

Abstract
Rapid evolution is a hallmark of centromeric DNA in eukaryotic genomes. Yet, the centromere itself has a conserved functional role that is mediated by the kinetochore protein complex. To broaden our understanding about both the DNA and proteins that interact at the functional centromere, we sought to gain a detailed view of the evolutionary events that have shaped the primate kinetochore. Specifically, we performed comparative mapping and sequencing of the genomic regions encompassing the genes encoding three foundation kinetochore proteins: Centromere Proteins A, B, and C (CENP-A, CENP-B, and CENP-C). A histone H3 variant, CENP-A provides the foundation of the centromere-specific nucleosome. Comparative sequence analyses of the CENP-A gene in 14 primate species revealed encoded amino-acid residues within both the histone-fold domain and the N-terminal tail that are under strong positive selection. Similar comparative analyses of CENP-C, another foundation protein essential for centromere function, identified amino-acid residues throughout the protein under positive selection in the primate lineage, including several in the centromere localization and DNA-binding regions. Perhaps surprisingly, the gene encoding CENP-B, a kinetochore protein that binds specifically to alpha-satellite DNA, was not found to be associated with signatures of positive selection. These findings point to important and distinct evolutionary forces operating on the DNA and proteins of the primate centromere.
Keywords: kinetochore, selection, evolution, centromere
Introduction
The conservation of both coding and noncoding genome sequence throughout evolution is a primary predictor of functional constraint. Comparisons of genome sequences from evolutionarily diverse species are routinely used to detect conservation, which in turn points to genomic regions likely to be functionally important (Pennacchio and Rubin 2001). In contrast, comparisons of genome sequences from closely related species can identify genomic regions that have diverged over short periods of evolution, perhaps due to selective pressure to change rapidly (Boffelli et al. 2003).
The centromere appears to be an enigma with respect to the paradigm of “conservation implies function” (Henikoff et al. 2001). Basic centromere function is required in all eukaryotic species, and, indeed, the mechanism of action of the proteins associated with centromeric DNA is well conserved (Saffery et al. 2000). Thus, the fundamentals of centromere biology would be expected to require extensive evolutionary conservation. Yet, centromeric DNA sequences vary markedly from species to species (Sullivan et al. 2001). Proteins associated with centromeric DNA—in particular, those of the kinetochore complex—have orthologs in most species examined to date (Cheeseman and Desai 2008); however, these, too, appear to be rapidly evolving (Malik and Henikoff 2001; Talbert et al. 2002, 2004).
A model for the evolution of centromeric DNA is emerging from comparisons of orthologous pericentromeric sequences in primates (Schueler and Sullivan 2006). Functional centromeres in primates consist of alpha-satellite DNA (a tandem 171-bp repeat), which exists in two forms: monomeric (simple, head-to-tail repetition of divergent monomers) and higher-order (amplified groups of monomers in tandem, head-to-tail configurations) (Alexandrov et al. 2001). There are notable differences in centromeric DNA among primates. Some Old World monkeys lack higher-order segments of alpha satellite, and some have higher-order segments that are the same among nonhomologous chromosomes. In contrast, the great apes (including human) have chromosome-specific, higher-order alpha-satellite repeat structures (Willard 1990). These features of alpha satellite suggest a change from genomewide to chromosome-specific homogenization of centromeres within the last 25–35 My of primate evolution (Alexandrov et al. 2001).
It has been proposed that rapidly evolving centromeric DNA drives the unequal transmission of chromosomes during female meiosis (Zwick et al. 1999; Henikoff et al. 2001). Specifically, meiotic drive results from changes in centromeric DNA that leads to more efficient, non-Mendelian transmission of chromosomes bearing these new sequences to the egg. Centromeres containing older sequences, and the chromosomes bearing them, would then be more likely lost in the first or second polar bodies (Pardo-Manuel de Villena and Sapienza 2001). Such a disparity would enhance the transmission of all genes linked to the improved centromere. To compensate for these imbalances, other factors may play a role in restoring parity. Inner kinetochore proteins that directly associate with the changing centromeric DNA are likely candidates for such a “balancer” role (Henikoff et al. 2001).
Centromere Proteins A, B, and C are foundation kinetochore proteins that directly bind to or closely associate with centromeric DNA (Amor et al. 2004). Centromere Protein A (CENP-A), a histone H3 variant found at active centromeres in all eukaryotic species examined, is responsible for forming a centromere-specific nucleosome upon which the kinetochore assembles (Allshire and Karpen 2008). The CENP-A gene has been shown to be under positive selection in Drosophila (Malik and Henikoff 2001) and Arabidopsis (Talbert et al. 2002), but no signature of positive selection was detected in mammals (Talbert et al. 2004). Centromere Protein B (CENP-B) binds to a 17-bp recognition sequence (CENP-B box) within alpha-satellite DNA (Masumoto et al. 1989) and is found at most, but not all, active mammalian centromeres (Earnshaw et al. 1989; Saffery et al. 2000). Human alpha-satellite DNA containing CENP-B boxes can form artificial chromosomes in vitro, whereas that containing altered CENP-B boxes cannot (Harrington et al. 1997; Ikeno et al. 1998; Masumoto et al. 1998; Ohzeki et al. 2002). Centromere Protein C (CENP-C) is closely associated with centromeric DNA, but no specific binding site within the DNA has been identified (Sugimoto et al. 1994; Yang et al. 1996). CENP-C cannot associate with centromeric DNA in the absence of CENP-A, and physical interaction between CENP-C and CENP-B has been demonstrated (Suzuki et al. 2004). Evidence for positive selection acting on CENP-C has been found in mammalian and plant lineages (Talbert et al. 2004).
In this study, we performed comparative sequence analyses of the genes encoding CENP-A, CENP-B, and CENP-C in primates. By sampling species from all major branches of the primate phylogenetic tree, we aimed to identify rapidly evolving regions of these centromere proteins that may be acting as catalysts for (or in response to) changes in centromeric DNA. Our findings provide new insights into the evolution of these important proteins and the roles that they play in centromere biology.
Materials and Methods
Sequence Generation and Annotation
The reference human-genome sequence (build hg18; genome.ucsc.edu) was used as the basis for all comparative analyses. Key features of the reference human sequence for the CENP-A-, CENP-B-, and CENP-C-containing regions (each ∼300 kb in size) are provided in table 1. Orthologous bacterial artificial chromosome (BAC) clones were isolated using our previously described methods (Thomas et al. 2003). Specifically, alignments between the human sequence and orthologous mouse sequence were used to identify regions of conservation, from which universal overgo probes were designed. The resulting probes were used to screen arrayed BAC libraries generated from nonhuman primates (chimpanzee [Pan troglodytes], CHORI 251; gorilla [Gorilla gorilla], CHORI 255; orangutan [Pongo abelii], CHORI 253; gibbon [Nomascus leucogenys], CHORI 271; macaque [Macaca mulatta], CHORI 250; baboon [Papio anubis], RPCI 41; vervet monkey [Cercopithicus aethiops], CHORI 252; colobus monkey [Colobus guereza], CHORI 272; squirrel monkey [Saimiri boliviensis], CHORI 254; dusky titi [Callicebus moloch], LBNL-5; owl monkey [Aotus nancymai], CHORI 258; marmoset [Callithrix jacchus], CHORI 259; spider monkey [Ateles geoffroyi], UC-1; mouse lemur [Microcebus murinus], CHORI 257; galago [Otolemur garnetti], CHORI 256; black lemur [Eulemur macaco], CHORI 273; and ring-tailed lemur [Lemur catta], LBNL 2). All BAC libraries and clones are available at the BACPAC Resources Center (cpac.chori.org). Restriction enzyme digest-based fingerprint analysis and probe-content mapping were used to assemble BAC contigs, from which minimal tiling paths of clones were selected (Marra et al. 1997).
