

Abstract
The evolutionarily conserved Notch signaling pathway plays broad and important roles during embryonic development and in adult tissue homeostasis. Unlike most other pathways used during animal development, Notch signaling does not rely on second messengers and intracellular signaling cascades. Instead, pathway activation results in the cleavage of the Notch intracellular domain and its translocation into the nucleus, where it functions as a transcriptional co-activator of the Notch target genes. To ensure tight spatial and temporal regulation of a pathway with such an unusually direct signaling transduction, animal cells have devised a variety of specialized modulatory mechanisms. One such mechanism takes advantage of decorating the Notch extracellular domain with rare types of O-linked glycans. In this review, we will discuss the genetic and biochemical data supporting the notion that carbohydrate modification is essential for Notch signaling and attempt to provide a brief historical overview of how we have learned what we know about the glycobiology of Notch. We will also summarize what is known about the contribution of specific nucleotide-sugar transporters to Notch biology and the roles—enzymatic and non-enzymatic—played by specific glycosyltransferases in the regulation of this pathway. Mutations in the Notch pathway components cause a variety of human diseases, and manipulation of Notch signaling is emerging as a powerful tool in regenerative medicine. Therefore, studying how sugar modification modulates Notch signaling provides a framework for better understanding the role of glycosylation in animal development and might offer new tools to manipulate Notch signaling for therapeutic purposes.
Keywords: glycans, glycosyltransferases, Notch signaling
Introduction
Intercellular communication is essential for the development and maintenance of multicellular organisms. During animal development, seven highly conserved signaling pathways mediate the majority of cell–cell interactions: Hedgehog, Jak-STAT, nuclear hormone receptor, Wnt, transforming growth factor β, receptor tyrosine kinase, and Notch (Barolo and Posakony 2002). The Notch signaling pathway is widely used during animal development and adult life, and regulates a variety of processes including cell fate specification, differentiation, left–right asymmetry, apoptosis, compartment boundary formation, somitogenesis and angiogenesis (Fortini 2009; Kopan and Ilagan 2009; Tien et al. 2009). Notch signaling also plays key roles in stem cell proliferation, the interaction between stem cells and their microenvironmental niche, and differential behavior of some stem cells in young versus old animals (Chiba 2006; Carlson and Conboy 2007; Wang et al. 2009). The Notch gene was first identified as a spontaneous dominant X-linked mutation in the fruit fly Drosophila melanogaster and takes its name from the “Notches” of tissue loss in the wing blade of animals lacking one functional copy of the gene (Dexter 1914; Morgan and Bridges 1916; Mohr 1919). During the 1930s, Poulson mapped the fly gene cytologically and reported that the loss of both copies of Notch is lethal and results in a massive hyperplasia of the embryonic nervous system (Poulson 1940). In the 1980s, the Artavanis-Tsakonas and Young groups cloned the gene independently and reported that the fly Notch protein contains 36 epidermal growth factor (EGF)-like motifs (Wharton et al. 1985; Kidd et al. 1986). Interestingly, cloning of the Drosophila Notch showed that it is homologous to the Caenorhabditis elegans gene lin-12, mutations in which were also reported to affect cell fate decisions in this nematode (Greenwald et al. 1983; Greenwald 1985). Since then, a large number of studies have shown important and broad roles for Notch signaling in the development of various animals. The importance of this pathway in animal biology is also highlighted by the observation that both gain- and loss-of-function mutations in components of the Notch signaling pathway play causative roles in a variety of human diseases, including developmental disorders and cancer (Table I).
Table I.
Human diseases caused by mutations in the components of the Notch pathway
| Disease | Mutated Notch pathway component |
|---|---|
| T-cell acute lymphoblastic leukemiaa | Notch1 |
| Alagille syndromeb | Jagged1, Notch2 |
| Tetralogy of Fallotc | Jagged1 |
| Aortic valve diseasesd | Notch1 |
| CADASIL syndromee | Notch3 |
| Spondylocostal dysostosisf | Delta-like3, Lunatic Fringe, Mesp2g, Hes7 |
| Diffuse large B-cell lymphomah | Notch2 |
| Hearing lossi | Jagged1 |
Mesp2 is a transcription factor which regulates Notch signaling in the context of somitogenesis (Takahashi et al. 2003)
Detailed evolutionary analysis of Notch pathway components and their conserved motifs in 35 different eukaryotic species has shown that Notch signaling is only used by metazoans, even though some of the pathway components have homologs in other eukaryotes including plants and fungi (Gazave et al. 2009). The core components of the canonical Notch pathway are the Notch receptors (in the signal-receiving cell), the Notch ligands (in the adjacent signal-sending cell), and the CSL family of transcription factors (CBF1/RBP-J proteins in vertebrates, Su(H) in flies and Lag1 in worms). The Drosophila genome encodes one receptor (Notch) and two ligands (Delta and Serrate), whereas mammals have four receptors (Notch1–4) and five classical ligands (Delta-like1, 3 and 4; Jagged1 and 2) (Fortini 2009; Kopan and Ilagan 2009). Both Notch receptors and their ligands are type I transmembrane proteins. The newly synthesized Notch protein undergoes its first proteolytic cleavage in the Golgi (the S1 cleavage) and reaches the plasma membrane as a heterodimer (Figure 1) (Logeat et al. 1998; Lake et al. 2009). Even though the predominant form of mammalian Notch receptors at the membrane is the heterodimeric form, a recent report indicates that the S1 cleavage might not be essential for productive signalng (Gordon, Vardar-Ulu, et al. 2009). Upon binding of ligands to the Notch extracellular domain (NECD), Notch is further cleaved at the S2 site by the ADAM10 metalloprotease Kuzbanian (Pan and Rubin 1997; Cagavi Bozkulak and Weinmaster 2009; van Tetering et al. 2009). This cleavage generates Notch extracellular truncation (NEXT), a membrane-bound form of Notch which lacks most of the extracellular domain (Figure 1). The γ-secretase complex will then cleave NEXT at S3 and S4 sites to release the Notch intracellular domain (NICD) (Schroeter et al. 1998; Struhl and Greenwald 1999). The NICD is translocated into the nucleus and participates in a transcriptional activator complex with CSL and Mastermind proteins to activate the expression of Notch target genes, including the members of the “Hairy-Enhancer of Split” (Hes) and Hairy-Enhancer of Split related with YRPW motif (Hey) families (Figure 1) (Jarriault et al. 1995; Lecourtois and Schweisguth 1995; Tamura et al. 1995; Petcherski and Kimble 2000; Wu et al. 2000; Fryer et al. 2002).
