

Abstract

The total synthesis of the highly degraded steroidal natural product, aplykurodinone-1 (1), has been accomplished. Key features include a one-flask hydrolysis/retro-aldol/iodolactonization sequence to excise the C8 hydroxymethylene functionality with retention of stereochemistry, and the stereoselective installation of the C13 methyl group through hydrogenation with homogeneous catalyst.
Of all of the classes of natural products, steroids have perhaps had the most enduring role in prompting new ideas in total synthesis.1 A not insignificant body of organic chemistry has been discovered in the context of pursuits directed to the total synthesis of steroids. Apparently, this interest continues to the present day.2
One of the vexing problems in steroid total synthesis is that of exercising control of the configuration at C20.3 The challenge is that of correlating the configuration of the presumably “freely rotating” C20, with the resident stereochemistry of the polycyclic domain. Though ingenious solutions to the “C20 problem” have been advanced, it is still an interesting and thought provoking matter.
It was in this setting that we took particular note of the aplykurodines, isolated from marine mollusks of the genus Aplysia.4 This family can well be viewed as a rare class of highly degraded marine steroids, where C13 in the aplykurodine setting corresponds to C20 in wild-type steroids. Also of note is the cis fusion of the hydrindane sector in contrast to the trans C-D fusion of hydrindanes, which is so prevalent in fully mature steroids.5
We note that various aplykurodines exhibit cytotoxic activity against a range of human cancer cell lines.6 Notwithstanding the potential elements of chemistry and medicinal chemistry-based points of interest, surprisingly little attention at the level of synthesis has been directed to the aplykurodines. Our laboratory took particular note of a recently isolated member of the family, aplykurodinone-1 (1).7 Following its isolation from the sea hare Synphonota geographica in 2005, the structure of aplykurodinone-1 (1) was elucidated through a combination of spectroscopic methods, X-ray crystallography, and chemical correlation. As shown in Scheme 1, 1 was found to possess a cis-fused C-D ring with epimeric C8 (steroidal numbering) and an unsaturated side chain (as compared to cholesterol).
Scheme 1.

Synthetic Strategy Toward Aplykurodinone-1 (1).
On the basis of considerations advanced above, a total synthesis effort directed to this “degraded” steroid was undertaken. Below is described the inaugural total synthesis of (±)-1, resulting from this initiative. As will be seen, the program potentially encompassed several chemistry-level initiatives of broader heuristic value.
Using ideas stemming from analysis by pattern recognition,8 we envisioned that, in principle, the hypothetical hydrindanone 4 contains an array of coordinated functionality, which could well enable access to the eventual target system, 1. The plan envisioned halolactonization as the instrumentality for fusing the γ-lactone to the cis-fused hydrindanone, thereby creating a ladder-like topography. Accordingly, we needed to gain secure control over the configurational relationship between strategic carbons 3 and 7 in 1 (vide infra). It occurred to us that an otherwise unnecessary quaternary function at C8 could orchestrate the required relationship of the angular methyl group at C7 with a soon-to-be-installed acetic acid moiety at C3 (see structures 5 and 6). Iodolactonization leading to the γ-lactone could then follow in the wake of excision of the now traceless C1 fragment.
Given the general line of thinking adumbrated above, and a long-term proclivity for Diels-Alder reactions, we hoped to advance along such lines. In the event, it was discouraging to find that attempted cycloaddition between 29 and 310 failed under various thermal and Lewis acid-mediated conditions. However, reaction of 2 with methyllithium generated an operational lithium enolate,11 which did undergo effective cycloaddition with 3, thereby providing a 73% yield of 4. It is well recognized that realization of the required cycloaddition does not, in and of itself, inform as to the limiting, mechanistic, extremi (two sequential Michael additions or an anionically mediated Diels-Alder reaction).12
The enone was regioselectively masked as a thioketal.13 The C9 ketone was then reduced, as shown, and the resultant alcohol protected as a MOM ether. Upon reduction of the ester and removal of the dithiane ketal,13a,14 compound 10 was in hand. The primary alcohol was next converted to its diazoacetyl derivative, 11. As shown in Scheme 2, intramolecular cyclopropanation onto the electron-deficient enone afforded the activated cyclopropane, 5, though only in moderate yield (40%).15
Scheme 2.