Table 1.
Comparative Sequence Data Sets for Genomic Regions Containing CENP-A, -B, and -C.
| Human Reference Sequence Data |
Comparative Sequence Data |
||||||
| Gene | Coordinates (hg18) | RefSeq Genes in Region (partial genes) | No. Speciesa | No. BACsb | No. BAC Gapsc | No. Sequence Gapsd | No. Basese |
| CENP-A | chr2: 26,741,969-27,041,969 | KCNK3, C2orf18, CENP-A, DPYSL5 | 13 | 40 | 0 | 173 | 5,728,210 |
| CENP-B | chr20: 3,503,000-3,802,999 | (ATRN),GFRA4, ADAM33, SIGLEC1, HSPA12B, C20orf27, SPEF1, CENP-B, CDC25B, C20orf29, VISA | 15 | 45 | 5 | 180 | 6,567,732 |
| CENP-C | chr4: 67,903,828-68,203,829 | CENP-C, STAP1, (UBA6) | 12 | 26 | 0 | 53 | 3,437,435 |
Number of nonhuman primate species for which DNA sequence orthologous to the targeted human genomic region was generated.
Total number of BACs sequenced from all species for the indicated genomic region.
Total number of gaps between BAC clones from all species for the indicated genomic region.
Total number of gaps within the generated sequence from all species for the indicated genomic region.
Total number of nonredundant bases of generated sequence from all species for the indicated genomic region.
The selected BACs were subjected to shotgun sequencing, with the nascent sequence assemblies then finished to “comparative-grade” standards in which all sequence contigs are ordered and oriented (Blakesley et al. 2004). Additional sequence-finishing efforts were applied to specific regions within assemblies where sequence gaps or poor sequence quality fell within the coding region of annotated genes. Following sequence finishing, multi-BAC sequence assemblies corresponding to the minimal tiling path of BACs for each species were generated and deposited into GenBank (table 1 and supplementary table 1, Supplementary Material online; CENP-A–containing region: DP000519, DP000521, DP000522, DP000524–DP000529, and DP000532–DP000535; CENP-B–containing region: DP000466–DP000473, DP000475–DP000477; and DP000480–DP000483; CENP-C–containing region: DP000603–DP000611 and DP000614–DP000616).
The final assembled sequences were annotated based on identified homologies with the reference human genome sequence, as represented on the UCSC Genome Browser (supplementary fig. 1, Supplementary Material online). Multisequence alignments were generated using PipMaker (pipmaker.bx.psu.edu/pipmaker) and VISTA (genome.lbl.gov/vista). To detect gross rearrangements, deletions, or insertions, each species' assembled sequence was used in turn as the reference sequence for aligning all other species' sequences (data not shown). For the known genes in each targeted genomic region, the human RefSeq mRNA sequences were aligned to the genomic sequence with Spidey (www.ncbi.nlm.nih.gov/IEB/Research/Ostell/Spidey) to deduce full-length mRNA sequences for genes in each species. All deduced coding sequences were checked for proper protein translation using Translate (www.expasy.ch/tools/dna.html) and Sequin (www.ncbi.nlm.nih.gov/Sequin); note that all amino-acid residue numbers are based on the human-protein sequence. Nucleic-acid and protein-sequence alignments between species were generated with ClustalW2 (Larkin et al. 2007) and used for downstream analyses with statistical (PAML and K-estimator), visualization (BoxShade; www.ch.embnet.org/software/BOX_form.html), and phylogenetic (MEGA4; Tamura et al. 2007) programs. Residual gaps in the coding-sequence alignments were manually adjusted to maintain the deduced in-frame amino-acid sequence. The deduced protein sequences were also examined for potential posttranslational modifications using NetPhos2.0 (www.cbs.dtu.dk/services/NetPhos), NetPhosK (with and without the ESS filter; www.cbs.dtu.dk/services/NetPhosK), KinasePhos (default HMM score; kinasephos.mbc.nctu.edu.tw), and DisPhos (default predictor; core.ist.temple.edu/pred/pred.html) (supplementary tables 2 and 3, Supplementary Material online). Conservation/divergence between species was determined using MEGA4 (Tamura et al. 2007) (supplementary tables 4–8, Supplementary Material online).
Testing for Evidence of Positive Selection
To test for evidence of positive selection, we compared the likelihood of models of neutral codon evolution to models of codon evolution allowing for selection using three different comparisons (table 2). First, the neutral model M1 had a class of codons with dN/dS = 1 and a class with dN/dS estimated from the data but limited to being between 0 and 1; we compared the neutral model M1 to a selection model that added an additional class of codons with a dN/dS ratio greater than one. Our second comparison had the neutral model M7 that limited the dN/dS ratio to follow a beta distribution limited to the interval between 0 and 1; we compared the neutral model M7 with a selection model M8 that adds an additional class of codons with a dN/dS ratio that is greater than 1. Our third comparison compared model M8 with a similar neutral model M8a in which the additional class of codons has the dN/dS ratio fixed at 1. In all models, we used the full F61 codon model, with the transition–transversion ratio estimated from the data. For all model comparisons, the negative of twice the difference between the selection and neutral models was compared with the χ2 distribution, with degrees of freedom equal to the difference between the numbers of parameters in each model. Convergence was checked by running all models from three different initial dN/dS ratios (0.5, 1, and 3). In all cases, the likelihoods and parameter estimates were identical between the different runs. All analyses were performed using the CODEML program of the PAML package (version 4.0). For sliding window analyses, we used K-estimator version 6.0 with default parameters (Comeron 1999) (supplementary tables 9–11, Supplementary Material online).
Table 2.
Results of PAML Model Comparisons.
| Gene | Na | Lcb | Sc | dN/dSd | −2ΔlM8 versus M8Ae | −2ΔlM1 versus M2e | −2ΔlM7 versus M8e | Parameter Estimates from M8 | Positively Selected Sitesf |
| CENP-A | 14 | 140 | 1.1 | 0.40 | 18.1** | 18.1** | 19.4** | p1 = 0.13, dN/dS = 3.5 | 7S, 17S, 18P, 35A, 39Q, 41S, 42R, 45Q, 46G, 62I, 65L, 76V |
| p0 = 0.87, β(19.2, 99) | |||||||||
| CENP-B | 16 | 618 | 0.9 | 0.06 | 0.0 | 0.0 | 0.0 | NA | NA |
| CENP-C | 13 | 950 | 1.1 | 0.75 | 24.6** | 24.4** | 24.8** | p1 = 0.07, dN/dS = 3.9 | 12G, 24R, 64R, 83P, 99F, 106A, 108N, 117H, 126S, 132D, 133S, 136I, 177S, 192M, 229D, 240S, 256R, 283A, 287P, 291C, 294D, 296T, 297K, 325G, 331T, 332I, 372T, 385Y, 391T, 395Y, 405K, 412R, 417I, 429P, 436V, 444I, 445H, 446T, 450T, 452D, 453E, 465H, 468M, 472C, 479P, 481V, 499R, 506N, 526R, 553H, 558R, 567S, 572R, 589Q, 594F, 613S, 614L, 633C, 650Q, 653P, 670N, 676H, 679S, 690N, 699N, 707H, 715Q, 769S, 771V, 777I, 778S, 787I, 791N, 834E, 841V, 891V |
| p0 = 0.93 β (0.15, 0.078) | |||||||||
| C2orf18g | 14 | 371 | 0.8 | 0.05 | 0.4 | 0.54 | 7.86* | p1 = 0.03, dN/dS = 1.4 | 32M, 73A, 167H, 169S, 264V, 350L |
| p0 = 0.97, β (0.66, 18.64) | |||||||||
| SPEF1g | 16 | 236 | 1.1 | 0.14 | 0.0 | 0.0 | 0.0 | NA | NA |
| STAP1g | 13 | 299 | 0.69 | 0.27 | 0.0 | 0.0 | 0.2 | NA | NA |
| Dpysl5g | 14 | 564 | 0.54 | 0.02 | 0.0 | 0.0 | 0.0 | NA | NA |
| CDC25g | 13 | 588 | 0.91 | 0.14 | 0.2 | 0.0 | 1.8 | NA | NA |
Number of taxa.