Fig. 1.

A simplified model for canonical Notch signaling. Notch is synthesized in the ER and glycosylated by the soluble ER enzymes Pofut1 and Rumi. In the Golgi, the O-linked glycans of Notch are extended by the membrane-bound glycosyltransferases like Fringe and GXylT. Also, the S1 cleavage generates a heterodimeric form of Notch, which is composed of the extracellular (NECD, black box) and intracellular (NICD, red box) domains of Notch. Binding of ligands to the NECD and endocytosis of ligand into the signal-sending cell is thought to result in dissociation of the Notch heterodimer and the S2 cleavage of Notch. Notch is further cleaved at S3/S4 sites. The intracellular domain of Notch (NICD) is then translocated into the nucleus. In the absence of Notch signaling, the CSL protein functions as a transcriptional repressor because of its association with co-repressor (CoR) proteins. NICD will displace the co-repressors and result in the formation of a transcriptional activator complex comprising CSL, NICD, Mastermind (Mam) and other co-activators (not shown)
Since most of the above-mentioned developmental signaling pathways use secreted ligands, they are able to activate their corresponding receptors in both neighboring and distant cells. Receptor activation in turn triggers the activation of cascades of kinases and other signaling proteins and ultimately affects the transcription of pathway target genes (Gerhart 1999). The Notch signaling pathway, however, functions differently from the rest of this evolutionarily conserved toolbox in two important ways. First, since Notch ligands are transmembrane proteins, in almost all cases only neighboring cells can use this pathway to communicate. Secondly, since the Notch protein functions both as the receptor (the ligand-responsive, extracellular domain of Notch) and the transcriptional co-activator (the NICD) of the pathway, Notch signaling does not rely on intermediate signaling cascades for the transmission of signal between cell surface and the nucleus. Perhaps due to these unique qualities, a variety of mechanisms, including ubiquitination, endocytosis, recycling and glycosylation, are employed by animal cells to ensure that the Notch pathway is precisely regulated in time and space (Bray 2006). Indeed, the degree to which animal cells dedicate their resources to control the Notch pathway has come as a surprise in some recent studies. For example, based on their yeast phenotypes, animal homologs of genes like epsin and sec15 were thought to have “housekeeping” roles in clathrin-mediated endocytosis and exocytic trafficking, respectively. However, sec15 mutations in flies and epsin mutation in both flies and mice do not show general defects in endocytosis and exocytosis but exhibit specific Notch signaling defects (Overstreet et al. 2004; Tian et al. 2004; Wang and Struhl 2004; Jafar-Nejad et al. 2005; Chen et al. 2009).
Given that carbohydrates cover the surface of all cells in nature, it is not surprising that glycans play important roles in multiple aspects of animal development and physiology (Varki et al. 2009). To our knowledge, one of the first reports on the role of glycosylation in animal development was published three decades ago by Surani, who showed that blocking the formation of N-glycans by tunicamycin results in severe defects in the early stages of mouse embryonic development (Surani 1979). Identification of the enzymes responsible for the formation and elongation of sugar chains, advances in genetics and molecular biology, and major improvements in methods for biochemical identification and analysis of carbohydrates allowed several groups to study the outcome of loss of specific glycosylation events in the development of model organisms. These studies have shown broad roles for a variety of carbohydrate modifications in the regulation of both vertebrate and invertebrate development (Haltiwanger 2002; Haltiwanger and Lowe 2004).
Early work on the Notch protein showed that it binds Lentil lectin (Johansen et al. 1989), indicating that it is decorated with N-glycans (Kornfeld et al. 1981). In addition to N-glycosylation, three forms of unusual O-linked glycosylation have been described on the EGF-like modules of the Notch proteins: O-fucosylation, O-glucosylation and O-GlcNAcylation (Moloney, Shair, et al. 2000; Matsuura et al. 2008). A growing body of literature indicates that glycosylation of the Notch receptors is a major regulator of Notch signaling (Stanley 2007; Okajima, Matsuura, et al. 2008; Luther and Haltiwanger 2009). There are also reports on the glycosylation of some other Notch pathway components, including the Notch ligands, the γ-secretase complex component Nicastrin, and the glycosyltransferase Pofut1 (Panin et al. 2002; Tomita et al. 2002; Loriol et al. 2007). In this review, we will discuss those types of Notch glycosylation for which a regulatory role has been established in animal model studies. We will also review what is known about the potential non-enzymatic roles of glycosyltransferases involved in Notch pathway regulation. Finally, we will cover the literature on the roles played by nucleotide-sugar transporters in Notch signaling.