Synthesis of 6 En Route to Aplykurodinone-1 (1).
aKey: (a) MeLi, DME/THF, −50 °C; TFA, CHCl3, reflux, 73%; (b) HSCH2CH2SH, AcOH, p-TSA, 74%; (c) NaBH4, CH2Cl2/MeOH, −78 °C→ −40 °C; (d) MOMCl, iPr2NEt, CH2Cl2, 84% over 2 steps; (e) DIBAL-H, CH2Cl2, −78 °C; (f) Tl(NO3)3, MeOH/THF/H2O, 95% over 2 steps; (g) TsNHN=CHCOCl, PhNMe2, Et3N, CH2Cl2, 0 °C, 88%; (h) bis-(N-t-butylsalicylaldiminato) copper (II), PhMe, reflux, 40%; (i) NaBH4, CeCl3•7H2O; (j) I2, PPh3, imidazole, PhMe, reflux, 78% over 2 steps; (k) SmI2, THF/MeOH, 85%; (l) HCl, THF/H2O; (m) DMP, CH2Cl2, 80% over 2 steps.
The next phase entailed the opening of the cyclopropane ring, with concomitant installation of unsaturation at C4–C5. Toward this end, the C5 ketone was converted to an iodide through a two-step sequence, commencing with Luche reduction,16 which provided a 2:1 (β:α) epimeric mixture of alcohols.17 These compounds were readily converted to iodide 12. In the event, upon exposure of 12 to SmI2,18 it readily suffered the hoped-for reductive cyclopropane opening, to deliver lactone 13 in good yield (85%). The latter was converted to 6 in a straightforward fashion, as shown.
The otherwise extraneous C8 oxymethylene functionality had indeed been exploited as a very useful C3 stereochemical linchpin (vide supra). Having served its function in mediating the stereoselective installation of the C3 center, the C1 fragment was cleaved via a hydrolysis–retro-aldol sequence following exposure of 6 to the action of K2CO3 in water at 100 °C. Subsequent iodolactonization19 of the resultant carboxylic acid (14) was accomplished in the same flask, to provide 15 in 75% yield from 6. The re-emergence of the cis-junction was confirmed through NOESY analysis of the methyl ester derivative of 14 and by X-ray diffraction of the subsequent deiodinated intermediate, 16.20 Finally, Saegusa oxidation21 of 16 afforded 7, presenting the tricyclic core of aplykurodinone-1, in the sense shown.
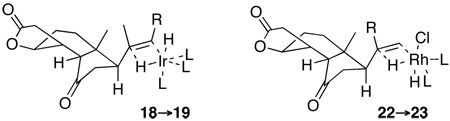
With compound 7 in hand, the synthesis entered its terminal phase, i.e. installation of the C11 side chain. A stepwise strategy was set in place. As outlined in Scheme 4, BF3-mediated conjugate addition of the cuprate derived from vinyl bromide 1722 was accomplished with a high degree of facial selectivity (10:1 dr) to provide intermediate 18. Given the difficulties surrounding the heterogeneous hydrogenation of the C20–C22 olefin as a means of installing the C20 stereocenter of trans-fused mature steroids,23 it was hoped that homogeneous hydrogenation protocols may be more productive.24 Thus, following exposure to Crabtree catalyst,25 under an atmospheric pressure of H2, the tri-substituted olefin was reduced in a diastereoselective fashion (>5:1) to furnish compound 19, which bears all of the stereocenters of aplykurodinone-1 (1). A single crystal of this advanced intermediate was obtained, and X-ray crystallographic analysis was found to support the proposed structure. Elaboration of the side chain was accomplished in a straightforward fashion, as shown. Thus, cleavage of the TBS group26 and subsequent oxidation27 delivered aldehyde 20, which readily underwent modified Julia olefination28 to furnish the target compound, aplykurodinone-1 (1). Its spectroscopic properties were the same as those reported for the isolated natural product. In the course of these synthetic studies, we also investigated a different route for the emplacement of the C11 side chain. The fully elaborated dienyl fragment could be appended to the core (7) in a single step. It was hoped that the intermediate thus obtained could then be converted, in one step, to aplykurodinone-1 (1), through regio- and stereo-selective hydrogenation. In the event, the conjugate addition of 2129 to 7 was accomplished, to provide 22 in moderate yield. Upon exposure to Wilkinson’s catalyst30 under atmospheric H2, the 13-epi-aplykurodinone-1 isomer (23) was selectively formed (>6:1 dr). Perhaps in each of the two hydrogenation transition states (18→19; 22→23), the double bond projects from the β-face of the molecular backbone. The bulky catalyst then approaches the olefin from the unhindered of the olefin, to provide the observed stereochemical outcome.31
Scheme 4.

aKey: (a) 17, t-BuLi, CuCN, BF3•OEt2, −78 °C→ −98 °C, 73%, 10:1 dr; (b) Crabtree catalyst, H2, CH2Cl2, 75% conversion, 50% yield (pure); (c) HF, CH3CN/THF; (d) DMP, CH2Cl2, 87% over two steps; (e) THF, LiHMDS, −78 °C, 68%.
In summary, the total synthesis of aplykurodinone-1 (1) has been accomplished. A key feature of the effort involved an anionically-mediated cycloaddition of a metallo-enolate derived from 2 with 3 (see formation of 4). The seemingly extraneous oxymethyl C1 function at C8, used to govern the configurational relationship between C3 and C7. The C8 function is ultimately excised. The stereochemistry at C13 is managed with good stereoselectivity based on suitable order of introduction of H and Me to this carbon.
Supplementary Material
Scheme 3.