Length of alignment in codons.
Tree length.
Ratio of nonsynonymous to synonymous nucleotide changes.
For model comparisons (see text), significance is indicated with one (P < 0.05) or two asterisks (P < 0.0001).
For positively selected residues, those in bold have posterior probabilities >0.90, underlined 0.70–0.89, and regular font 0.50–0.69.
Non-CENP genes listed here are included to evaluate potential regional selection. C2orf18 and DPYSL5 flank CENP-A; SPEF1 and CDC25B flank CENP-B; and STAP1 is the only other complete gene within the analyzed CENP-C–containing region (see supplementary fig. 1, Supplementary Material online).
Results
Comparative Genome Sequencing
We generated the sequences of the genomic regions encompassing the CENP-A, -B, and -C genes in multiple primate species. Details about the resulting comparative sequence data sets are provided in table 1 and supplementary table 1, Supplementary Material online. Sequence data from 13, 15, and 12 nonhuman primate species were generated for CENP-A, -B, and –C, respectively (supplementary table 1, Supplementary Material online, and fig. 1); in aggregate, over 15 Mb of high-quality primate genome sequence was generated. For all three genomic regions, no gross rearrangements were detected in any species relative to the human reference sequence. Most of the detected interspecies variation reflects insertions and deletions of transposable elements. mRNA sequences for the CENP and flanking genes were deduced at each locus, allowing for the annotation of three genes in the CENP-A–containing region in each of 13 nonhuman primates, three genes in the CENP-B–containing region in each of 15 nonhuman primates, and two genes in the CENP-C–containing region in each of 12 nonhuman primates. All eight of these genes could be annotated in a common set of nine nonhuman primates that together provide representation of the four major branches of the primate phylogenetic tree.
FIG. 1.

Alignment of the deduced CENP-A protein sequences from 14 primates. (A) N-terminal tail region of CENP-A (positions 1–44; residue numbers are based on the human CENP-A sequence). Deviations from the human sequence are indicated by a different amino acid (single-letter abbreviations are depicted); a dot indicates that the amino acid is the same as in the human sequence, and a dash reflects insertion/deletion of an amino acid. Ser, Thr, and Tyr residues predicted to be phosphorylated (table 3 and supplementary table 2, Supplementary Material online) are shown in red, green, and blue, respectively. Deviations from the predicted phosphorylation status of a conserved amino acid are indicated by replacing the black dot by the single-letter amino-acid symbol of the conserved residue in black. Colored letters within the body of the alignment indicate residues predicted to be phosphorylated in that species but not in others. Black asterisks along the top indicate residues under positive selection. The green and blue boxes highlight protein kinase C motifs and cAMP- or cGMP-dependent protein kinase motifs, respectively. SPKK motifs and dipeptide motifs observed in other CENP-A homologs (Malik et al. 2002) are underlined in black or indicated by arrowheads, respectively. Red horizontal lines separate species in each of the four major branches of the primate phylogenetic tree. (B) Histone-fold domain of CENP-A (positions 45–140). Predicted protein structural features (Regnier et al. 2003) are indicated along the top; the CENP-A-targeting domain (CATD; Black et al. 2004) is also indicated. Other features of the alignment are as indicated in A.
Comparative Sequence Analysis of CENP-A
Just over 5.7 Mb of comparative sequence data were generated for the genomic region encompassing CENP-A (table 1 and supplementary table 1, Supplementary Material online). The final assembled sequence for each species reflects data from 2 to 4 BACs, with no gaps in the BAC contigs and an average of 13 sequence gaps. We generated a set of multispecies sequence alignments in which each species' sequence was used in turn as the reference; analysis of these alignments revealed significant interspecies variation upstream of CENP-A and within the gene's first intron (supplementary fig. 2, Supplementary Material online). In all species, there is notable evidence for extensive insertions and deletions of transposable elements, particularly between the end of the upstream gene (C2orf18) and approximately 500 bp upstream of CENP-A exon 1. Such extensive variation is not seen elsewhere in this genomic region and perhaps points to differences in the transcriptional control of CENP-A among species. Of note, immediately downstream of the last CENP-A exon, an SVA element (Shen et al. 1994), which is a retrotransposon currently active in the human genome, resides in the human sequence but is absent in all other species' sequences (supplementary fig. 1, Supplementary Material online).
Alignment of the predicted primate CENP-A protein sequences (fig. 1) reveals a great deal of interspecies divergence in the N-terminal tail region (supplementary table 4, Supplementary Material online) and general conservation in the histone-fold domain (supplementary table 5, Supplementary Material online). The N-terminal tail region is enriched for potential phosphorylation sites; consequently, these sites vary greatly among species, with 11 distinct species-specific phosphorylation-site profiles seen among the 14 primates (including human; fig. 1 and table 3). Of note, human and gorilla share a single potential phosphorylation-site profile (table 3, profile 1). Baboon and squirrel monkey share a profile (profile 5) as do macaque and vervet monkey (profile 6); the other eight primate species examined each have a unique combination of potential phosphorylation sites.
Table 3.
Predicted Phosphorylation-Site Profiles of Primate CENP-A Proteins.
 |
Other notable features of the N-terminal tail region include conserved SPKK motifs (underlined in fig. 1) and an expanded dipeptide motif (arrowheads in fig. 1) (Malik et al. 2002). SPKK motifs bind to DNA through the minor groove and are thought to do so based upon the local DNA conformation of A/T-rich DNA (Churchill and Suzuki 1989; Suzuki 1989). The SPKK motifs are the most conserved N-terminal sequences within this collection of primates. Our data also reveal an expansion of the dipeptide motif, XP. A total of four such motifs are seen in the N-terminal tail of mouse Cenp-A, whereas there are six of these motifs in human (Malik et al. 2002). Comparison of this region in primates reveals a stepwise expansion of copy number through primate evolution. Like human, gorilla and orangutan have six copies of the repeat, whereas chimpanzee, gibbon, the Old World monkeys, and squirrel monkey all have five copies. The prosimians, like mouse, have only four copies.
