

Abstract
Reductive lithiation of N-Boc α-amino nitriles generated α-amino alkyllithium reagents with unexpected selectivity. The intermediate radical prefers to align with the nitrogen lone pair, and this interaction leads to an A1,3-strain effect that biases the conformation of the radical. In cyclohexane rings with α-substituents the net effect is an inversion of configuration on reductive lithiation. In the presence of a tethered electrophile the alkyllithium cyclizes to produce a spiro compound, again with inversion of configuration. The overall result is retention of configuration in the cyclization reaction. The same overall selectivity is found with α-oxygen alkyllithium cyclizations, but in this case both steps proceed with retention. The difference can be explained by careful consideration of the intermediate geometries. The α-amino spirocyclization was utilized in a concise and stereoselective synthesis of lepadiformine C.
Stereoselective construction of fully-substituted carbon atoms is a challenge that is often at the heart of a successful complex natural product synthesis. Several years ago we reported a synthesis of spirocycles that was based on stereoselective generation of alkyllithium reagents arising from anomeric stabilization of radical intermediates.1,2 Herein we describe studies on the generation of α-hetero alkyllithium reagents where classic anomeric stabilization is not relevant, but that still show highly stereoselective transformations. This new stereoselective strategy leads to fully substituted carbon atoms and is illustrated with a concise synthesis of lepadiformine C.3
Reduction of N-protected α-amino nitriles with Freeman's reagent (LiDBB)4 is an effective strategy to generate tertiary α-amino alkyllithium reagents.5 An N-alkyl 2-cyano piperidine generally leads to the alkyllithium with high selectivity, but when the nitrogen atom is protected as a Boc carbamate the alkyllithium is produced as a 1.5:1 mixture of isomers.6 The reductive lithiation of N-Boc piperidine 1 in Scheme 1 was instigated to probe the reactivity of the Boc-protected α-amino alkyllithium reagent. Although the lithiation is not stereoselective, both alkyllithium isomers cyclize effectively at −78 °C to produce the bicycle 2 in excellent yield. These tertiary N-Boc α-amino alkyllithium reagents are expected to be conformationally stable at −78 °C, with inversion barriers ca. 20 kcal/mol.7 This experiment demonstrated that tertiary N-Boc alkyllithium reagents may cyclize with retention (SEret) or inversion (SEinv), and the suitably constrained systems can produce single isomers in high yields.8,9
Scheme 1.
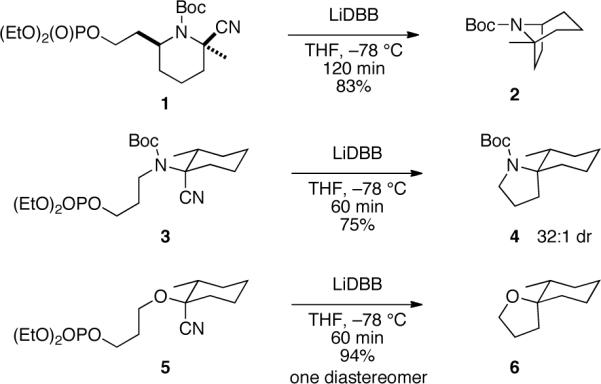
Stereoselective reductive spirocyclizations
The spirocyclizations of nitriles 3 and 5 were highly stereoselective. Both cyclizations proceed with overall retention at the fully substituted center,10 and the heteroatom ends up in the equatorial position. The rationale for the selectivity is not obvious, but the transformation could be useful in synthesis.
Lepadiformine C is an alkaloid isolated from the tunicate Clavelina moluccensis along with lepadiformine B.3 These alkaloids have an effect on the cardiovascular system and block the cardiac inward rectifying K+ channel. Lepadiformine C is the simplest of the alkaloids in this class11 and appears to be a suitable synthetic target for the spirocyclization reaction described above. Our synthetic analysis is shown in Figure 1. Reductive lithiation and cyclization of nitrile 9 would generate the spirocycle 8. Compound 9 could arise from a Strecker reaction of the corresponding cyclohexanone, but preliminary investigation suggested that the equilibrium was unfavorable.12 A more interesting approach would generate the cyclohexane ring by sequential alkylation2a,2d,13 of amino nitrile 10 with a dibromide 11. This strategy rapidly introduces all three rings from acyclic precursors.
