

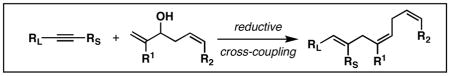
Abstract
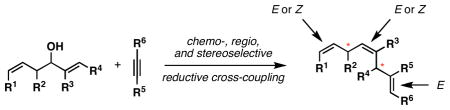
A convergent synthesis of highly substituted and stereodefined skipped polyenes is described from the reductive cross-coupling of substituted 1,5-diene-3-ols with alkynes. The control of site-selectivity in functionalization of the substituted diene is a central feature of this complex fragment union reaction.
Skipped polyenes are structural motifs that are present in a vast array of natural products of biological significance. While stereochemically homogeneous skipped-polyenes are a central feature of a variety of natural polyunsaturated fatty acids,1 skipped polyenes of diverse stereochemistry and substitution are also abundant, with examples including natural products that are of potential value as antibiotic, antifungal and anticancer agents (Figure 1).2 Despite their frequent occurrence in natural products, and potential utility in organic chemistry, there remains a great dearth of efficient methods for the synthesis of stereodefined skipped polyenes.3 We describe here a highly selective convergent pathway to skipped trienes via chemo-, regio-, and stereoselective reductive cross-coupling of substituted 1,5-dienes and alkynes.
Figure 1.

Natural products housing skipped polyenes.
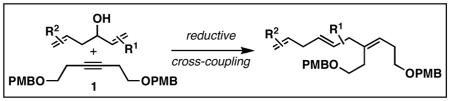
In efforts aimed at providing a solution to problems associated with the synthesis of stereochemically heterogeneous skipped polyenes, we previously described a reductive cross-coupling of substituted vinylcyclopropanes with alkynes.4 This reaction, depicted in Figure 2A, furnishes skipped trienes bearing a central (E)-alkene, with the potential to install a flanking (E)-or (Z)-disubstituted alkene. We hypothesized that a related reductive cross-coupling of 1,5-dienes with alkynes may define a more convenient and stereochemically complementary pathway to related skipped trienes (Figure 2B). That said, such a process would require control of chemoselectivity defined by the competition between reaction paths depicted in Figure 2C: 1) Site selective reductive cross-coupling at the allylic alcohol (Path A),5 2) site selective reductive cross-coupling via the homoallylic alcohol (Path B),6 or 3) reaction at both alkenes of the 1,5-diene.
Figure 2.

Convergent coupling reactions for skipped triene synthesis.
Our initial investigations were focused on determining whether structural features on the 1,5-diene could be used to control site selective C–C bond formation. As depicted in entry 1 of Table 1, coupling of the parent unsubstituted diene 2 with symmetrical alkyne 1 (by treatment of the alkyne with the combination of Ti(Oi-Pr)4 and c-C5H9MgCl,7 followed by addition of the lithium alkoxide of 2 and subsequent hydrolysis with a saturated aqueous solution of NaHCO3) led to the formation of the skipped triene 3. While we were delighted to isolate a skipped triene from this coupling reaction, both the site- and stereoselectivity for this process were poor (1.4:1 and 1:1, respectively). Moving on, reaction of the Me-substituted 1,5-diene 4 with alkyne 1 delivered the 1,7-diene 5 in 50% yield, with no trace of a skipped triene being produced (entry 2).
Table 1.
 | |||||
|---|---|---|---|---|---|
| entry | 1,5-diene | yielda (%) | path selectivityb | stereoselectivity | major productd |
| 1 |
2 |
- | 1.4:1 | 1:1 |
3 |
| 2 |
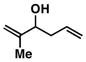 4 |
50 | ≥ 20:1 | - |
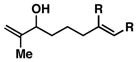 5 |
| 3 |
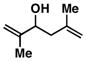 6 |
57 | ≥ 20:1 | ≥ 20:1c |
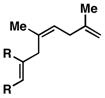 7 |
| 4 |
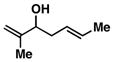 8 |
57 | ≥ 20:1 | ≥ 20:1c |
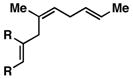 9 |
| 5 |
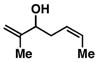 10 |
76 | ≥ 20:1 | ≥ 20:1c |
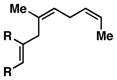 11 |
| 6 |
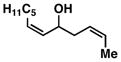 12 |
53e | ≥ 20:1 | ≥ 20:1c |
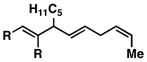 13 |
Reaction conditions: 1 (2–3 equiv.), Ti(Oi-Pr)4, c-C5H9MgCl, PhMe (−78 to −35 °C), then cool to −78 °C and add Li alkoxide of the allylic alcohol as a solution in THF (warm to 0 °C).