Regulation of Notch signaling by enzymes involved in the formation and elongation of O-linked fucose
In 1990, Sarin and colleagues published the first report on the presence of O-linked fucose on the EGF-like repeat of a protein (urinary-type plasminogen activator) (Kentzer et al. 1990). Since then, several other proteins were shown to be O-fucosylated, including tissue-type plasminogen activator and human coagulation factors VII and XII (Harris and Spellman 1993). Originally, it was proposed that O-fucosylation occurs at a serine or threonine immediately preceding the third cysteine of those EGF-like repeats that conform to the C2–X–X–G–G–(S/T)–C3 consensus sequence (X can be any amino acid; G, S, T are glycine, serine and threonine, respectively) (Harris and Spellman 1993). The more flexible C2–X–X–X–(A/G/S)–(S/T)–C3 consensus sequence was later proposed to account for the growing number of confirmed O-fucosylated EGF-like repeats (A stands for alanine) (Haines and Irvine 2003). The extracellular domain of the Notch proteins contains between 29 and 36 EGF-like repeats, three Lin12-Notch Repeat (LNR) motifs and a heterodimerization domain (HD) (Figure 2). Each EGF-like repeat is a 38- to 40-amino-acid module with six cysteine residues, which form three disulfide bonds and thereby give the EGF-like repeat its specific three-dimensional structure (Figure 2) (Harris and Spellman 1993). All mammalian Notch proteins and ligands and their Drosophila homologs contain EGF-like repeats with consensus O-fucosylation motifs (Figure 3) (Rampal et al. 2007). The presence of O-linked fucose on Drosophila Notch, Delta and Serrate and mammalian Notch1, Delta-like1 and Jagged1 has experimentally been shown by the Irvine and Haltiwanger labs (Moloney, Panin, et al. 2000; Moloney, Shair, et al. 2000; Panin et al. 2002). However, in vivo and co-culture experiments strongly suggest that O-fucosylation is required in the Notch-expressing signal-receiving cell but not in the ligand-expressing signal-sending cell (Irvine and Wieschaus 1994; Kim et al. 1995; Hicks et al. 2000; Moloney, Panin, et al. 2000). The O-linked fucose can be elongated by sequential addition of three other sugar residues: N-acetylglucosamine (GlcNAc), galactose (Gal) and sialic acid (SA) (Figure 2). This process is protein- and EGF-like repeat specific, in the sense that some proteins/EGF-like repeats only contain O-fucose monosaccharides, while others are decorated with a combination of mono-, di- and oligosaccharides (Moloney et al. 1997; Moloney, Shair, et al. 2000; Shao et al. 2003; Rampal, Li, et al. 2005).
Fig. 2.
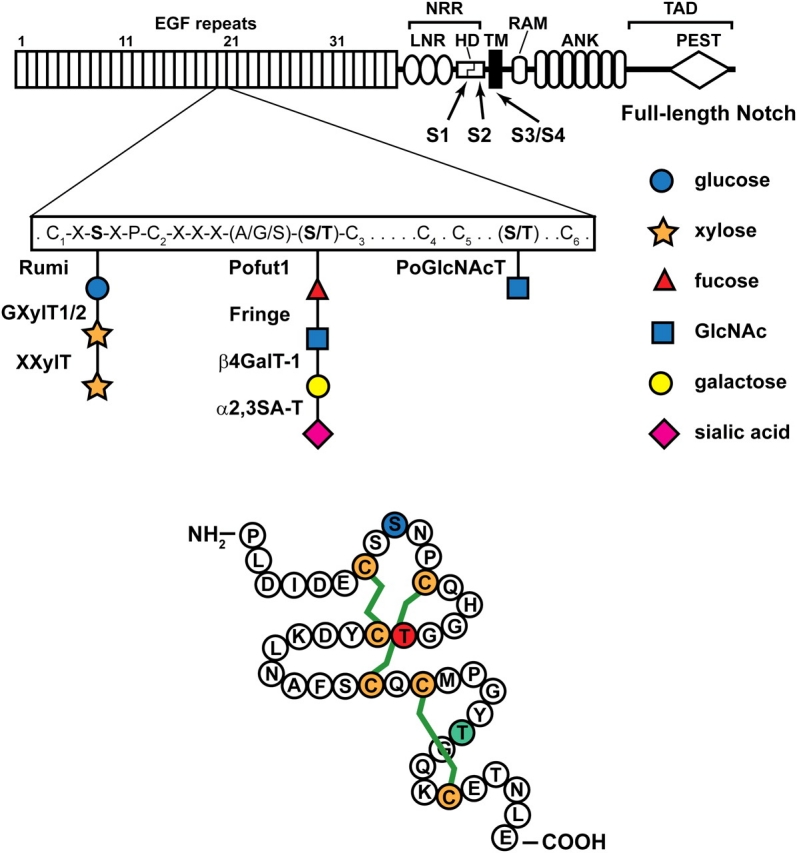
A schematic representation of the full-length Drosophila Notch is shown. The extracellular domain of Notch contains 36 EGF-like repeats, followed by the Negative Regulatory Region (NRR), which consists of three LNR motifs and a heterodimerization domain (HD). The intracellular domain contains an RBP-J association module (RAM), two nuclear localization signals (NLS), seven Ankyrin repeats (ANK) and a PEST domain. The C-terminal part of the intracellular domain contains a trans-activation domain (TAD). Each EGF repeat has six cysteine residues (C1 to C6). The consensus motifs for O-linked glucose and O-linked fucose and the enzymes responsible for their formation and elongation are shown. O-GlcNAc is added to a serine or threonine between C5 and C6, but the exact consensus motif is not known yet. S1–S4 show the position of Notch cleavage sites. TM, transmembrane. The lower drawing is based on the crystal structure of one of the EGF-like repeats of human coagulation factor IX (Huang et al. 1989). The shown amino acid sequence is from EGF-like repeat 20 of the Drosophila Notch, which has experimentally been shown to harbor all three forms of O-linked glycans discussed here (Matsuura et al. 2008). The green lines depict the three disulfide bridges which form between pairs of cysteines in EGF-like repeats (C1–C3, C2–C4, and C5–C6). Blue S, red T and green T show the amino acids to which O-linked glucose, fucose and GlcNAc are attached, respectively
Fig. 3.