Although most potential phosphorylation sites reside in the N-terminal tail of the CENP-A protein, there are notable sites within the histone-fold domain. Serine 57 (S57) in loop 0 is a predicted phosphorylation site in all species except squirrel monkey, and S68 in helix 1 is predicted to be phosphorylated in lemur only. The region encompassing these two sites has been shown to exhibit fast deuterium exchange when CENP-A is associated with H4 in a heterotetramer but shows slower exchange when CENP-A is in the more typical structure of a histone octomer (Black et al. 2007). The gorilla and marmoset CENP-A proteins each contain an additional predicted phosphorylated serine (S75), whereas lemur and galago each contain an additional predicted phosphorylated threonine (T79 and T71, respectively). These reside in the CENP-A-targeting domain (CATD) that is likely to be exposed to solvent (Black et al. 2004) and has been demonstrated to confer centromere-localization capability to an otherwise typical histone H3 (Black et al. 2004). Exposure of the CATD to solvent may be critical for association between CENP-A and HJURP, a recently reported chaperone responsible for shuttling CENP-A to the centromere (Dunleavy et al. 2009; Foltz et al. 2009). Finally, T120 is predicted to be phosphorylated in all primate species examined (supplementary table 2, Supplementary Material online).
Comparative Sequence Analysis of CENP-B
Just over 6.5 Mb of comparative sequence data were generated for the genomic region encompassing CENP-B (table 1 and supplementary table 1, Supplementary Material online). The final assembled sequence for each species reflects data from 2 to 4 BACs, with five gaps in the BAC contigs and an average of 12 sequence gaps. This genomic region is relatively gene dense, containing nine complete and one partial gene in addition to the single exon CENP-B (table 1 and supplementary fig. 1, Supplementary Material online). Multispecies alignments of the generated genomic sequences reveal little remarkable interspecies variation aside from evidence of insertions and deletions of transposable elements.
Alignment of the predicted primate CENP-B protein sequences reveals an overall highly conserved protein (supplementary fig. 3, supplementary tables 6 and 7, Supplementary Material online). Pairwise comparisons between species from separate phylogenetic branches or species within the same major branch reveal only 1–3% divergence of the proteins, except for the prosimian proteins (where divergence reaches 3–5%). Notably, although the 125-amino-acid DNA-binding domain of CENP-B (residues 1–125; supplementary fig. 3, Supplementary Material online) is highly similar among the 16 primates studied, 8 amino-acid differences are seen in baboon, and the same single amino-acid difference is seen in both lemur and black lemur. In the case of the baboon protein, five of the differences occur in helix 1, whereas helixes 3 and 4 each have one difference; however, none of these differences occur at previously identified functional sites (Iwahara et al. 1998). The divergent site in lemur and black lemur (position 25) is thought to play a role in the association between CENP-B protein and alpha-satellite DNA (Iwahara et al. 1998). The amino-acid sequence of the DNA-binding domain that is shared among the other primates is also 100% conserved in the mouse and muntjac CENP-B orthologs (data not shown).
CENP-B is related to the Pogo family of transposases and shares amino-acid sequence features with this family throughout the N-terminal half of the protein (Kipling and Warburton 1997). The D35E motif that accomplishes strand cleavage in the transposases is a G29E motif in CENP-B but lacks strand-transfer capability (Kipling and Warburton 1997). Our data indicate that this motif is 100% identical among all 16 primates studied (supplementary fig. 3, Supplementary Material online).
The CENP-C-interaction domain of CENP-B (residues 404–470; fig. 2 and supplementary fig. 3, Supplementary Material online [Suzuki et al. 2004]) exhibits a great deal of interspecies variation relative to the remainder of the protein (supplementary fig. 3, Supplementary Material online). Of the 16 primates examined, only 2 (dusky titi and owl monkey) share 100% amino-acid sequence identity across this domain. The variation seen among the remaining 14 primates includes 10 conservative amino-acid substitutions and 12 insertions/deletions. Interestingly, the region between the CENP-C-interaction domain and the dimerization domain (residues 471–540; supplementary fig. 3, Supplementary Material online) is also less conserved than other regions of the protein; however, this region has yet to be implicated in a specific function.
FIG. 2.

Alignment of the deduced CENP-C-interaction domain sequences of the CENP-B protein from 16 primates. The multispecies alignment of the entire CENP-B protein sequence is provided in supplementary figure 3, Supplementary Material online, with positions 404–470 shown here. Features of the alignment are as in figure 1.
Comparative Sequence Analysis of CENP-C
Just over 3 Mb of comparative sequence data were generated for the genomic region encompassing CENP-C (table 1 and supplementary table 1, Supplementary Material online). The final assembled sequence for each species reflects data from two to three BACs, with no gaps in the BAC contigs and an average of 5 sequence gaps. Only two complete genes, CENP-C and STAP1, and one partial gene, UBA6, reside in this sequenced region (supplementary fig. 1, Supplementary Material online). Multispecies alignments of the generated genomic sequences reveal little remarkable interspecies variation aside from evidence of insertions and deletions of transposable elements.
Alignment of the predicted primate CENP-C protein sequences reveals significant interspecies divergence across the protein (fig. 3; supplementary fig. 4 and supplementary tables 6 and 8, Supplementary Material online). This is comparable with that seen in the N-terminal tail of CENP-A and far greater than that observed within CENP-B or the histone-fold domain of CENP-A (supplementary tables 4–7, Supplementary Material online). This was found when comparing primates from different major phylogenetic branches as well as those within the same major branch (supplementary tables 4–8, Supplementary Material online).
FIG. 3.
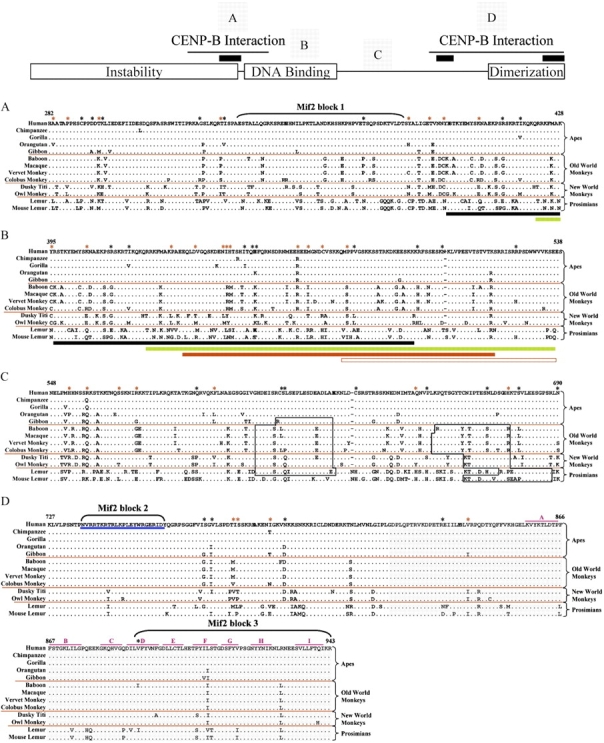
Alignment of the deduced protein sequence of the major functional domains of CENP-C from 13 primates. The major functional domains of the CENP-C protein are indicated in the model at the top. Specifically, the Instability, DNA Binding, and Dimerization domains are shown along with the two CENP-B-Interaction domains (thin black lines) and the three Mif2-homology domains (black bars). The multispecies protein sequence alignment of selected regions (A–D) is shown below the model; the labeled squares above the model show the relative positions of each of these regions. Black asterisks along the top indicate residues under positive selection with posterior probabilities of greater than 0.5; red asterisks reflect residues under positive selection with posterior probabilities of greater than 0.7. Other features of the alignment are as in figure 1. (A) N-Terminal CENP-B-interaction domain (residues 282–428; Suzuki et al. 2004), which overlaps the instability domain (residues 1–373; Lanini and McKeon 1995), contains a Mif2-homology domain (Mif2 block 1, residues 336–383; Brown 1995), and overlaps the start of the DNA-binding domain (residues 395–538; Yang et al. 1996; Sugimoto et al. 1997; Cohen et al. 2008). Black and green bars indicate overlap with the start of the DNA-binding domain (see B). (B) DNA-binding domain (residues 395–538). Highlighted below the alignment are portions of the DNA-binding domain, as determined by previous studies (black bar, residues 396–498 [Sugimoto et al. 1997]; green bar, residues 422–537 [Cohen et al. 2008]; and red bar, residues 433–520 [Yang et al. 1996]). The open red bar indicates the minimal CATD (residues 478–537; Yang et al. 1996). (C) Region containing potential PEST sequences (open black boxes), as determined in this study. (D) C-terminal CENP-B-interaction domain (residues 727–943; Suzuki et al. 2004) and dimerization domain (gray highlighted residues 820–943; Sugimoto et al. 1997), which encompass the other Mif2-homology domains (residues 736–759 and 890–943; Brown 1995), the CENP-C-signature domain (underlined in blue, residues 736–759; Meluh and Koshland 1995), and the 9 (A–I) domains of the β-jelly roll (indicated with pink lines; Dunwell et al. 2001; Cohen et al. 2008).