aKey: (a) K2CO3, H2O, 100 °C; (b) NIS, CH2Cl2, 75%; (c) Ra-Ni, CH2Cl2/EtOH, 90%; (d) TMSOTf, Et3N, CH2Cl2, 0 °C; Pd(OAc)2, CH3CN, 76% (90% brsm).
Scheme 5.

aKey: (a) 17, t-BuLi, CuCN, Et2O, −78 °C, 51%; (b) Wilkinson catalyst, H2, benzene, 67%.
Acknowledgments
Support was provided by the NIH (HL25848 and CA103823 to S.J.D.). Aaron Sattler, Wesley Sattler from the Parkin group (Columbia University) are thanked for their help with X-ray diffraction experiments (CHE-0619638 from the NSF). We thank Prof. M. Carbone (Instituto di Chimica Biomolecolare, CNR, Italy) for providing NMR spectra of the aplykurodinones. Valuable discussion with Ms. Rebecca Wilson throughout the course of this project is gratefully acknowledged.
Footnotes
Supporting Information Available Experimental procedures, copies of spectral data, and characterization data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kametani T, Nemoto H. Tetrahedron. 1981;37:3–16. [Google Scholar]; (b) Skoda-Foldes R, Kollar L. Chem. Rev. 2003;103:4095–4129. doi: 10.1021/cr020075g. [DOI] [PubMed] [Google Scholar]; (c) Chapelon A-S, Moraleda D, Rodriguez R, Ollivier C, Santelli M. Tetrahedron. 2007;63:11511–11616. [Google Scholar]; (d) Ibrahim-Ouali M. Steroids. 2009;74:133–162. doi: 10.1016/j.steroids.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 2.(a) Yueng Y-Y, Chein R-J, Corey EJ. J. Am. Chem. Soc. 2007;129:10346–10347. doi: 10.1021/ja0742434. [DOI] [PubMed] [Google Scholar]; (b) Giroux S, Corey EJ. J. Am. Chem. Soc. 2007;129:9866–9867. doi: 10.1021/ja074306i. [DOI] [PubMed] [Google Scholar]
- 3.(a) Piatak DM, Wicha J. Chem. Rev. 1978;78:199–241. [Google Scholar]; (b) Redpath J, Zeelen FJ. Chem. Soc. Rev. 1983;12:75–98. [Google Scholar]
- 4.(a) Miyamoto T, Higuchi R, Komori T. Tetrahedron Lett. 1986;27:1153–1156. [Google Scholar]; (b) Spinella A, Gavagnin M, Crispino A, Cimino G, Martinez E, Ortea J, Sodano G. J. Nat. Prod. 1992;55:989–993. doi: 10.1021/np50085a027. [DOI] [PubMed] [Google Scholar]; (c) Ortega MJ, Zubia E, Salva J. J. Nat. Prod. 1997;60:488–489. doi: 10.1021/np9607464. [DOI] [PubMed] [Google Scholar]
- 5.Hill RA, Kirk DN, Makin HLJ, Murphy GM. Dictionary of Steroids, Chemical Data, Structures and Biographies. London: Chapman and Hall; 1991. pp. xiv–xxix. [Google Scholar]
- 6.(a) De Riecardis F, Spinella A, Izzo I, Giordano A, Sodano G. Tetrahedron Lett. 1995;36:4303–4306. [Google Scholar]; (b) Izzo I, Meduri G, Avallone E, De Riccardis F, Sodano G. Eur. J. Org. Chem. 2000:439–448. [Google Scholar]
- 7.Gavagnin M, Carbone M, Nappo M, Mollo E, Roussis V, Cimino G. Tetrahedron. 2005;61:617–621. [Google Scholar]
- 8.For a discussion of the value of pattern anaylsis as a supplement to strategic bond disconnection in retrosynthetic analysis, see: Wilson RM, Danishefsky SJ. J. Org. Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s.
- 9.Danishefsky SJ. Acc. Chem. Res. 1981;14:400–406. [Google Scholar]
- 10.Hua DH, Venkataraman S, King RCY, Paukstelis JV. J. Am. Chem. Soc. 1988;110:4741–4748. [Google Scholar]
- 11.(a) Stork G, Hudrlik PF. J. Am. Chem. Soc. 1968;90:4464–4465. [Google Scholar]; (b) Baker R, Selwood DL, Swain CJ, Webster NMH. J. Chem. Soc, Perkin Trans 1. 1988:471–480. [Google Scholar]
- 12.(a) Ihara M, Fukumoto K. Angew. Chem. Int. Ed. Engl. 1993;32:1010–1022. [Google Scholar]; (b) Kanemasa S, Kumegawa M, Wada E, Nomura M. Bull. Chem. Soc. Jpn. 1991;64:2990–3004. [Google Scholar]