Distribution of EGF-like repeats with consensus O-fucosylation and/or O-glucosylation motifs in Notch receptors and their ligands. Alignment of extracellular domains of Drosophila Notch (dNotch), its human homologs (hNotch1–4) and their ligands are shown. Jag is Jagged; Dll is Delta-like. Blue and yellow boxes indicate EGF-like repeats with a consensus motif for O-glucosylation and O-fucosylation, respectively. The red lines denote Ca+//0-binding EGF-like repeats. hNotch3 and hNotch4 have 34 and 29 EGF-like repeats, respectively. The dotted lines in the schematic of these two proteins show the EGF-like repeats missing compared to dNotch, hNotch1 and hNotch2 (Uyttendaele et al. 1996). Note that the distribution of O-glucosylation, O-fucosylation and Ca+//0-binding EGF-like repeats is well conserved in Notch receptors. dSerrate and hDll3 do not have a predicted O-glucosylation motif. We note that mouse and rat Dll3 have a single predicted O-glucosylation motif (not shown). LNR, Lin12-Notch repeat; HD, heterodimerization; TM, transmembrane; DSL, Delta-Serrate-Lag2; CR, cysteine-rich. S1–S4 show the position of Notch cleavage sites
The enzymes responsible for the addition of O-fucose to EGF-like repeats and its elongation have been identified and extensively studied (Figure 2) (Stanley 2007). Addition of fucose residues to the hydroxyl group of serine and threonine occurs in the endoplasmic reticulum (ER) and is catalyzed by “protein O-fucosyltransferase” or Pofut1 (Ofut1 is Drosophila) (Wang et al. 2001; Luo and Haltiwanger 2005; Okajima et al. 2005). GlcNAc can then be added to the O-linked fucose by the Fringe family of Golgi β1,3N-acetylglucosaminyltransferases (Fringe in Drosophila; Lunatic, Manic and Radical Fringe proteins in mammals) (Bruckner et al. 2000; Moloney, Panin, et al. 2000). In Drosophila Schneider 2 (S2) cells, only fucose monosaccharide and GlcNAc-Fuc disaccharides have been reported (Xu et al. 2007). However, studies in Chinese hamster ovary (CHO) cells indicate that two other enzymes—β4galactosyltransferase-1 (β4GalT-1) and α2,3sialyltransferase (α2,3SA-T)—elongate the O-linked disaccharide on the mammalian Notch1 to its longest form, the sia-α2,3-Gal-β1,4-GlcNAc-β1,3-Fuc-α-O-Ser/Thr tetrasaccharide (Stanley 2007; Luther and Haltiwanger 2009).
Fringe proteins regulate Notch signaling in specific contexts
Out of the above-mentioned enzymes, Fringe was the first to be linked to the Notch signaling pathway. Several years before the discovery of Fringe’s biochemical activity, fringe was found to be essential for the formation of the dorsal–ventral compartment boundary in Drosophila wing and was suggested to be involved in the regulation of the Notch signaling pathway (Irvine and Wieschaus 1994; Kim et al. 1995; Haines and Irvine 2003). Further genetic studies indicated that both Drosophila Fringe and its mammalian homologs are involved in the regulation of Notch signaling in a variety of developmental processes, including vertebrate somitogenesis and the establishment of the dorsal–ventral boundary in the Drosophila eye and leg (Cho and Choi 1998; Dominguez and de Celis 1998; Evrard et al. 1998; McGrew et al. 1998; Zhang and Gridley 1998; Rauskolb and Irvine 1999). Importantly, among the well-known paradigms through which Notch signaling functions during animal development—lateral inhibition, asymmetric division and “inductive signaling”—only “inductive signaling” requires the function of Fringe (Haines and Irvine 2003). The distant homology of Fringe to a bacterial glycosyltransferase, the cell-autonomous requirement for Fringe in the signal-receiving cell and the presence of conserved O-glycosylation motifs in the extracellular domain of Drosophila and mammalian Notch proteins suggested that Fringe might regulate the Notch pathway by glycosylating Notch (Panin et al. 1997; Yuan et al. 1997; Moloney, Shair, et al. 2000). Indeed, it was later demonstrated by the Cohen, Haltiwanger, Irvine, Stanley and Vogt groups that Fringe is a glycosyltransferase that adds GlcNAc to O-linked fucose on some of the Notch EGF-like repeats and is required for Notch signaling (Bruckner et al. 2000; Moloney, Panin, et al. 2000). By further showing that the enzymatic activity of Fringe is essential for the regulation of the Notch pathway, these studies provided the first direct evidence for the importance of O-linked glycosylation of the Notch receptor in Notch signaling (Bruckner et al. 2000; Moloney, Panin, et al. 2000).
Loss of function studies indicate that Fringe promotes Delta-to-Notch signaling and inhibits Serrate-to-Notch signaling during Drosophila wing development (Panin et al. 1997). How does the addition of GlcNAc to O-linked fucose on Notch EGF-like repeats mediate differential response of the Notch receptor to Delta and Serrate? Fleming and colleagues showed that replacing the Notch-binding domain of Serrate with the Notch-binding domain of Delta prevents Fringe from blocking the Serrate function and suggested that Fringe regulates Notch signaling at the level of ligand binding (Fleming et al. 1997). This notion was later confirmed by assays developed in the Cohen lab. Specifically, by examining the effects of Fringe expression on the binding of tagged, soluble forms of the Notch extracellular domain to the surface of cells stably expressing Delta or Serrate (and reciprocal experiments with soluble ligands and Notch-expressing cells), the Cohen and Irvine groups showed that Fringe promotes Notch-Delta binding and opposes Notch-Serrate binding (Bruckner et al. 2000; Okajima et al. 2003). These effects have also been confirmed by using purified components in in vitro Notch–ligand binding assays (Xu et al. 2007).