Figure 1.
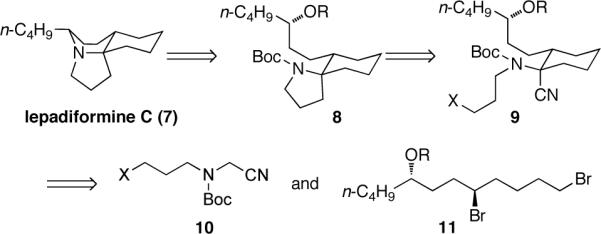
Synthetic analysis of lepadiformine C
The synthesis of lepadiformine C is presented in Scheme 2. The known optically pure alcohol 1214,15 was coupled with 5-((tert-butyldimethylsilyl)oxy)pentanal (13) and oxidized to generate ketone 14. Enantioselective reduction of the propargyl ketone using Noyori's method,15 hydrogenation, deprotection and bromination produced the dibromide 16 in good overall yield. The assembly of the ring system began with alkylation of amino nitrile 17 with dibromide 16. The two reagents were combined and treated with an LDA solution in portions until no more of the monoalkylated product was visible by TLC analysis. Cyclohexane 18 was isolated as a single isomer with the nitrile in the axial position.16 The TBS ether was exchanged for a diethylphosphate leaving group, and subsequent reduction of phosphate 19 with Freeman's reagent (LiDBB)4 produced the cyclic product as a single isomer in excellent yield. Deprotection of the TIPS group led to the crystalline alcohol 21, and X-ray analysis confirmed the expected relative configuration of the stereogenic centers.17 The synthesis was completed by converting the alcohol to a mesylate and removing the Boc group.18 Upon neutralization, the amine cyclized to form the final ring of lepadiformine C, which was isolated as its hydrochloride salt. The characterization data (1H and 13C NMR, HRMS, optical rotation, IR) for lepadiformine C matched that reported for the natural product.3a The synthesis features a rapid and stereoselective assembly of the tricyclic structure from acyclic dibromide 16 and amino nitrile 17.
Scheme 2.

Synthesis of lepadiformine C
The origin of the selectivity in the reductive lithiation and cyclization of nitriles 3, 5 and 19 was explored (Scheme 3). All of the alkyllithium intermediates are expected to be configurationally stable under the reaction conditions.7 The configuration of the alkyllithium intermediates was assessed by protonation, which is assumed to proceed with retention of configuration.7b Reductive lithiation and protonation of TBS ether 18 gave the cis disubstituted cyclohexane 26.19 The lithiation proceed with an unanticipated inversion of configuration. One explanation is that the initially formed radical intermediate 23 is stabilized by interaction with the nitrogen electrons, and resulting steric crowding of the equatorial R' group generates a conformational isomerization to produce radical 24.20 Further reduction of radical 24 would produce alkyllithium 25, which upon protonation would lead to carbamate 26. The lithiation of nitrile 19 presumably follows the same course, with intermediate alkyllithium 27 arising from inversion of configuration. The subsequent cyclization of 27 also proceeds with clean inversion of configuration to produce spirocycle 20. The efficient cyclization of nitrile 1 to bicycle 2 demonstrates that these N-Boc alkyllithium reagents may cyclize efficiently with either retention21 or inversion7 of configuration. Coordination of the Boc nitrogen to the lithium atom in 27 would restrict the conformation, and the phosphate is well positioned to react from the back of the alkyllithium in an SEinv pathway. The spirocyclization is highly stereoselective and proceeds through an unanticipated double inversion sequence.
Scheme 3.

