

Abstract
Many plants, fungi, and bacteria catabolize allantoin as a mechanism for nitrogen assimilation. Recent reports have shown that in plants and some bacteria the product of hydrolysis of allantoin by allantoinase is the unstable intermediate ureidoglycine. While this molecule can spontaneously decay, genetic analysis of some bacterial genomes indicates that an aminotransferase may be present in the pathway. Here we present evidence that Klebsiella pneumoniae HpxJ is an aminotransferase that preferentially converts ureidoglycine and an α-keto acid into oxalurate and the corresponding amino acid. We determined the crystal structure of HpxJ, allowing us to present an explanation for substrate specificity.
The uric acid catabolic pathway is the final stage of purine catabolism and functions in plants and some bacteria to provide a source of nitrogen, particularly when other nitrogen sources are depleted (1, 2). Uric acid catabolism begins with the oxidation of uric acid by uricase followed by ring opening and decarboxylation reactions catalyzed by 5-hydroxyisourate hydrolase and 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase, respectively (3-6). The latter two steps lead to the production of allantoin, a process that also occurs non-enzymatically after oxidation of uric acid (1, 4). Allantoin can then be further degraded to allantoate by an allantoinase enzyme (1).
The further catabolism of ureides follows one of two pathways. The degradation of allantoate is catalyzed by either allantoicase or allantoate amidohydrolase. The former produces urea and ureidoglycolate, both of which require further catabolism for full nitrogen assimilation. The latter pathway, which is found in plants and some bacteria (1, 2), also results in ureidoglycolate, but has been demonstrated to proceed via ureidoglycine, a molecule now known to be the product of the allantoate amidohydrolase catalyzed reaction (7).
Characterization of the fate of ureidoglycine has been complicated by the fact that it is unstable in aqueous environments and is subject to rapid non-enzymatic decay (7, 8). A recent report by Werner et al., however, demonstrated that ureidoglycine can be hydrolyzed by ureidoglycine aminohydrolase (9). The resulting ureidoglycolate can be consumed by ureidoglycolate amidohydrolase or the urea-releasing ureidoglycolate hydrolase (ureidoglycolate lyase) (2, 9). Alternatively, in some organisms, ureidoglycolate can be oxidized to oxalurate which can be used in subsequent reactions to generate ATP and ammonia (1, 10).
Two recent genetic studies have identified a gene cluster encoding a purine degradative pathway in Klebsiella sp. that proceeds through similar intermediates as those outlined above, but does so using different catalytic strategies (11, 12). Scheme 1 outlines the proposed enzymes and intermediates in this pathway (7, 9, 12). One surprising finding in the gene cluster encoding these enzymes was the presence of a gene for a protein that belongs to the pyridoxal-5′-phosphate (PLP)1-dependent aspartate aminotransferase superfamily. An examination of the intermediates along the degradative pathway reveals that only ureidoglycine has the requisite amino group needed to participate in an aminotransfer reaction. Also notable is the absence of an enzyme capable of oxidizing ureidoglycolate to oxalurate. Taken together, this suggests that the pathway could proceed from ureidoglycine to oxalurate directly in some organisms. The presence of an aminotransferase associated with purine degradation is not unique to Klebsiella sp. In a recent cross-organism protein association analysis, a gene encoding an aminotransferase was found linked to allantoate amidohydrolase in several organisms, particularly those lacking a ureidoglycine aminohydrolase (9).
Scheme 1.
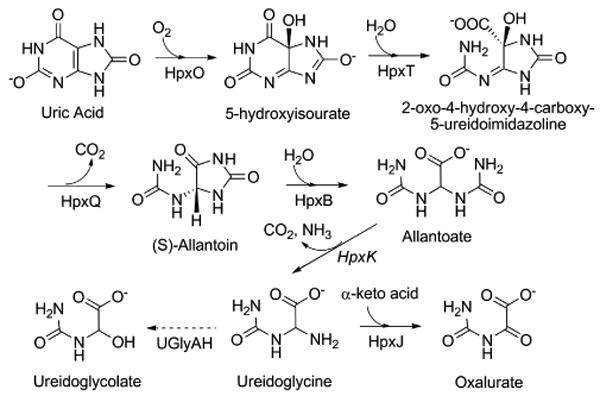
Uric acid catabolic pathway in Klebsiella pneumoniae. The enzyme names are taken from the K. pneumoniae degradative pathway. The dashed line indicates an alternative route that operates in other organisms.