In cases where selectivity is reported as ≥20:1, no evidence was found for products derived from C–C bond formation by a different path.
In cases where selectivity is reported as ≥ 20:1, no evidence was found for the formation of stereoisomeric products.
Olefin geometry of the major products was assigned by analogy to previous examples.
Yield reported is after HPLC purification.
To our delight, when both alkenes of the 1,5-diene are substituted, reductive cross-coupling reactions uniformly proceed with high selectivity for the formation of stereodefined skipped trienes. The generality of this process can be seen in entries 3–6, where skipped trienes bearing two or three stereodefined alkenes (7, 9, 11 and 13) are produced with ≥ 20:1 selectivity for C–C bond formation at the allylic alcohol terminus of the 1,5-diene. Notably, these coupling reactions proceed with very high levels of stereoselectivity, do not disturb the preset stereochemistry of the 1,5-diene starting material, and provide a foundation of evidence that supports the ability to control site-selectivity in reductive cross-coupling reactions of dienes with alkynes.
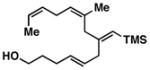
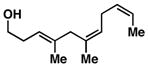
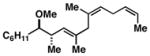
Advancing beyond a simple exploration of the 1,5-diene partner, we shifted our attention to employing this bond construction for regioselective coupling of unsymmetrical alkynes.8 While TMS-alkynes were expected to behave in a predictable manner, with C–C bond formation occurring distal to the TMS group, we were delighted to see that TMS-substituted enynes (14 and 16) were compatible substrates in this chemoselective reductive cross-coupling reaction (entries 1–2). Here, selective activation of the alkyne of 14 and 16 is followed by chemo- and stereoselective C–C bond formation with substituted 1,5-dienes. Even alkynes that boast a modest level of steric differentiation (Me- vs TIPSOEt-) result in regioselective cross-coupling (entry 3); here, the stereodefined triene 19 is produced in 54% yield (rs = 9:1). Use of a more sterically differentiated internal alkyne results in skipped trienes with even higher levels of regioselection. As depicted in entry 4, union of alkyne 20 with diene 10 produces triene 21 in 65% yield with ≥20:1 rs. Substitution is also tolerated in the 1,5-diene partner. As illustrated in entry 5, union of alkyne 18 with diene 22 results in the formation of triene 23 – a stereodefined skipped polyene possessing one (E)-trisubstituted-, one (Z)-trisubstituted- and one (Z)-disubstituted alkene, while also establishing a 1,4-diene bearing a central stereodefined alkyl substituent.
In conclusion, we have described a convenient synthetic pathway to a range of stereochemically defined skipped trienes by the union of 1,5-dienes with disubstituted alkynes. This achievement documents the ability to control chemoselectivity in reductive cross-coupling reactions of dienes, where alkene substitution and location of a pendant alkoxide combine to dictate the course of C–C bond formation.9 A mechanistic feature that has been revealed in the course of these studies is the increased sensitivity of homoallylic alcohol–alkyne reductive cross-coupling reactions to alkene substitution in comparison to allylic alcohol-alkyne coupling processes. In all cases explored, where each alkene of a 1,5-diene is disubstituted, chemoselective reductive cross-coupling proceeds by a pathway that exploits the enhanced reactivity of an allylic alcohol in preference to a homoallylic alcohol – a mechanistic rationale for this divergent reactivity awaits further experimentation. Finally, we have demonstrated that this coupling reaction is useful for the selective union of unsymmetrical coupling partners, including substrates that display only minor steric differences (i.e. 18) – this observation was unexpected and points to a potentially powerful aspect of the present reductive cross-coupling reaction. We look forward to advances that follow from these initial findings.
Supplementary Material
Table 2.
 | ||||
|---|---|---|---|---|
| entry | alkyne | 1,5-diene | yield (%)a | skipped triene product |
| 1 |
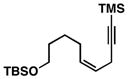 14 |
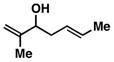 8 |
60b |
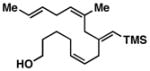 15 |
| 2 |
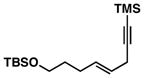 16 |
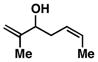 10 |
51b |
 17 |
| 3 |
18 |
10 | 54b |
 19 |
| 4 |
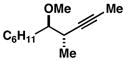 20 |
10 | 65 |
 21 |
| 5 |
18 |
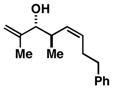 22 |
58b |
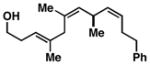 23 |
Reaction conditions: alkyne (2–3 equiv.), Ti(Oi-Pr)4 or ClTi(Oi-Pr), c-C5H9MgCl, PhMe (−78 to −35 °C), then cool to −78 °C and add Li alkoxide of the allylic alcohol as a solution in THF (warm to 0 °C).