Several regions of the CENP-C protein have been shown to serve specific functional roles (fig. 3 and supplementary fig. 4, Supplementary Material online; Lanini and McKeon 1995; Yang et al. 1996; Sugimoto et al. 1997; Song et al. 2002; Suzuki et al. 2004). Of these, only the dimerization domain (residues 820–943; Sugimoto et al. 1997) and the more C-terminal Mif2-homology domain (the C-signature domain; Meluh and Koshland 1995) are highly conserved across primates (fig. 3). The reported N-terminal instability domain (Lanini and McKeon 1995) shows moderate divergence (supplementary fig. 4, Supplementary Material online) but does not contain sequences associated with rapidly degraded proteins (PEST; Rogers et al. 1986) by our analysis (fig. 3). Interestingly, the central DNA-binding/CENP-B-interaction domain and an adjacent region that does contain potential PEST sequences are highly diverged among primates (fig. 3).
Evidence for Positive Selection: General Findings
We investigated whether the divergence of genes in the three studied genomic regions was promoted by positive selection using maximum likelihood methods to analyze the variation in the dN/dS ratio among sites (Yang et al. 2000). These methods are robust at detecting positive selection acting on particular codon positions, and are more sensitive than analyzing the dN/dS ratio averaged across all sites (Anisimova et al. 2001). As described in Materials and Methods, we used three distinct tests for these analyses (table 2). The most robust test (M8 vs. M8a) basically determines if the dN/dS ratio for the class of codons under selection is significantly greater than the neutral expectation of dN/dS = 1 (Swanson et al. 2003). Of the eight genes studied, only two (CENP-A and CENP-C) show evidence of positive selection by this robust method; this evidence remains significant after correcting for multiple tests using a Bonferroni–Holm step-down correction (Bonferroni 1936; Holm 1979). C2orf18, which resides upstream of CENP-A, was significant for one test (M7 vs. M8) but not the more robust M8 versus M8A test; further, the M8 versus M8A test does not hold up for C2orf18 upon correction for multiple tests. The detection of positive selection acting on CENP-A and CENP-C can be used to detect which codon positions have been the target of selection, potentially indicating functionally important regions of the encoded proteins (Swanson et al. 2001). We confirmed the maximum likelihood results for CENP-C using a simpler approach involving a sliding window dN/dS analysis (supplementary fig. 5 and supplementary table 11, Supplementary Material online). By this analysis, CENP-C shows a robust signal of positive selection; however, CENP-A and CENP-B do not (supplementary fig. 5, supplementary table 9 and 10, Supplementary Material online). The maximum likelihood method uses all of the phylogenetic information contained in our data set, thus providing greater power to detect positive selection in CENP-A (Schmid and Yang 2008).
Evidence for Positive Selection: CENP-A
The highly diverged N-terminal tail of CENP-A (residues 1–44) contains 7 of the 12 amino-acid residues that are under positive selection in the protein (table 2 and fig.1A). Of these, six are residues that differ between histone H3 and CENP-A, and four are likely to be involved in posttranslational modifications. S7 and S17 are under positive selection and lie within cAMP- or cGMP-dependent kinase phosphorylation motifs (RRRS; see blue shading in fig. 1A). S7 has been shown to be phosphorylated in a cell cycle–dependent manner and to lie within a motif similar to that of S10 in histone H3 (Zeitlin et al. 2001); phosphorylation of the latter has been associated with chromatin condensation (Hsu et al. 2000). Both S41 and R42 in the N-terminal tail are also under positive selection and may be subject to posttranslational phosphorylation and methylation, respectively; S41 is predicted to be phosphorylated by protein kinase C and, along with R42, resides in one of three protein kinase C phosphorylation motifs (RRR; see green shading in fig. 1A). In total, 20 of the 44 amino acids that comprise the N-terminal tail of the human CENP-A protein are potential sites for phosphorylation, methylation, or glycosylation. Other amino-acid residues in the CENP-A N-terminal tail that are under positive selection (fig. 1A) may contribute to differences in the protein structure among species.
In the histone-fold domain of CENP-A (residues 45–140), there are five residues under positive selection (fig. 1B). Of these, two (Q45 and V76) are at residue positions that differ from human histone H3. Both of these residues reside at functionally significant junctions: Q45 at the junction between the N-terminal tail and the histone-fold domain and V76 at the start of the centromere-targeting domain (CATD; fig. 1B). Interestingly, though different than the corresponding residues in histone H3, the amino acids found at these two positions are conserved across all studied primate CENP-A proteins except those in the prosimians. The CATD-containing region has been shown to exhibit slow deuterium exchange (Black et al. 2007), suggesting that V76 participates in intramolecular interactions; selection for a hydrophobic residue at position 76 may help to ensure the physical integrity of the helical structure. The other three residues under selection within the histone-fold domain (G46, I62, and L65) are conserved with human histone H3 but vary among primates. Structural constraints at the start of the N-terminal helix may require small or nonpolar amino acids at residue 46 between the highly conserved flanking residues—a strongly hydrophilic (and under selection) Gln at position 45 and a large hydrophobic Trp at position 47. Likewise, variation at residues 62 and 65 near the junction of loop 0 and helix 1 may impact the length of helix 1 (or at least the characteristics of its N-terminal end).
Interestingly, there is evidence for lineage- and species-specific combinations of amino acids at positions 46 and 65 of CENP-A. Both prosimians examined have an Ala at position 65 but differ with respect to their amino acid at position 46. All the studied New and Old World monkeys have a Tyr at position 65; however, the 3 New World monkeys have a Gly at position 46, whereas the four Old World monkeys have either an Ala or Ser at this position. All four great apes have a Leu at position 65, whereas at position 46, gibbon has a Ser, orangutan has an Ala, and the remaining great apes have a Gly. Like positions 46 and 65, the patterns of variation at the two N-terminal tail residues under positive selection (A35 and Q39) also reflect lineage- and species-specific combinations.
Evidence for Positive Selection: CENP-B
No specific amino-acid residues of the CENP-B protein were found to be associated with evidence of positive selection (table 2). The only region of the protein with significant differences among primate species is the CENP-C-interaction domain (residues 404–470; fig. 2); this region shows interspecies length polymorphism, resulting largely from insertion/deletion of Glu residues (fig. 2). Our analyses failed to reveal evidence for selection related to these length differences or a significant correlation between the rate of change of this region and residues within the CENP-B-interaction domain of the CENP-C protein (data not shown).