Given that mammals have four Notch proteins, five ligands and three Fringe homologs, it is not surprising that studies on the molecular mechanism of mammalian Notch pathway regulation by Fringe proteins have drawn a more complicated and sometimes controversial picture. Reports from the Weinmaster lab indicate that all three mammalian Fringe proteins are able to increase the binding between Delta-like1 and Notch1 proteins, and also Delta-like1-induced Notch1 signaling in luciferase reporter assays in NIH3T3 and C2C12 cells (Hicks et al. 2000; Yang et al. 2005). These studies also showed that expression of Manic or Lunatic Fringe in the Notch1-expressing cell decreased Jagged1-induced signaling. But in contrast to the Drosophila reports, binding between Notch1 and Jagged1 was not decreased upon Manic or Lunatic Fringe expression. Accordingly, Weinmaster and colleagues suggested that rather than disrupting Jagged1-Notch1 binding, Fringe proteins might reduce signaling by decreasing the binding strength between Jagged1 and Notch1 (Hicks et al. 2000; Yang et al. 2005). These authors also provided evidence that Lunatic Fringe can enhance Jagged1-Notch2 signaling, suggesting that upon Fringe activity, the same ligand might elicit opposite responses from different Notch receptors (Hicks et al. 2000). Results from the Stanley and Hirai groups, however, do not fully agree with some of the above-mentioned observations. A recent report from the Stanley lab indicates that Lunatic Fringe can decrease the binding of Jagged1 to the surface of Notch1-expressing, Lec1 CHO cells (Stahl et al. 2008). Also, Hirai and colleagues report that not only do Lunatic and Manic Fringe decrease Jagged1-Notch2 signaling, but they also decrease the binding between these two proteins (Shimizu et al. 2001). Considering the above discrepancies, the exact mechanism for the modulation of signaling between various mammalian ligand–Notch pairs by Fringe proteins will need further exploration.
Even though at a biochemical level all three mammalian Fringe proteins perform the same function (Rampal, Li, et al. 2005), several recent studies have highlighted the non-redundant roles played by these proteins in different developmental contexts (Visan et al. 2006; Ryan et al. 2008; Benedito et al. 2009; Moran et al. 2009; Tan et al. 2009). These observations are in line with the notion that amino acids other than the O-fucosylated S/T might determine which EGF-like repeats are good substrates for the function of Fringe proteins and which Fringe protein is the optimal enzyme for modification of any given EGF-like repeat (Rampal, Li, et al. 2005; Luther and Haltiwanger 2009). Understanding the molecular mechanisms underlying the differences among the functions of the Fringe proteins promises to uncover another layer of complexity in the way Notch signaling is regulated in mammals.
Protein O-fucosyltransferase 1 and O-fucose in fly Notch signaling
Fringe proteins specifically add a GlcNAc residue to O-fucosylated EGF-like repeats, and the enzymatic activity of Fringe is essential for its effects on the Notch pathway in both Drosophila and mammalian systems (Bruckner et al. 2000; Moloney, Panin, et al. 2000; Chen et al. 2001; Yang et al. 2005). Accordingly, the enzyme responsible for the addition of O-linked fucose to Notch EGF-like repeats was also predicted to be required for Notch signaling. Indeed, genetic experiments by Irvine, Matsuno and Stanley labs have demonstrated that the function of Pofut1/Ofut1 is essential for Notch signaling in flies and mice (Okajima and Irvine 2002; Sasamura et al. 2003; Shi and Stanley 2003). As mentioned above, fringe mutations only affect some aspects of Notch signaling. However, Pofut1/Ofut1 mutants show a full-blown loss of Notch signaling in all aspects reported so far in both vertebrates and invertebrates (Okajima and Irvine 2002; Sasamura et al. 2003; Shi and Stanley 2003). These observations indicate that either O-linked fucose on Notch EGF-like repeats has functions independent of its elongation by Fringe or that Pofut1/Ofut1 uses an additional non-enzymatic mechanism to regulate Notch signaling. Of course, these possibilities are not mutually exclusive.
Reports by Okajima and Irvine provide strong support for a simple model for Ofut1 function in flies (Okajima et al. 2005; Okajima, Reddy, et al. 2008). In their model, Ofut1 plays two roles in the regulation of Notch signaling: an enzymatic role essential for the function of Fringe but dispensable for Fringe-independent modes of Notch signaling, and a non-catalytic role required for proper folding and cell surface expression of the Notch receptor. In support of their model, they have shown that Notch does not reach the cell surface in Ofut1 mutant clones and Ofut1 knock-down Schneider 2 cells. They have also reported that overexpression of the R245A mutant form of Ofut1—which lacks enzymatic activity in vitro—can restore the cell surface expression of Notch caused by loss of Ofut1 (Okajima et al. 2005; Okajima, Reddy, et al. 2008). Furthermore, they demonstrate that the severe neurogenic phenotype observed in embryos lacking the maternal contribution of Ofut1 can be rescued by a genomic construct encoding an enzymatically inactive form of Ofut1 (Okajima, Reddy, et al. 2008). They also generated embryos lacking guanosine diphosphate (GDP)-fucose by making germline mutant clones of gmd, which encodes GDP-d-mannose dehydratase (Figure 4). Gmd is essential for generation of GDP-fucose in flies due to the absence of a salvage pathway for GDP-fucose synthesis in these animals (Okajima, Reddy, et al. 2008). Indeed, staining the mutant embryos with a fucosylation-specific antibody confirmed lack of fucosylation in gmd mutants. However, these embryos did not show a classical neurogenic phenotype, strongly suggesting that O-fucose per se is not required for Notch signaling except for contexts like the wing margin development, where Fringe plays an essential role (Okajima, Reddy, et al. 2008).
Fig. 4.