In order to determine if a ureidoglycine aminotransferase participates in the purine degradative pathway, we have biochemically and structurally characterized the K. pneumoniae enzyme with this putative function. To establish if HpxJ had aminotransferase activity, we first examined its ability to catalyze the transfers of an amino group between aspartate and glyoxylate. To quantitate the amino acid content in the reaction mixtures, we treated the mixtures with DABS-Cl prior to HPLC analysis. Figure S1 shows HPLC chromatograms of the aminotransfer reaction in the presence and absence of HpxJ. A clear peak that coelutes with the glycine standard appears in the enzyme catalyzed reaction while the aspartate peak diminishes in size. The production of glycine was enzyme dependent and also required the presence of an amino donor.
Having established that HpxJ was capable of catalyzing aminotransfer, we then wished to determine if ureidoglycine would be a substrate for this enzyme. Since this molecule is known to be unstable in solution we employed E. coli allantoate amidohydrolase (AAH) to synthesize ureidoglycine in situ. This enzyme and the chemistry of the AAH reaction have been recently characterized (7). The production of ureidoglycine by AAH reaction can be followed by monitoring the production of ammonia with a glutamate dehydrogenase coupled assay (Figure S4).
The amino acid products of the HpxJ-catalyzed aminotransfer reaction between ureidoglycine and pyruvate were followed by HPLC after derivatization with DABS-Cl. A peak corresponding to alanine was observed in the presence of the enzyme when pyruvate was the amino-acceptor (Figure 1A, bottom panel). We also observed a small amount of glycine being produced both in the presence and absence of added pyruvate (Figure 1A, bottom two panels). This can be explained by the non-enzymatic decay of ureidoglycine. The decay of this molecule produces glyoxylate (7, 8) which can then be used as a substrate for the reaction. HPLC traces of standards and additional aminotransfer reactions are provided in the supporting material.
Figure 1.
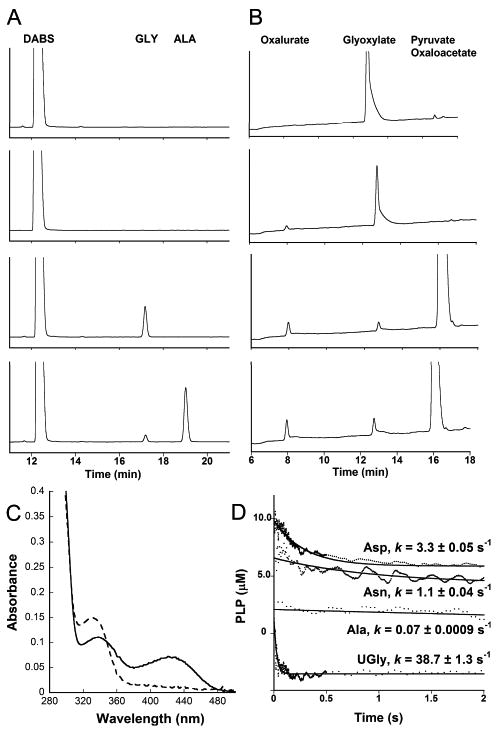
Characterization of K. pneumoniae HpxJ. A) HPLC traces of the amino products of the HpxJ aminotransfer reactions. From top to bottom: control reaction run with HpxJ in the absence of ureidoglycine, control reaction run in the absence of HpxJ with all other components present, the reaction with HpxJ and ureidoglycine in the absence of exogenous amino acceptor, and the HpxJ catalyzed reaction run in the presence of ureidoglycine and pyruvate. B) HPLC traces of the keto-acid products of the HpxJ aminotransfer reaction. From top to bottom: control in the absence of HpxJ, reaction with HpxJ and ureidoglycine in the absence of exogenous amino acceptor, transfer reaction in the presence of pyruvate, and the transfer reaction in the presence of oxaloacetate. Note that any unreacted allantoate in the reaction is hydrolyzed to glyoxylate during the derivatization work-up. C) UV absorbance spectra of native HpxJ (solid line) and reduced HpxJ (dashed line). D) Pre-steady state kinetics of the first half reaction catalyzed by HpxJ and rates from a fit to an exponential function. Note that the alanine and ureidoglycine traces have been shifted down the y-axis for clarity.