Yield reported is over two steps: chemoselective reductive cross-coupling and silyl deprotection with TBAF in THF.
Acknowledgments
The National Institutes of Health –NIGMS supported this study (GM80266 and GM80266-04S1).
Footnotes
Supporting Information Available: Experimental procedures and tabulated spectroscopic data for new compounds (PDF) are available free of charge via the Internet at http://pubs.acs.org/paragonplus/submission/jacsat/.
References
- 1.a) Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, Wahli W. Mol Endocrinol. 1997;11:779. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]; b) Ringbom T, Huss U, Stenholm Å, Flock S, Skattebol L, Perara P, Bohlin L. J Nat Prod. 2001;64:745. doi: 10.1021/np000620d. [DOI] [PubMed] [Google Scholar]; c) Hamilton JA. Prostag Leukotr Ess. 2002;62:65. [Google Scholar]; d) Yaqoob P. Trends Immunol. 2003;24:639. doi: 10.1016/j.it.2003.10.002. [DOI] [PubMed] [Google Scholar]; e) Lie Ken Jie MSF, Pasha MK, Syed-Rahmatullah MSK. Nat Prod Rep. 1997;14:163. [Google Scholar]
- 2.a) Irschik H, Augustiniak H, Gerth K, Hoefle G, Reichenback H. J Antibiot. 1995;48:787. doi: 10.7164/antibiotics.48.787. [DOI] [PubMed] [Google Scholar]; b) Gerth K, Washausen P, Hofle G, Irschik H, Reichenback H. J Antibiot. 1996;49:71. doi: 10.7164/antibiotics.49.71. [DOI] [PubMed] [Google Scholar]
- 3.For a review of methods for the synthesis of (Z,Z)-1,4-dienes, see: Durand S, Parrain JL, Santelli M. J Chem Soc, Perkin Trans 1. 2000:253.For carbonyl olefination-based methods to access skipped alkenes, see: Fürstner A, Larionou O, Flügge S. Angew Chem Int Ed. 2007;46:5545. doi: 10.1002/anie.200701640.For recent reviews, see: Aïssa C. Eur J Org Chem. 2009:1831.Demeunier R, Marko IE. In: Modern Carbonyl Olefination. Takeda T, editor. Wiley–VCH; Weinheim: 2004. p. 104.For a route to 1,4-dienes by 1,5-hydrogen migrations in cis-vinylcyclopropanes, see: Ellis RJ, Frey HM. P Chem Soc London. 1964:221.Parziale PA, Berson JA. J Am Chem Soc. 1990;112:1650.Parziale PA, Berson JA. J Am Chem Soc. 1991;113:4595.
- 4.Macklin TK, Micalizio GC. Nature Chem. 2010;2 doi: 10.1038/nchem.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112. doi: 10.1021/ja075678u.See also: Lysenko IL, Kim K, Lee HG, Cha JK. J Am Chem Soc. 2008;130:15997. doi: 10.1021/ja806440m.
- 6.Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440. doi: 10.1002/anie.200603515. [DOI] [PubMed] [Google Scholar]
- 7.For reviews of metal-π complexes from Ti(Oi-Pr)4, see: Kulinkovich OG, Meijere A. Chem Rev. 2000;100:2789. doi: 10.1021/cr980046z.Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835. doi: 10.1021/cr990277l.
- 8.Regioselection in reductive cross-coupling is often challenging to control. For a recent review, see: Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2010:391. doi: 10.1002/ejoc.200901094.For a recent example of ligand-controlled regioselective reductive cross-coupling of internal alkynes with aldehydes, see: Makik HA, Sormunen GJ, Montgomery J. J Am Chem Soc. 2010;132:6304. doi: 10.1021/ja102262v.
- 9.In Ni-catalyzed reductive cross-coupling of enynes with aldehydes, Jamison has demonstrated that the pendent alkene plays a role in regioselective functionalization of the alkyne; see: Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799.The method described herein does not rely on similar alkene direction and is tolerant of diverse alkene substitution and stereochemistry.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.