4-methylumbelliferyloleate (4MUO) dissolved in 4 % (w/v) Triton X-100. Reactions were incubated at 37 °C, and fluorescence (355 nm excitation/450 nm emission) was monitored using a SpectraMax M2 fluorometer (MDS Inc., Toronto, Canada). For the IC50 determinations, the reaction time was 1 h. IC50's for selected compounds were determined by fitting (MATLAB, Non-linear least squares Trust-Region algorithm) dose-response curves to the rectangular hyperbola y=m/(x+b)+c, solved for y = 50, where y = normalized enzymatic activity (%), x = compound concentration (nM), and m, b, c are coefficients.
Decarbamoylation rates (k3) were determined as follows: phLAL was incubated with 10 μM compound for 30 min, diluted 250× to 40 nM with substrate (125 μM). The reaction was monitored at 10 min intervals for 2 h. The apparent decarbamoylation rates were calculated in MATLAB via linear regression of the plots of the equation as described previously16: ln(Fraction Inhibited) = −k*t, Fraction Inhibited = (ActivityDMSO-ActivityCompound)/ActivityDMSO for each time point.
phLAL carbamoylation was assessed by pre-incubating phLAL with the various compounds at 833 nM for indicated times at 37 °C. The reaction was started by addition of 125 μM (final) 4-methylumbelliferyloleate (4MUO) dissolved in 4 % (w/v) Triton X-100 and monitored for 30 min at 37 °C. The final compound concentration in the assay was 500 nM. LAL activity in lysed cells was determined mostly as described previously.12
Human fibroblasts were grown in 384-well assay plates (Corning), incubated with compounds for 3–4 days at indicated concentrations. At the end of the incubation, the cells were washed 3× with PBS, then washed once with 150 mM sodium acetate pH 5.5, and then lysed with 1 % (w/v) Triton X-100 for 30 min at 37 °C. The reaction was started by the addition of 1 mM (final) 4MUO dissolved in 4 % Triton X-100. The reaction was monitored as described above. Enzymatic activity was normalized to protein concentration in the reaction mixture as determined by the bicinchoninic acid assay. No significant variation in protein concentration between the different conditions was observed.
Automated fluorescence microscopy and image analysis
Filipin and LipidTOX green staining and automated image analysis (LSO filipin assay) were done as described.12 Images were acquired using an ImageXpressMICRO imaging system from Molecular Devices Devices (MDS Analytical Technologies, Downington, PA, USA) equipped with a 300 W Xenon-arc lamp. A CoolSnapHQ camera (1,392 × 1,040 pixels) from Roper Scientific (Tucson, AZ, USA) was used to acquire images. Filipin images were acquired using 377/50 nm excitation and 447/60 nm emission filters with a 415 nm dichroic filter using a 10× Plan Fluor 0.3 numerical aperture (NA) objective from Nikon (Melville, NY, U.S.A). LipidTOX green images of neutral lipid accumulation were acquired using 482/35 nm excitation and 536/40 nm emission band-pass filters with a 513 nm dichroic filter using a 20× Plan Fluor 0.5 NA objective from Nikon (Melville, NY, U.S.A). Images were acquired at either four (10×) or nine (20×) sites per well using 2 × 2 pixel binning. Each site was individually focused using a high-speed laser autofocus comprised of a 690 nm diode laser and a dedicated 8-bit CMOS camera. 696 × 520 pixel images were acquired at 12 intensity bits per pixel.
Images were background corrected as described and either the filipin or the LipidTOX green LSO ratio was quantified as integrated thresholded fluorescence power of filipin or LipidTOX green fluorescence within the LSOs, normalized to the total cell area as defined by a low threshold using filipin fluorescence.10
Software
MetaExpress and MetaMorph were from Molecular Devices (MDS Analytical Technologies, Downington, PA, USA). MATLAB was from The MathWorks (Natick, MA). Compound structures were generated using ChemDraw (CambridgeSoft, Cambridge, MA).
Supplementary Material
Acknowledgments
The authors thank the Ara Parseghian Medical Research Foundation and NIH grant R37-DK27083 (to FRM) for financial support of this research. We thank Dr. Peter Lobel (Rutgers University) for helpful discussions and Harold Ralph (Weill Cornell Cell Screening Core Facility) for help with HTS image acquisition and analysis. We also thank Drs. Gregory Grabowski and Hong Du (Cincinnati Children's Hospital Medical Center) for a gift of phLAL.