Stereoselective reductive lithiation and protonation control experiments
The cyclization of nitrile 5 to spirocycle 6 shows the same sense of selectivity, but the underlying sequence diverges. In this case reductive lithiation of the corresponding TBS ether 28, followed by protonation proceeds with retention of configuration.19,22 Presumably the axial radical 29 is stabilized by the lone pairs on the adjacent oxygen, with the alkyl substituent adopting a gauche alignment reminiscent of an exo-anomeric effect. Cyclization also takes place with retention of configuration1,23 in the reaction with nitrile 5. The oxygen and N-Boc substrates present an interesting contrast, where superficially similar selectivities result from stereochemically complementary intermediates.
These stereoselective spirocyclization reactions provide an interesting new tool for assembling fully substituted carbon centers. In the case of lepadiformine C, the nitrile in substrate 17 facilitates two alkylation reactions followed by a stereoselective reductive lithiation and cyclization. Thus all the carbon-carbon bonds in the most highly substituted atom were assembled late in the synthesis in an effective but unconventional strategy.
Supplementary Material
ACKNOWLEDGMENT
This project was supported by a generous gift from the Schering-Plough Research Institute. Initial studies were supported by NIGMS GM-65338. SAW acknowledges Amgen for a pre-doctoral fellowship. MAP acknowledges Vertex Pharmaceuticals for a Vertex Scholar fellowship. We thank Prof. Jean-Francois Biard for providing authentic spectra for lepadiformine C and Dr. Joe Ziller for solving the crystal structure of carbamate 21.
Footnotes
Supporting Information Available: Experimental procedures for all compounds, X-ray analysis of compound 21 and configuration assignments for compound 26 and 31 are included.
References
- 1.(a) Rychnovsky SD, Takaoka LR. Angew. Chem. Int. Ed. 2003;42:818–820. doi: 10.1002/anie.200390218. [DOI] [PubMed] [Google Scholar]; (b) Takaoka LR, Buckmelter AJ, La Cruz TE, Rychnovsky SD. J. Am. Chem. Soc. 2005;127:528–529. doi: 10.1021/ja044642o. [DOI] [PubMed] [Google Scholar]
- 2.Cyclization of α-amino alkyllithiums: Guillaume D, Brum-Bousquet M, Aitken DJ, Husson H-P. Bull. Soc. Chim. Fr. 1994;131:391–396.. Coldham I, Hufton R, Snowden DJ. J. Am. Chem. Soc. 1996;118:322–5323.. Coldham I, Fernandez J-C, Price KN, Snowden DJ. J. Org. Chem. 2000;65:3788–3795. doi: 10.1021/jo000088z.. Bahde RJ, Rychnovsky SD. Org. Lett. 2008;10:4017–4020. doi: 10.1021/ol801523r..
- 3.isolation: Sauviat M-P, Vercauteren J, Grimaud N, Juge M, Nabil M, Petit J-Y, Biard J-F. J. Nat. Prod. 2006;69:558–562. doi: 10.1021/np050215s.. synthesis: Meyer AM, Katz CE, Li S-W, Vander Velde D, Aube J. Org. Lett. 2010;12:1244–1247. doi: 10.1021/ol100113r..
- 4.Lithium di-tert-butylbiphenylide (LiDBB): Freeman PK, Hutchinson LL. J. Org. Chem. 1980;45:1924–1930..
- 5.Wolckenhauer SA, Rychnovsky SD. Org. Lett. 2004;6:2745–2748. doi: 10.1021/ol049039p. [DOI] [PubMed] [Google Scholar]
- 6.Wolckenhauer SA, Rychnovsky SD. Tetrahedron. 2005;61:3371–3381. [Google Scholar]
- 7.We are not aware of any data on the racemization of tertiary N-Boc α-amino alkyllithium reagents, but secondary reagents have been studied: Ashweek NJ, Brandt P, Coldham I, Dufour S, Gawley RE, Haeffner F, Klein R, Sanchez-Jimenez G. J. Am. Chem. Soc. 2005;127:449–457. doi: 10.1021/ja048090l.. Faibish NC, Park YS, Lee S, Beak P. J. Am. Chem. Soc. 1997;119:11561–11570.. Effect of TMEDA on RLi racemization: Coldham I, Leonori D, Beng TK, Gawley RE. Chem. Commun. 2009:5239–5241. doi: 10.1039/b911024k.. Beng TK, Yousaf TI, Coldham I, Gawley RE. J. Am. Chem. Soc. 2009;131:6908–6909. doi: 10.1021/ja900755a..