Evidence for Positive Selection: CENP-C
A total of 76 amino-acid residues in the CENP-C protein appear to be under positive selection. Of these, 10 show posterior probabilities of >0.90, and another 29 show posterior probabilities of >0.70 (table 2). These 39 residues (indicated by red asterisks in fig. 3 and supplementary fig. 4, Supplementary Material online) have the following attributes: 1) 13 (R64, F99, H117, I136, S177, M192, D229, S240, R256, A283, P287, T296, and T331) are in the N-terminal instability region (residues 1–373; fig. 3A and supplementary fig. 4, Supplementary Material online), with the last four of these also sitting within the N-terminal CENP-B-interaction domain (residues 283–429; fig. 3A); 2) five additional residues (Y385, T391, Y395, K405, and P429) lie within the N-terminal CENP-B-interaction domain, with the last of these also sitting within the DNA-binding domain (residues 396–540; fig. 3B); 3) eight additional residues (V436, I444, H445, T446, H465, M468, C472, and P479) are within the DNA-binding domain; 4) 7 (H553, R558, S567, R572, F594, Q650, and H676) are within the region containing potential PEST sequences (residues 548–690; fig. 3C); and 5) four (I777, S778, I787, and V841) are within the N-terminal CENP-B-interaction domain (residues 727–943; fig. 3D), with the last of these also sitting within the dimerization domain (residues 820–943; fig. 3D).
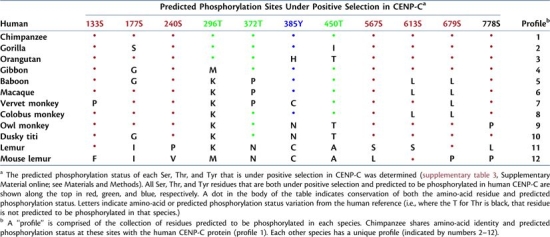
Among the CENP-C amino-acid residues under positive selection, 11 are predicted phosphorylation sites in human CENP-C (S133, S177, S240, T296, T372, Y385, T450, S567, S613, S679, and S778). Comparison of the predicted phosphorylation status at just these 11 residues reveals species-specific profiles for each of the 12 studied nonhuman primates (table 4). Only chimpanzee has the same phosphorylation-site profile as human CENP-C at these sites. Each of the other 11 species has a unique profile. Among the predicted phosphorylation sites, five reside within the N-terminal instability domain (S133, S177, S240, T296, and T372; fig. 3 and supplementary fig. 4, Supplementary Material online), three within the N-terminal CENP-B-interaction domain (T296, T372, and Y385; fig. 3A), one within the DNA-binding domain (T450; fig. 3B), two within predicted PEST sequences (S613 and S679; fig. 3C), and one in the C-terminal CENP-B-interaction domain (S778; fig. 3D).
Table 4.
Predicted Phosphorylation-Site Profiles at Positively Selected Residues in Primate CENP-C Proteins.
 |
Discussion
Centromeric DNA is highly variable among species, and yet, it is essential for chromosome segregation. The proteins that form the kinetochore machinery must interact with this DNA for proper centromere function. The dynamic nature of this relationship is thought to result from female meiotic drive, in which the expanding centromere sequences act selfishly to exploit the asymmetric production of female gametes (Zwick et al. 1999). To balance the resulting skewed transmission of genomic loci, kinetochore proteins adapt to ameliorate drive by restoring epigenetic control of centromere function and, thus, the random segregation of chromosomes (Henikoff et al. 2001; Malik and Henikoff 2002; Dawe and Henikoff 2006; Malik and Bayes 2006).
Comparative genome sequencing can be used to identify genomic regions that have been conserved or have diverged throughout evolution. Broad comparisons of genome sequences from species residing on distant branches of the evolutionary tree can reveal loci that have remained relatively unchanged over time (Pennacchio and Rubin 2001). Conservation implies essential (or universal) function, and such sequences are thought to be constrained by negative selection. Conversely, genomic regions associated with elevated rates of divergence among closely related species indicate positive selection (Boffelli et al. 2003). We have applied these principles to perform evolutionary analyses of the genomic regions containing the foundation kinetochore protein genes CENP-A, -B, and -C. These proteins provide part of the interface between centromeric DNA and the outer kinetochore (Amor et al. 2004) and, as such, are potential mediators of meiotic drive (Henikoff et al. 2001; Malik and Henikoff 2002; Dawe and Henikoff 2006; Malik and Bayes 2006).
By generating and analyzing orthologous genomic sequences from a diverse set of primates, we have, for the first time, demonstrated positive selection acting on mammalian CENP-A. This protein is a histone H3 variant that plays a central role in the centromere-specific nucleosome (Allshire and Karpen 2008). Prior studies found positive selection acting on Drosophila (Malik and Henikoff 2001; Malik et al. 2002) and Arabidopsis (Talbert et al. 2004) homologs of human CENP-A. However, a broad evolutionary comparison of mammalian (human, chimpanzee, mouse, rat, and bovine) CENP-A homologs failed to reveal evidence of positive selection (Talbert et al. 2004).
Throughout evolution, the CENP-A histone-fold domain has been highly conserved, unlike the highly variable (with respect to length and sequence) N-terminal tail (Yoda et al. 2000; Henikoff et al. 2001). Although very little data exist regarding posttranslational modification of CENP-A (Zeitlin et al. 2001), extensive characterization of the closely related histone H3 protein has identified modifications of one threonine, seven lysine, four arginine, and two serine residues within the N-terminal tail (Kouzarides 2007). Interestingly, the identity and modification status of only one of these sites (serine 10 in histone H3 and serine 7 in CENP-A) is conserved in the CENP-A protein. In fact, at each of the other modified histone H3 positions, a different amino-acid residue is present and predicted to be modified in CENP-A.
The major feature of CENP-A evolution highlighted by our comparative analyses is the presence of species-specific DNA sequences, especially in the N-terminal tail. Such variation affects potential posttranslational modification sites and points to the intriguing possibility that each species has a unique combination of centromeric DNA and CENP-A protein sequence. This feature of CENP-A evolution supports both an ongoing genetic conflict at the centromere that is linked with speciation (Henikoff et al. 2001) and epigenetic compensation for rapidly evolving DNA (Dawe and Henikoff 2006).
Homologs of human CENP-C have been shown to be subject to positive selection in all species examined, including some mammals (Talbert et al. 2004). Residues under positive selection in mammalian CENP-C were shown to lie within the central DNA-binding region. Interestingly, we found signatures of positive selection throughout the CENP-C protein; in fact, each region of this protein that has been previously demonstrated to be functionally important was found to contain residues under positive selection.
Although we did not detect evidence of positive selection acting on CENP-B, our analyses highlight an intriguing relationship between CENP-B and CENP-C. The CENP-C-interaction domain of the CENP-B protein is highly variable among primate species, yet the rest of the protein is otherwise highly conserved. Positioned in the central portion of the protein, the CENP-C-interaction domain appears to have been subjected to numerous insertion and deletion events throughout evolution. CENP-B binds to DNA via its N-terminus and forms homodimers at its C-terminus. It is thus intriguing that the one evolutionarily dynamic region of CENP-B represents the portion of the protein that interacts with other kinetochore components.