Mechanisms for the synthesis of GDP-fucose and its transport into the secretory pathway. In the cytoplasm, GDP-mannose is converted into GDP-fucose by GMD and FX enzymes. In mammalian cells, an alternative, salvage pathway exists, which uses the free fucose transported from the extracellular space or from the lysosome to make GDP-fucose (Smith et al. 2002). FK stands for fucose kinase; GPP stands for GDP-fucose pyrophosphorylase. The salvage pathway allows the rescue of GMD and Fx mutations by providing fucose in the medium. Drosophila cells lack the salvage pathway. Efr and Gfr transport GDP-fucose into the ER and Golgi lumens, respectively. Genetic and biochemical experiments indicate that Gfr is essential for the addition of fucose to N-glycans on Notch and other proteins in Drosophila larvae. For the addition of O-linked fucose to Notch—which occurs in the ER—Gfr and Efr function redundantly. Since Gfr resides in the Golgi, retrograde transport of GDP-fucose from Golgi to the ER (the dashed arrow) is likely to underlie the ability of Gfr to compensate for the lack of Efr
Work in the Matsuno lab suggests that Ofut1 might also play a role in the trafficking of Notch, and that O-fucose plays roles beyond serving as a substrate for the addition of GlcNAc by Fringe (Sasaki et al. 2007; Sasamura et al. 2007). These authors report that exocytosis of Notch to the plasma membrane does not require the function of Ofut1 but that transcytosis of Notch from the plasma membrane to adherens junctions requires the O-fucosylation of Notch by Ofut1 (Sasaki et al. 2007). They further show that Ofut1 is required for the endocytosis of Notch and that this role is independent of Ofut1’s enzymatic activity because the endocytic defects were not observed in a gmd null mutant lacking any detectable GDP-fucose (Sasamura et al. 2007). The discrepancy between some of the findings of the Irvine and Matsuno groups might in part result from different techniques used to follow Notch trafficking and localization (Vodovar and Schweisguth 2008). In our opinion, the in vivo rescue experiments and analysis of gmd mutant embryos by Okajima and Irvine (Okajima, Reddy, et al. 2008) provide very strong evidence for the conclusion that the enzymatic activity of Ofut1 is not required for Notch signaling during embryonic neurogenesis. However, we cannot rule out the possibility that later during development, O-fucosylation of Notch plays a role in regulating Fringe-independent aspects of signaling. This issue can be addressed by testing whether gmd mutants show defects in the lateral inhibition process during adult sensory organ development. fringe mutants do not exhibit lateral inhibition defects in adult peripheral nervous system (Haines and Irvine 2003). Therefore, lateral inhibition defects in adult Drosophila with gmd mutant clones would strongly suggest that in some Fringe-independent contexts, O-fucose might be required for optimal Notch signaling (lack of fucose in other types of glycans, like N-glycans, might contribute to this phenotype as well). Also, it is worth mentioning that to our knowledge, the glycosylation status of the Notch protein has not been tested in gmd mutant flies or Ofut1 mutant flies rescued by R245A Ofut1 transgene. Therefore, one can speculate that in gmd mutants a sugar other than fucose might be added to the O-fucosylation sites that can partially compensate for the lack of fucose but does not allow the addition of GlcNAc by Fringe or that although the R245A mutant Ofut1 does not show enzymatic activity in vitro, it could still have residual activity in vivo, which might transfer low levels of O-linked fucose to Notch and mask the phenotypes that a complete loss of O-fucose would show. We note that a requirement for O-fucosylation of Notch in Fringe-independent contexts would be consistent with earlier experiments from Irvine, Matsuno and Baker labs indicating that the presence of O-linked fucose on Drosophila Notch affects its binding to Delta and Serrate in the absence of Fringe (Li et al. 2003; Okajima et al. 2003; Sasamura et al. 2003).
Protein O-fucosyltransferase 1 and O-fucose in mammalian Notch signaling
Genetic studies from the Stanley lab showed that mouse Pofut1 is essential for Notch signaling (Shi and Stanley 2003). Studies on an independent Pofut1 null allele recently generated by the Saga group showed similar results and genetically placed the function of Pofut1 upstream of the Notch intracellular domain (Okamura and Saga 2008). Similar to Drosophila Ofut1 mutants, Pofut1 null allele mouse embryos show Notch pathway defects much broader than those of the Fringe mutant animals (Shi and Stanley 2003; Okamura and Saga 2008). Interestingly, analysis of a hypomorphic allele of Pofut1 by the Gossler group indicates that somitogenesis is much more sensitive to Pofut1 levels compared to other Notch-dependent processes in the mouse embryo (Schuster-Gossler et al. 2009). This observation suggests that Fringe-dependent aspects of Notch signaling require higher levels of Pofut1. Although loss of Ofut1/Pofut1 results in global loss of Notch signaling in both flies and mice, there might be differences between the molecular mechanisms of Notch pathway regulation by Pofut1 in flies and mice. In Pofut1 mutant mouse embryos, Notch1 shows decreased cell surface expression and ER accumulation (Okamura and Saga 2008) similar to what has been observed in Ofut1 mutant Drosophila cells and tissues (Okajima et al. 2005; Okajima, Reddy, et al. 2008). However, another report indicates that the total level and cell surface expression of Notch1–3 receptors are normal in Pofut1 null mouse embryonic stem (ES) cells, even though these cells fail to bind the Notch ligands Jagged1 and Delta1 and do not show ligand-induced Notch signaling (Shi et al. 2005; Stahl et al. 2008). To examine whether mouse Pofut1 also plays a non-enzymatic role similar to its Drosophila homolog, Stahl and colleagues overexpressed an enzymatically inactive form of Pofut1 in Pofut1 null ES cells and observed a partial rescue of ligand binding and signaling. To test the specificity of this effect, they overexpressed an enzymatically inactive form of an unrelated ER α-glucosidase in Pofut1 null cells and observed a similar partial rescue of Notch signaling (Stahl et al. 2008). These results cast doubt on the specificity of the observed non-enzymatic activity of Pofut1 in mouse ES cells and highlight a potential difference between the ways Drosophila Ofut1 and mouse Pofut1 may regulate Notch signaling.