Footnotes
Abbreviations: ACAT, acyl Co-A: cholesteryl acyl transferase; CE, LDL-associated cholesterol esters; DMAP, 4-dimethylaminopyridine; EDC, N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride; LAL, lysosomal acid lipase; LDA, lithium diisopropyl amide; LE/LY, late endosomes/lysosomes; LiHMDS, lithium hexamethyldisilazide; LSO, lysosomal storage organelle; NPC, Niemann-Pick type C disease; phLAL, purified human lysosomal acid lipase; SAR, structure-activity relationship; TFA, trifluoroacetic acid.
Supporting Information: Detailed experimental procedures and characterization data for all intermediates, products, and procedures for biochemical assays are available. This material is provided free of charge from http://pubs.acs.org.
References
- (1).For recent reviews, see: Sturley SL, Patterson MC, Balch W, Liscum L. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2004;1685:83–87. doi: 10.1016/j.bbalip.2004.08.014.. Mukherjee S, Maxfield FR. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2004;1685:28–37. doi: 10.1016/j.bbalip.2004.08.009.. Maxfield FR, Tabas I. Nature. 2005;438:612–621. doi: 10.1038/nature04399..
- (2).Cataldo AM, Nixon RA. Neuronal Protein Trafficking in Alzheimer's Disease and Niemann-Pick Type C Disease. In: Bean AJ, editor. Protein Trafficking in Neurons. Academic Publishing; New York, NY: 2007. pp. 391–411. [Google Scholar]
- (3).Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low-density lipoprotein derived cholesterol is defective in Niemann-Pick type C fibroblasts. J. Cell Biol. 1989;108:1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Patterson MC, Wraith JF, Mengel E, Sedel F, Hwu W-L, Rohrbach M, Bembi B, Walterfang M, Korenke C, Marquardt T, Luzy C, Giorgino R, Pineda M. A multicentre retrospective cohort study of miglustat in patients with Niemann-Pick disease type C. Mol. Gen. Metab. 2009;98:61–61. doi: 10.1016/j.ymgme.2009.07.003. [DOI] [PubMed] [Google Scholar]
- (5).(a) Liu B, Turley SD, Burns DK, Miller AM, Reppa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. U. S. A. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sturley SL, Patterson MC, Pentchev P. Unraveling the sterol-trafficking defect in Niemann-Pick C disease. Proc. Natl. Acad. Sci. U. S. A. 2009;106:2093–2094. doi: 10.1073/pnas.0812934106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Abi-Mosleh L, Infante RE, Radhakrishnan A, Goldstein JL, Brown MS. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U. S. A. 2009;106:19316–19321. doi: 10.1073/pnas.0910916106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease Amerliorates Neuronal Cholesterol and Glycosphingolipid Storage and Disease Progression. PLoS One. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of β-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. U. S. A. 2010;107:5477–5482. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 1975;250:8487–8495. [PubMed] [Google Scholar]; (b) Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- (7).Mesmin B, Maxfield FR. Intracellular sterol dynamics. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2009;1791:636–645. doi: 10.1016/j.bbalip.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sokol J, Blanchette-Mackie J, Kruth HS, Dwyer NK, Amende LM, Butler JD, Robinson E, Patel S, Brady RO, Comley ME, anier MT, Pentchev PG. Type C Niemann Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J. Biol. Chem. 1988;263:3411–3417. [PubMed] [Google Scholar]
- (9).Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM. J. Lipid Res. doi: 10.1194/jlr.M000257. Doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pipalia NH, Huang A, Ralph H, Rujoi M, Maxfield FR. Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. J. Lipid Res. 2006;47:284–301. doi: 10.1194/jlr.M500388-JLR200. [DOI] [PubMed] [Google Scholar]