Species-specific length variation within the central domain of the CENP-B protein may enable the resulting dimer to “reach” binding sites within alpha-satellite DNA that are uniquely positioned within each species. Emergence of the CENP-B box within alpha-satellite DNA 15–25 Ma in the primate lineage (Haaf et al. 1995) has been followed by continued evolution of the frequency and organization of CENP-B boxes within centromeric regions (Schueler et al. 2001, 2005). Recent coevolution of CENP-B and CENP-C (or other kinetochore proteins) may account for improved artificial chromosome formation by alpha-satellite DNA that contains CENP-B boxes versus that which lacks them (Harrington et al. 1997; Ikeno et al. 1998; Masumoto et al. 1998; Ohzeki et al. 2002). Although CENP-B does not appear to be necessary (Hudson et al. 1998; Kapoor et al. 1998; Perez-Castro et al. 1998) or sufficient (Sullivan and Schwartz 1995; Sullivan and Willard 1998) for centromere activation, it may have recently evolved an important role in current centromere function. CENP-B-binding sites may represent the most recent efforts of “selfish centromeric DNA” to gain genetic control. In the ongoing conflict between genetic and epigenetic control of centromere function (Dawe and Henikoff 2006), kinetochore proteins may continuously be evolving to compensate for improved centromere function via the emergence of new CENP-B-binding sites within centromeric DNA.
In summary, our comparative genomic studies provide new insights relevant to the evolution of primate centromeres and the epigenetic mechanisms controlling chromosome transmission. The latter involves a complex cellular choreography, with centromeric DNA and the kinetochore proteins that associate with it being central elements. Studying the evolution of these elements continues to reveal many interesting species- and lineage-specific findings. Further understanding the basis for these evolutionary changes may provide valuable clues about centromere function and, perhaps, speciation.
Supplementary Material
Supplementary figures 1–4 and supplementary tables 1–11 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
We thank all participants of the NISC Comparative Sequencing Program (in particular, Bob Blakesley, Gerry Bouffard, Alice Young, Jenny McDowell, Morgan Park, Baishali Maskeri, Jyoti Gupta, Shelise Brooks, Betty Barnabas, Karen Schandler, and Shi-Ling Ho) for generating the comparative sequence data reported here. This work was supported in part by the Intramural Research Program of the National Human Genome Research Institute of the National Institutes of Health.
References
- Alexandrov I, Kazakov A, Tumeneva I, Shepelev V, Yurov Y. Alpha-satellite DNA of primates: old and new families. Chromosoma. 2001;110:253–266. doi: 10.1007/s004120100146. [DOI] [PubMed] [Google Scholar]
- Allshire RC, Karpen GH. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet. 2008;9:923–937. doi: 10.1038/nrg2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor DJ, Kalitsis P, Sumer H, Choo KH. Building the centromere: from foundation proteins to 3D organization. Trends Cell Biol. 2004;14:359–368. doi: 10.1016/j.tcb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- Black BE, Brock MA, Bedard S, Woods VL, Jr., Cleveland DW. An epigenetic mark generated by the incorporation of CENP-A into centromeric nucleosomes. Proc Natl Acad Sci U S A. 2007;104:5008–5013. doi: 10.1073/pnas.0700390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Foltz DR, Chakravarthy S, Luger K, Woods VL, Jr., Cleveland DW. Structural determinants for generating centromeric chromatin. Nature. 2004;430:578–582. doi: 10.1038/nature02766. [DOI] [PubMed] [Google Scholar]
- Blakesley RW, Hansen NF, Mullikin JC, et al. (22 co-authors) An intermediate grade of finished genomic sequence suitable for comparative analyses. Genome Res. 2004;14:2235–2244. doi: 10.1101/gr.2648404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- Bonferroni CE. Teoria statistica delle classi e calcolo delle probabilit `a. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze. 1936;8:3–62. [Google Scholar]
- Brown MT. Sequence similarities between the yeast chromosome segregation protein Mif2 and the mammalian centromere protein CENP-C. Gene. 1995;160:111–116. doi: 10.1016/0378-1119(95)00163-z. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Churchill ME, Suzuki M. ‘SPKK’ motifs prefer to bind to DNA at A/T-rich sites. Embo J. 1989;8:4189–4195. doi: 10.1002/j.1460-2075.1989.tb08604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RL, Espelin CW, De Wulf P, Sorger PK, Harrison SC, Simons KT. Structural and functional dissection of Mif2p, a conserved DNA-binding kinetochore protein. Mol Biol Cell. 2008;19:4480–4491. doi: 10.1091/mbc.E08-03-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM. K-estimator: calculation of the number of nucleotide substitutions per site and the confidence intervals. Bioinformatics. 1999;15:763–764. doi: 10.1093/bioinformatics/15.9.763. [DOI] [PubMed] [Google Scholar]
- Dawe RK, Henikoff S. Centromeres put epigenetics in the driver's seat. Trends Biochem Sci. 2006;31:662–669. doi: 10.1016/j.tibs.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–497. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, 3rd, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma. 1989;98:1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–484. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf T, Mater AG, Wienberg J, Ward DC. Presence and abundance of CENP-B box sequences in great ape subsets of primate-specific alpha-satellite DNA. J Mol Evol. 1995;41:487–491. doi: 10.1007/BF00160320. [DOI] [PubMed] [Google Scholar]
- Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, Willard HF. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat Genet. 1997;15:345–355. doi: 10.1038/ng0497-345. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001;293:1098–1102. doi: 10.1126/science.1062939. [DOI] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Hsu JY, Sun ZW, Li X, et al. (13 Co-authors) Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- Hudson DF, Fowler KJ, Earle E, et al. (15 co-authors) Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J Cell Biol. 1998;141:309–319. doi: 10.1083/jcb.141.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, McGill NI, Cooke H, Masumoto H. Construction of YAC-based mammalian artificial chromosomes. Nat Biotechnol. 1998;16:431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- Iwahara J, Kigawa T, Kitagawa K, Masumoto H, Okazaki T, Yokoyama S. A helix-turn-helix structure unit in human centromere protein B (CENP-B) Embo J. 1998;17:827–837. doi: 10.1093/emboj/17.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Montes de Oca Luna R, Liu G, Lozano G, Cummings C, Mancini M, Ouspenski I, Brinkley BR, May GS. The cenpB gene is not essential in mice. Chromosoma. 1998;107:570–576. doi: 10.1007/s004120050343. [DOI] [PubMed] [Google Scholar]
- Kipling D, Warburton PE. Centromeres, CENP-B and Tigger too. Trends Genet. 1997;13:141–145. doi: 10.1016/s0168-9525(97)01098-6. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lanini L, McKeon F. Domains required for CENP-C assembly at the kinetochore. Mol Biol Cell. 1995;6:1049–1059. doi: 10.1091/mbc.6.8.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. (13 co-authors) Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Malik HS, Bayes JJ. Genetic conflicts during meiosis and the evolutionary origins of centromere complexity. Biochem Soc Trans. 2006;34:569–573. doi: 10.1042/BST0340569. [DOI] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. Adaptive evolution of Cid, a centromere-specific histone in Drosophila. Genetics. 2001;157:1293–1298. doi: 10.1093/genetics/157.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. Conflict begets complexity: the evolution of centromeres. Curr Opin Genet Dev. 2002;12:711–718. doi: 10.1016/s0959-437x(02)00351-9. [DOI] [PubMed] [Google Scholar]