- 13.(a) Bosch MP, Camps F, Coll J, Guerrero A, Tatsuoka T, Meinwald J. J. Org. Chem. 1986;51:773–784. [Google Scholar]; (b) Golinski M, Brock CP, Watt DS. J. Org. Chem. 1993;58:159–164. [Google Scholar]
- 14.Jones PS, Ley SV, Simpkins NS, Whittle AJ. Tetrahedron. 1986;42:6519–6534. [Google Scholar]
- 15. Corey EJ, Myers AG. Tetrahedron Lett. 1984;25:3559–3562. (b) An alternative approach to compound 12, involving a high-yielding cyclopropanation of an electron rich olefin was also investigated. See Supporting Information for details.
- 16.Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- 17.The stereochemistry of the 5-β-epimer was confirmed by X-ray analysis of its thiocarbonyl imidazole derivative. See Supporting Information. Barton DH, McCombie SW. J. Chem. Soc., Perkin Trans. 1. 1975:1574–1585.
- 18.Abad A, Agullo C, Cunat AC, Navarro I. Tetrahedron Lett. 2001;42:8965–8968. [Google Scholar]
- 19.(a) Kitagawa O, Sato T, Taguchi T. Chem. Lett. 1991:177–180. [Google Scholar]; (b) Devaux JF, O’Neil SV, Guillo N, Paquette LA. Collect. Czech. Chem. Commun. 2000;65:490–510. [Google Scholar]
- 20.(a) Kamano Y, Pettit GR. J. Org. Chem. 1973;38:2202–2204. [Google Scholar]; (b) Piccinini P, Vidari G, Zanoni G. J. Am. Chem. Soc. 2004;126:5088–5089. doi: 10.1021/ja0493796. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011–1013. [Google Scholar]
- 22.(a) Le Monez P, Fageas V, Poisson J, Ardisson J. Tetrahedron Lett. 1994;35:7767–7770. [Google Scholar]; (b) Tiefenbacher K, Arion VB, Mulzer J. Angew. Chem. Int. Ed. 2007;46:2690–2693. doi: 10.1002/anie.200604781. [DOI] [PubMed] [Google Scholar]
- 23.(a) Narwid TA, Cooney KE, Uskokovic MR. Helv. Chim. Acta. 1974;57:771–781. doi: 10.1002/hlca.19740570331. [DOI] [PubMed] [Google Scholar]; (b) Schow SR, McMorris TC. J. Org. Chem. 1979;44:3760–3765. [Google Scholar]; (c) Kametani T, Suzuki K, Nemoto H. J. Am. Chem. Soc. 1981;103:2891–2892. [Google Scholar]
- 24.Harmon RE, Gupta SK, Brown DJ. Chem. Rev. 1973;73:21–52. [Google Scholar]
- 25.Crabtree R. Acc. Chem. Res. 1979;12:331–337. [Google Scholar]
- 26.When Bu4NF was used, extensive decomposition of the starting material was observed.
- 27.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- 28.(a) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]; (b) Marti C, Carreira EM. J. Am. Chem. Soc. 2005;127:11505–11515. doi: 10.1021/ja0518880. [DOI] [PubMed] [Google Scholar]
- 29.Mushti CS, Kim JH, Corey EJ. J. Am. Chem. Soc. 2006;128:14050–14052. doi: 10.1021/ja066336b. [DOI] [PubMed] [Google Scholar]
- 30.(a) Young JF, Osborn JA, Jardine FH, Wilkinson G. Chem. Commun. 1965:131–132. [Google Scholar]; (b) O’Connor C, Wilkinson G. J. Chem. Soc. (A) 1968:2665–2671. [Google Scholar]
-
31.Proposed transition states of the homogeneous hydrogenation with Crabtree and Wilkinson catalyst are shown below. In each case, the bulky catalyst is believed to approach the double bond from its less hindered face. However, as described above, different stereochemical outcomes were observed in these two systems.
 For mechanistic study of these two types of hydrogenation reaction, See:
Brown JM, Derome AE, Hughes GD, Monaghan PK. Aust. J. Chem. 1992;45:143–153.
Nelson DJ, Li R, Brammer C. J. Org. Chem. 2005;70:761–767. doi: 10.1021/jo048968r.
For mechanistic study of these two types of hydrogenation reaction, See:
Brown JM, Derome AE, Hughes GD, Monaghan PK. Aust. J. Chem. 1992;45:143–153.
Nelson DJ, Li R, Brammer C. J. Org. Chem. 2005;70:761–767. doi: 10.1021/jo048968r.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.