- 8.(a) Gawley RE, Low E, Zhang Q, Harris R. J. Am. Chem. Soc. 2000;122:3344–3350. [Google Scholar]; (b) Gawley RE. Tetrahedron Lett. 1999;40:4297–4300. [Google Scholar]; (c) Beak P, Kerrick ST, Wu SD, Chu JX. J. Am. Chem. Soc. 1994;116:3231–3239. [Google Scholar]
- 9.These experiments do not completely exclude a possible slow isomerization of alkyllithium stereoisomers from compound 1 followed by cyclization of only one of them.
- 10.Assignment of 4 is presented in the Supporting Information. Assignment of 6: Negri JT, Paquette LA. J. Am. Chem. Soc. 1992;114:8835–8841..
- 11.(a) Weinreb SM. Chem. Rev. 2006;106:2531–2549. doi: 10.1021/cr050069v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weinreb SM. Acc. Chem. Res. 2003;36:59–65. doi: 10.1021/ar0200403. [DOI] [PubMed] [Google Scholar]; (c) Abe H, Aoyagi S, Kibayashi C. Angew. Chem. Int. Ed. 2002;41:3017–3020. doi: 10.1002/1521-3773(20020816)41:16<3017::AID-ANIE3017>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Morin MD. PhD Thesis. University of California; Irvine: 2008. [Google Scholar]
- 13.(a) Fleming FF, Shook BC. Tetrahedron. 2002;58:1–23. [Google Scholar]; (b) Fleming FF, Gudipati S. Eur. J. Org. Chem. 2008:5365–5374. doi: 10.1002/ejoc.200800715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Overman LE, Bell KL, Ito F. J. Am. Chem. Soc. 1984;106:4192–201. [Google Scholar]; (b) Aoyagi S, Wang TC, Kibayashi C. J. Am. Chem. Soc. 1993;115:11393–409. [Google Scholar]
- 15.Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J. Am. Chem. Soc. 1997;119:8738–8739. [Google Scholar]
- 16.Assigned by Fleming's correlation method: Fleming FF, Wei G. >J. Org. Chem. 2009;74:3551–3553. doi: 10.1021/jo900286f..
- 17.See supporting information for details of the X-ray structure of 21.
- 18.Abe H, Aoyagi S, Kibayashi C. J. Am. Chem. Soc. 2005;127:1473–1480. doi: 10.1021/ja040213e. [DOI] [PubMed] [Google Scholar]
- 19.The structures of cyclohexane 26 and 31 are discussed in the Supporting Information section.
- 20.A similar A1,3-strain explanation has been involved in the alkylation of cyclohexane carboxylate esters: Caine D. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Heathcock CH, editors. Vol. 3. Pergamon Press; New York: 1991. pp. 39–40.. Krapcho AP, Dundulis EA. J. Org. Chem. 1980;45:3236–3245..
- 21.Beak P, Lee WK. J. Org. Chem. 1993;58:1109–17. [Google Scholar]
- 22.Stereoselective decyanation: Fabre C, Welvart Z. C. R. Acad. Sci., Ser C. 1970;270:1887–1889..
- 23.(a) Woltering MJ, Frohlich R, Hoppe D. Angew. Chem., Int. Ed. 1997;36:1764–1766. [Google Scholar]; (b) Christoph G, Hoppe D. Org. Lett. 2002;4:2189–2192. doi: 10.1021/ol026068w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.