Genetic manipulation of the glycosyltransferases involved in the formation and elongation of O-linked fucose on Notch is not the only experimental setting used to demonstrate the importance of O-fucosylation in mammalian Notch signaling. Indeed, studies on cell lines and mice deficient in the synthesis of GDP-fucose have provided independent evidence for the critical role played by O-linked fucose in the regulation of Notch signaling in mammals (Stahl et al. 2008; Zhou et al. 2008). The Lec13 CHO cells have a mutation in Gmd and therefore lack GDP-fucose if cultured in the absence of fucose (Stahl et al. 2008). Co-culture studies have indicated that Lec13 cells are defective in Delta- and Jagged-mediated Notch signaling, despite having normal levels of Pofut1 and surface Notch expression (Stahl et al. 2008). Moreover, the Lowe group has generated a mouse strain mutant for the Fx gene, whose product functions downstream of the Gmd protein to generate GDP-fucose (Figure 4) (Smith et al. 2002). In mice, a salvage pathway for the generation of GDP-fucose from dietary fucose exists that does not depend on the functions of Gmd and Fx (Smith et al. 2002). Because of this salvage pathway, the Fx−/− strain has been used as an elegant model in which the fucosylation of Notch and other proteins can be abolished in a conditional and reversible manner (Smith et al. 2002; Zhou et al. 2008; Liu et al. 2009). For example, the Fx−/− model has recently been used to show that the loss of Notch fucosylation results in aberrant myleoproliferation (Zhou et al. 2008) and intestinal secretory cell hyperplasia (Waterhouse et al. 2010).
Does elongation of O-linked fucose beyond the disaccharide play a role in Notch signaling?
As mentioned above, the GlcNAcβ1,3Fuc-O-S/T disaccharide is further elongated via the sequential addition of galactose and sialic acid residues (Figure 2). However, several lines of evidence from Irvine and Haltiwanger labs indicate that the disaccharide form is sufficient for the full activity of the Notch receptor in flies. Drosophila homologs of β4GalTs—the enzymes which add galactose to GlcNAc—are dispensable for normal development and do not seem to be required for Notch signaling (Haines and Irvine 2005). Indeed, in vitro reconstitution studies indicate that addition of GlcNAc to the O-linked fucose is sufficient to explain the differential effects of Fringe on the binding of Notch to its ligands (Xu et al. 2007). Together, these observations demonstrate that Drosophila Notch signaling does not depend on the elongation of the O-fucose glycans on Notch beyond the GlcNAcβ1,3Fuc disaccharide.
Work from the Stanley lab indicates that at least in some contexts, elongation of the GlcNAcβ1,3Fuc disaccharide might contribute to optimal Notch signaling in vertebrates, although it does not seem to be an essential step in Notch signaling in vivo. In CHO cells, β4GalT-1 is required to add galactose to GlcNAcβ1,3Fuc-O-S/T disaccharides on Notch1 EGF-like repeats. Loss of β4GalT-1 blocks the modulatory effects of Fringe on Jagged1-induced Notch signaling in these cells, indicating that in CHO cells Fringe can only affect Notch signaling if a galactose is added to the disaccharide generated by sequential activities of Pofut1 and Fringe on Notch1 EGF-like repeats (Chen et al. 2001). However, in E9.5 β4GalT-1-null mutant embryos, the number and morphology of somites are identical to their wild-type littermates, indicating that the function of Lunatic fringe is not perturbed in the absence of β4GalT-1 (Chen et al. 2006). The mutant embryos do exhibit a mild increase in the number of lumbar vertebrae at E18.5, a phenotype which might result from defective activation of Hox gene expression due to decreased Notch signaling (Zakany et al. 2001). It is possible that in mouse embryos, loss of β4GalT-1 is compensated by other mammalian β4GalTs (Chen et al. 2006).
Drosophila Notch signaling depends on a protein O-glucosyltransferase
More than 20 years ago, Hase and colleagues reported the presence of a trisaccharide containing two xyloses and an O-linked glucose on the first EGF-like repeat of the bovine blood coagulation factors VII and IX (Hase et al. 1988). Chemical analysis determined the structure of the trisaccharide on factor IX to be Xyl-α1,3-Xyl-α1,3-Glc-β1-O-Serine (Hase et al. 1990). Subsequent studies identified Xyl–Glc disaccharide and Xyl–Xyl–Glc trisaccharides on several other secreted proteins including human factors VII and IX, human and bovine protein Z and bovine Thrombospondin (Nishimura et al. 1989, 1992). By comparing the sequences of the confirmed O-glucosylated EGF-like repeats, it was proposed that the O-linked glucose consensus site is located between the first and second cysteines of those EGF-like repeats harboring the C1–X–S–X–P–C2 consensus sequence (X represents any amino acid, S is the glucosylated serine) (Figure 2) (Harris and Spellman 1993). Using database searches on this consensus sequence, the Haltiwanger lab identified the Notch receptors as proteins with the largest number of predicted O-glucosylation sites and initiated a detailed biochemical analysis of O-glycans on the mammalian Notch1 proteins (Figure 3) (Moloney, Shair, et al. 2000). Radioactive labeling experiments showed that endogenous Notch1 in CHO cells is O-glucosylated and that the major form of the O-glucose glycans on Notch1 is a trisaccharide (Moloney, Shair, et al. 2000). Although the presence of xylose residues was not formally shown by Moloney and colleagues in the O-linked trisaccahrides found on Notch1, a recent report strongly suggests that the O-linked glucose on Notch1 is extended by two xylose residues (Bakker et al. 2009). Indeed, the identity of the trisaccharides on mouse Notch1 was recently confirmed to be Xyl-α1,3-Xyl-α1,3-Glc-β1-O-Serine (Figure 2) (Whitworth et al. 2010).