- Malik HS, Vermaak D, Henikoff S. Recurrent evolution of DNA-binding motifs in the Drosophila centromeric histone. Proc Natl Acad Sci U S A. 2002;99:1449–1454. doi: 10.1073/pnas.032664299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra MA, Kucaba TA, Dietrich NL, Green ED, Brownstein B, Wilson RK, McDonald KM, Hillier LW, McPherson JD, Waterston RH. High throughput fingerprint analysis of large-insert clones. Genome Res. 1997;7:1072–1084. doi: 10.1101/gr.7.11.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Ikeno M, Nakano M, Okazaki T, Grimes B, Cooke H, Suzuki N. Assay of centromere function using a human artificial chromosome. Chromosoma. 1998;107:406–416. doi: 10.1007/s004120050324. [DOI] [PubMed] [Google Scholar]
- Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol. 1989;109:1963–1973. doi: 10.1083/jcb.109.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meluh PB, Koshland D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol Biol Cell. 1995;6:793–807. doi: 10.1091/mbc.6.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Nakano M, Okada T, Masumoto H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J Cell Biol. 2002;159:765–775. doi: 10.1083/jcb.200207112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Manuel de Villena F, Sapienza C. Nonrandom segregation during meiosis: the unfairness of females. Mamm Genome. 2001;12:331–339. doi: 10.1007/s003350040003. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Rubin EM. Genomic strategies to identify mammalian regulatory sequences. Nat Rev Genet. 2001;2:100–109. doi: 10.1038/35052548. [DOI] [PubMed] [Google Scholar]
- Perez-Castro AV, Shamanski FL, Meneses JJ, Lovato TL, Vogel KG, Moyzis RK, Pedersen R. Centromeric protein B null mice are viable with no apparent abnormalities. Dev Biol. 1998;201:135–143. doi: 10.1006/dbio.1998.9005. [DOI] [PubMed] [Google Scholar]
- Regnier V, Novelli J, Fukagawa T, Vagnarelli P, Brown W. Characterization of chicken CENP-A and comparative sequence analysis of vertebrate centromere-specific histone H3-like proteins. Gene. 2003;316:39–46. doi: 10.1016/s0378-1119(03)00768-6. [DOI] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- Saffery R, Irvine DV, Griffiths B, Kalitsis P, Wordeman L, Choo KH. Human centromeres and neocentromeres show identical distribution patterns of >20 functionally important kinetochore-associated proteins. Hum Mol Genet. 2000;9:175–185. doi: 10.1093/hmg/9.2.175. [DOI] [PubMed] [Google Scholar]
- Schmid K, Yang Z. The trouble with sliding windows and the selective pressure in BRCA1. PLoS One. 2008;3:e3746. doi: 10.1371/journal.pone.0003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler MG, Dunn JM, Bird CP, Ross MT, Viggiano L, Rocchi M, Willard HF, Green ED. Progressive proximal expansion of the primate X chromosome centromere. Proc Natl Acad Sci U S A. 2005;102:10563–10568. doi: 10.1073/pnas.0503346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF. Genomic and genetic definition of a functional human centromere. Science. 2001;294:109–115. doi: 10.1126/science.1065042. [DOI] [PubMed] [Google Scholar]
- Schueler MG, Sullivan BA. Structural and functional dynamics of human centromeric chromatin. Annu Rev Genomics Hum Genet. 2006;7:301–313. doi: 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- Shen L, Wu LC, Sanlioglu S, Chen R, Mendoza AR, Dangel AW, Carroll MC, Zipf WB, Yu CY. Structure and genetics of the partially duplicated gene RP located immediately upstream of the complement C4A and the C4B genes in the HLA class III region. Molecular cloning, exon–intron structure, composite retroposon, and breakpoint of gene duplication. J Biol Chem. 1994;269:8466–8476. [PubMed] [Google Scholar]
- Song K, Gronemeyer B, Lu W, Eugster E, Tomkiel JE. Mutational analysis of the central centromere targeting domain of human centromere protein C, (CENP-C) Exp Cell Res. 2002;275:81–91. doi: 10.1006/excr.2002.5495. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Kuriyama K, Shibata A, Himeno M. Characterization of internal DNA-binding and C-terminal dimerization domains of human centromere/kinetochore autoantigen CENP-C in vitro: role of DNA-binding and self-associating activities in kinetochore organization. Chromosome Res. 1997;5:132–141. doi: 10.1023/a:1018422325569. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Yata H, Muro Y, Himeno M. Human centromere protein C (CENP-C) is a DNA-binding protein which possesses a novel DNA-binding motif. J Biochem (Tokyo) 1994;116:877–881. doi: 10.1093/oxfordjournals.jbchem.a124610. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Blower MD, Karpen GH. Determining centromere identity: cyclical stories and forking paths. Nat Rev Genet. 2001;2:584–596. doi: 10.1038/35084512. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Schwartz S. Identification of centromeric antigens in dicentric Robertsonian translocations: CENP-C and CENP-E are necessary components of functional centromeres. Hum Mol Genet. 1995;4:2189–2197. doi: 10.1093/hmg/4.12.2189. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Willard HF. Stable dicentric X chromosomes with two functional centromeres. Nat Genet. 1998;20:227–228. doi: 10.1038/3024. [DOI] [PubMed] [Google Scholar]
- Suzuki M. SPKK, a new nucleic acid-binding unit of protein found in histone. Embo J. 1989;8:797–804. doi: 10.1002/j.1460-2075.1989.tb03440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Nakano M, Nozaki N, Egashira S, Okazaki T, Masumoto H. CENP-B interacts with CENP-C domains containing Mif2 regions responsible for centromere localization. J Biol Chem. 2004;279:5934–5946. doi: 10.1074/jbc.M306477200. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Yang Z, Wolfner MF, Aquadro CF. Positive Darwinian selection drives the evolution of several female reproductive proteins in mammals. Proc Natl Acad Sci U S A. 2001;98:2509–2514. doi: 10.1073/pnas.051605998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Bryson TD, Henikoff S. Adaptive evolution of centromere proteins in plants and animals. J Biol. 2004;3:18. doi: 10.1186/jbiol11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Masuelli R, Tyagi AP, Comai L, Henikoff S. Centromeric localization and adaptive evolution of an Arabidopsis histone H3 variant. Plant Cell. 2002;14:1053–1066. doi: 10.1105/tpc.010425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thomas JW, Touchman JW, Blakesley RW, et al. (71 co-authors) Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- Willard HF. Centromeres of mammalian chromosomes. Trends Genet. 1990;6:410–416. doi: 10.1016/0168-9525(90)90302-m. [DOI] [PubMed] [Google Scholar]
- Yang CH, Tomkiel J, Saitoh H, Johnson DH, Earnshaw WC. Identification of overlapping DNA-binding and centromere-targeting domains in the human kinetochore protein CENP-C. Mol Cell Biol. 1996;16:3576–3586. doi: 10.1128/mcb.16.7.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda K, Ando S, Morishita S, Houmura K, Hashimoto K, Takeyasu K, Okazaki T. Human centromere protein A (CENP-A) can replace histone H3 in nucleosome reconstitution in vitro. Proc Natl Acad Sci U S A. 2000;97:7266–7271. doi: 10.1073/pnas.130189697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin SG, Barber CM, Allis CD, Sullivan KF. Differential regulation of CENP-A and histone H3 phosphorylation in G2/M. J Cell Sci. 2001;114:653–661. doi: 10.1242/jcs.114.4.653. [DOI] [PubMed] [Google Scholar]
- Zwick ME, Salstrom JL, Langley CH. Genetic variation in rates of nondisjunction: association of two naturally occurring polymorphisms in the chromokinesin nod with increased rates of nondisjunction in Drosophila melanogaster. Genetics. 1999;152:1605–1614. doi: 10.1093/genetics/152.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.