The products of the aminotransfer reaction between ureidoglycine and an α-keto acid are an amino acid and oxalurate. For further evidence that HpxJ catalyzes the aminotransfer reaction, we derivatized the keto-acid products of the reaction with o-phenylenediamine and followed them by HPLC. Figure 1B shows that oxalurate is produced in the presence of either pyruvate (Figure 1B, third panel) or oxaloacetate (Figure 1B, fourth panel) as amino acceptors. As with the amino products of the reaction, a small oxalurate peak was also observed in the absence of added amino-acceptor (Figure 1B, second panel).
One of the conserved features of the aspartate aminotransferase family is the dependence upon the cofactor PLP. The absorbance spectrum of HpxJ (Figure 1C) is characteristic of enzymes with bound PLP in the imine form. Treatment of this enzyme with sodium borohydride causes the loss of the peak at 420 nm and a shift of the 333 nm peak to a slightly lower wavelength. This is consistent with the reduction of the PLP-imine to the amine form, a phenomenon that is also observed in the first half of the transfer reaction as PLP is converted to PMP (13). It was also observed that this reduced form of HpxJ was unable to catalyze the aminotransferase reaction for any of the amino-donor/acceptor pairs that we tried (data not shown).
Aminotransferases are known to be promiscuous in regards to the identity of amino donor and acceptors (14, 15). To examine the specificity of HpxJ for different amino acids, we measured the pre-steady state rate for the first half reaction of the transfer by monitoring the conversion of PLP to PMP. The curves and calculated rates for the different amino acids are shown in Figure 1D. It is clear from this data that ureidoglycine is the favored substrate, turning over an order of magnitude faster than the next fastest substrate, aspartate.
In order to further examine this novel aminotransferase and its preference for ureidoglycine, we crystallized and determined the structure of HpxJ using molecular replacement with PDB 1VJ0 (16) as a search model. Details about the structure solution and model building, including collection and refinement statistics, can be found in the supplemental material. The crystal structure of HpxJ showed a homotetramer as the biologically relevant unit (Figure S2), an observation that was verified by size exclusion chromatography. Since this enzyme was highly similar to other PLP-dependent aminotransferases, it was not surprising to find a covalently bound PLP molecule in the active site (Figure 2A). The electron density allowed for the unambiguous placement of the PLP in an orientation that superimposed well with PLP in homologous structures.
Figure 2.
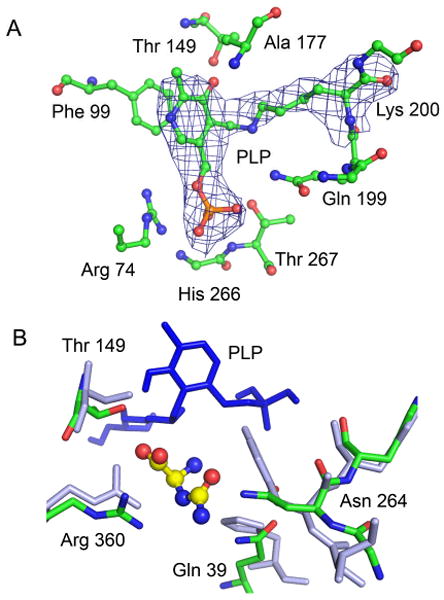
The active site of HpxJ. A) The structure of the PLP binding site of HpxJ showing a molecule of PLP covalently bound to lysine 200. FO - FC density is shown contoured at 3σ around the PLP and lysine residue. B) Superposition of the active site of HpxJ (green) and A. aegypti alanine glyoxylate aminotransferase (grey). Ureidoglycine is shown docked in the active site (yellow, ball and stick).
While the members of the alanine-glyoxylate aminotransferase superfamily are highly structurally conserved, there is a relatively large degree of variation amongst active site residues. With the exception of the highly conserved amino acids that interact with the PLP molecule, differences in active site residues account for the observed differences in substrate selectivity (see figure S3 for a sequence alignment). A superposition of HpxJ with structurally homologous aminotransferases shows several obvious differences in the active sites of these proteins. The most notable of these is an asparagine (Asn264) in place of a tyrosine residue and a glutamine (Gln39) in place of a histidine. The side chains of these residues point into the active site and are well positioned to make contacts with incoming substrates.