- (11).Cosner CC, Markiewicz JT, Bourbon P, Mariani CJ, Wiest O, Rujoi M, Rosenbaum AI, Huang AY, Maxfield FR, Helquist P. Investigation of N-Aryl-3-alkylidenepyrrolinones as Potential Niemann-Pick Type C Disease Therapeutics. J. Med. Chem. 2009;52:6494–6498. doi: 10.1021/jm900707n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rosenbaum AI, Rujoi M, Huang AY, Du H, Grabowski GA, Maxfield FR. Chemical screen to reduce sterol accumulation in Niemann-Pick C disease cells identifies novel lysosomal acid lipase inhibitors. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2009;1791:1155–1165. doi: 10.1016/j.bbalip.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Imanaka T, Moriyama Y, Ecsedi GG, Aoyagi T, Amanuma-Muto K, Ohkuma S, Takano T. Esterastin – a potent inhibitor of lysosomal acid lipase. J. Biochem. 1983;94:1017–1020. doi: 10.1093/oxfordjournals.jbchem.a134399. [DOI] [PubMed] [Google Scholar]
- (14).Koutentis PA. Product class 11: 1,2,5-thiadiazoles and related compounds. Sci. Synth. 2004;13:297–348. [Google Scholar]
- (15).(a) Bymaster FP, Shannon HE. Combination Therapy for Treatment of Psychoses. 2004/0023951 A1 U.S. Patent. 2004 February 5;; (b) Quimby SJ, Shannon HE, Bymaster FP, Sauerberg P, Olesen PH, Sheardown MJ, Suzdak PD, Mitch CH. Synthesis and Structure Activity Relationships of Alkyl Substituted Analogues of the Functional M1 Selective Muscarinic Receptor Agonist Xanomeline. Bioorg. Med. Chem. Lett. 1994;4:2205–2210. [Google Scholar]
- (16).Wilson IB, Hatch MA, Ginsburg S. Carbamylation of Acetylcholinesterase. J. Biol. Chem. 1960;235:2312–2315. [PubMed] [Google Scholar]
- (17).Bar-On P, Millard CB, Harel M, Dvir H, Enz A, Sussman JL, Silman I. Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine. Biochemistry. 2002;41:3555–3564. doi: 10.1021/bi020016x. [DOI] [PubMed] [Google Scholar]
- (18).Carolan CG, Dillon GP, Gaynor JM, Reidy S, Ryder SA, Khan D, Marquez JF, Gilmer JF. Isosorbide-2-carbamate Esters: Potent and Selective Butyrylcholinesterase Inhibitors. J. Med. Chem. 2008;51:6400–6409. doi: 10.1021/jm800564y. [DOI] [PubMed] [Google Scholar]
- (19).De Munno A, Bertini V, Picci N. C-alkylation, alkenylation, and arylation of 1,2,5-thiadiazoles. Heterocycles. 1986;24:1131–1136. [Google Scholar]
- (20).Kouvetakis J, Grotjahn D, Becker P, Moore S, Dupon R. Synthesis of Ethynyl-Substituted Precursors to Carbon-Nitrogen-Sulfur Extended Structures: Reactions of C3N3F3 and C2N2SCl2 with Alkali-Metal (Trimethylsilyl)acetamides. Chem. Mater. 1994;6:636–639. [Google Scholar]
- (21).Merschaert A, Boquel P, Gorissen H, Van Hoeck J-P, Borghese A, Antoine L, Mancuso V, Mockel A, Vanmarsenille M. The ring opening 3,4-dichloro-1,2,5-thiadiazole with metal amides. A new synthesis of 3,4-disubstituted-1,2,5-thiadiazoles. Tetrahedron Lett. 2006;47:8285–8288. [Google Scholar]
- (22).(a) Cao Y, Zhang M, Wu C, Lee S, Wroblewski ME, Whipple T, Nagy PI, Takács-Novák K, Balázs A, Torös S, Messer WS., Jr. Synthesis and biological characterization of 1-methyl-1,2,5,6-tetrahydropyridyl-1,2,5-thiadiazole derivatives as muscarinic agonists for the treatment of neurological disorders. J. Med. Chem. 2003;46:4273–4286. doi: 10.1021/jm0301235. [DOI] [PubMed] [Google Scholar]; (b) Sabb AL, Vogel RL, Kelly MG, Palmer Y, Smith DL, Andree TH, Schechter LE. 1,2,5-Thiadiazole derivatives are potent and selective ligands at human 5-HT1A receptors. Bioorg. Med. Chem. Lett. 2001;11:1069–1071. doi: 10.1016/s0960-894x(01)00151-2. [DOI] [PubMed] [Google Scholar]; (c) Biju P, Taveras AG, Yu Y, Zheng J, Hipkin RW, Fossetta J, Fan X, Fine J, Lundell D. 3,4-Diamino-1,2,5-thiadiazole as potent and selective CXCR2 antagonists. Bioorg. Med. Chem. Lett. 2009;19:1434–1437. doi: 10.1016/j.bmcl.2009.01.027. [DOI] [PubMed] [Google Scholar]
- (23).Wilson IB, Harrison MA, Ginsburg S. Carbamyl derivatives of acetylcholinesterase. J. Biol. Chem. 1961;236:1498–1500. [PubMed] [Google Scholar]
- (24).Crampton MR, Robotham IA. Acidities of Some Substituted Ammonium Ions in Dimethyl Sulfoxide. J. Chem. Research (S) 1997:22–23. [Google Scholar]
- (25).Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum. Mol. Genet. 2001;10:1639–1648. doi: 10.1093/hmg/10.16.1639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.