To better understand the selectivity of HpxJ for ureidoglycine over other amino acids, we modeled the ligand into the active site of our solved structure. Despite starting from several different conformers of ureidoglycine, all of the simulations converged to a similar orientation of the ligand in the active site. The structure of the HpxJ active site with the energy minimized ligand bound is shown in Figure 2B. What is apparent from this model is that, in addition to interactions with the amino and acid groups made by the PLP molecule, backbone atoms of a conserved proline-proline (Pro19 and Pro20) and a conserved arginine (Arg360), the ureidoglycine tail makes several hydrogen bonding contacts with Asn 264 and Gln39. These amino acids, which replace bulkier tyrosine and histidine residues seen in other aminotransferases, not only allow space for the larger substrate but also appear to directly interact with the substrate.
In summary, we have confirmed that HpxJ is an aminotransferase that catalyzes a novel transfer reaction between ureidoglycine and an α-keto acid. We have provided biochemical and structural evidence for this enzyme's specificity for ureidoglycine. Our data provide an explanation for the presence of this gene in the K. pneumoniae Hpx gene cluster and elucidate a novel branch in the ureide catabolic pathway.
Supplementary Material
Acknowledgments
The authors acknowledge Dr. Cynthia Kinsland at the Cornell Protein Production Facility for assistance with molecular biology, Kenneth Gee for assistance with HPLC experiments, and Leslie Kinsland for assistance with the preparation of the manuscript. We also thank the NE-CAT staff at beamline 24-ID-C of the Advanced Photon Source for assistance with data collection. JBF acknowledges funding from the Tri-Institutional Program in Chemical Biology.
Footnotes
This work was supported by National Institutes of Health Grant GM73220.
Abbreviations: PLP, pyridoxal-5-phosphate; PMP, pyridoxamine phosphate; DABS-Cl, dimethylaminobenzenesulfonyl chloride; OPD, o-phenylenediamine; AAH, allantoate amidoydrolase, UGly, ureidoglycine.
SUPPORTING INFORMATION AVAILABLE
Experimental details, data collection and refinement statistics and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vogels GD, Van der Drift C. Bacteriol Rev. 1976;40:403–468. doi: 10.1128/br.40.2.403-468.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zrenner R, et al. Annu Rev Plant Biol. 2006;57:805–836. doi: 10.1146/annurev.arplant.57.032905.105421. [DOI] [PubMed] [Google Scholar]
- 3.Modric N, et al. Tet Lett. 1992;33:6691–6694. [Google Scholar]
- 4.Kahn K, et al. J Am Chem Soc. 1997;119:5435–5442. [Google Scholar]
- 5.Kahn K, Tipton PA. Biochemistry. 1998;37:11651–11659. doi: 10.1021/bi980446g. [DOI] [PubMed] [Google Scholar]
- 6.Ramazzina I, et al. Nat Chem Biol. 2006;2:144–148. doi: 10.1038/nchembio768. [DOI] [PubMed] [Google Scholar]
- 7.Serventi F, et al. ACS Chem Biol. 2009;5:203–214. doi: 10.1021/cb900248n. [DOI] [PubMed] [Google Scholar]
- 8.van der Drift C, et al. Arch Biochem Biophys. 1970;136:273–279. doi: 10.1016/0003-9861(70)90351-6. [DOI] [PubMed] [Google Scholar]
- 9.Werner AK, et al. Nat Chem Biol. 2009;6:19–21. doi: 10.1038/nchembio.265. [DOI] [PubMed] [Google Scholar]
- 10.Cusa E, et al. J Bacteriol. 1999;181:7479–7484. doi: 10.1128/jb.181.24.7479-7484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Riva L, et al. J Bacteriol. 2008;190:7892–7903. doi: 10.1128/JB.01022-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pope SD, et al. J Bacteriol. 2009;191:1006–1017. doi: 10.1128/JB.01281-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMurry, J., and Begley, T. (2005), Roberts and Company, Englewood, Colorado.
- 14.Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 15.Islam MM, et al. J Biochem. 2003;134:277–285. doi: 10.1093/jb/mvg141. [DOI] [PubMed] [Google Scholar]
- 16.Han GW. Proteins. 2005;58:971–975. doi: 10.1002/prot.20360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.