An enzymatic activity able to covalently attach an O-linked glucose to EGF-like repeats has been found in cell extracts from a variety of metazoan cell and tissue extracts (Shao et al. 2002). The invertebrate cell extracts were able to catalyze the glucosylation of a vertebrate EGF-like repeat, indicating the evolutionary conservation of this process (Shao et al. 2002). Also, the Hase group characterized two distinct enzymatic activities mediating the Xyl-to-Glc and Xyl-to-Xyl linkages in a human cell line and purified the glucoside xylosyltransferase (GXylT) activity from bovine liver (Minamida et al. 1996; Omichi et al. 1997; Ishimizu et al. 2007). However, none of the three enzymes responsible for the generation of this trisaccharide was identified in these studies. On a functional level, a serine-to-alanine mutation in the consensus O-glucosylation motif of the recombinant human factor VII resulted in a 40% decrease in its clotting activity but did not alter its proteolytic activity toward factors IX and X or its interaction with tissue factor (Bjoern et al. 1991). The same mutation was also reported to decrease the secretion of recombinant factor VII from COS-7 cells by 50% (Bolt et al. 2007). In summary, despite the evolutionary conservation of O-linked glucose and its extended versions, until recently, the biological significance of this posttranslational modification or the enzymes responsible for it remained largely unknown.
Rumi: a protein O-glucosyltransferase required for Notch signaling in flies
The first in vivo evidence for the functional importance of protein O-glucosylation came from identification of the gene encoding the Drosophila protein O-glucosyltransferase, rumi, in a collaborative effort between Drosophila geneticists and glycobiologists (Acar et al. 2008). rumi mutations were isolated in a chemical mutagenesis screen for regulators of the Notch pathway in Drosophila. Mutations in rumi result in a temperature-sensitive loss of Notch signaling. When raised at 18°C, rumi homozygous mutant animals are viable and only show mild Notch signaling defects in certain developmental contexts. As the temperature at which the mutant animals grow increases, the Notch pathway defects become more severe and widespread. When raised at 28–30°C, rumi homozygous animals do not reach adulthood. Furthermore, clonal analysis shows that at 28–30°C, the phenotypes of rumi homozygous tissues are equivalent to null phenotypes of Notch itself. These data indicate that at the restrictive temperature, the function of Rumi is essential for Drosophila Notch signaling. Interestingly, a small deletion that removes ∼95% of the coding region of rumi also displays temperature-sensitive phenotypes (Acar et al. 2008). This observation demonstrates that the temperature sensitivity of rumi mutants is not a characteristic of specific missense alleles but is rather a quality of the step(s) during the Notch signal transduction which Rumi regulates.
rumi encodes a soluble ER protein with a C-terminal ER-recycling KDEL motif (Munro and Pelham 1987) and a CAP10 domain. This domain takes its name from the CAP10 protein, one of the four capsule-associated proteins (CAP10, CAP59, CAP60, and CAP64) required for the synthesis of the polysaccharide capsule and the virulence of the pathogen Cryptococcus neoformans (Chang and Kwon-Chung 1999; Okabayashi et al. 2007). Although the biochemical function of the CAP proteins is not known, both CAP10 and CAP59 show homology to other cryptococcal glycosyltransferases (Sommer et al. 2003; Klutts et al. 2007). Moreover, using sophisticated homology searches, Ponting and colleagues found similarity between the CAP10 domain and glycosyltransferases from Bacillus anthracis and Synechocystis sp. (Ponting et al. 2001). Together, these reports suggested that Rumi might be a glycosyltransferase. Indeed, biochemical experiments demonstrated that Rumi is able to catalyze the transfer of a glucose residue from UDP-Glc to an EGF-like repeat from human factor VII, which contains the consensus O-glucosylation motif (Acar et al. 2008). As mentioned above, Ofut1 regulates Drosophila Notch signaling via both enzymatic and non-enzymatic mechanisms (Okajima et al. 2005; Sasamura et al. 2007; Okajima, Reddy, et al. 2008). However, a point mutation in the CAP10 domain which abolishes the Rumi enzymatic activity but does not alter its expression level or stability also results in severe loss of Notch signaling (Acar et al. 2008). This observation strongly suggests that the main role of Rumi in Drosophila Notch signaling is mediated via its enzymatic activity. Altogether, these experiments resulted in the identification of a protein O-glucosyltransferase enzyme and suggested that O-glucosylation of one or more of the Notch pathway components is required for the regulation of this pathway.
Out of the 36 EGF-like repeats present in the extracellular domain of the Drosophila Notch, 18 contain the C1–X–S–X–P–C2 motif (Figure 3). One of the EGF-like repeats of the ligand Delta has this motif, but none of the EGF-like repeats of the ligand Serrate is predicted to be O-glucosylated (Figure 3). When overexpressed in rumi mutant clones, Delta and Serrate are able to signal to their neighboring cells, suggesting that the function of Rumi is not essential for the activity of Notch ligands. However, loss of rumi fully suppresses the overexpression phenotype of full-length Drosophila Notch (Acar et al. 2008). Moreover, while the level of Delta and Serrate are normal in rumi mutant clones, Notch protein accumulates in rumi mutant tissues in a cell-autonomous manner. Finally, RNAi-mediated knock-down of Rumi in a Drosophila cell line results in a severe decrease in the level of O-linked glucose on Notch (Acar et al. 2008). Altogether, these data strongly suggest that the Notch protein itself is the biologically relevant target of Rumi. It should be noted that the epistasis analysis between the Notch ligands and rumi has been performed in overexpression studies. Therefore, it is possible that at endogenous levels, Delta signals better when it is O-glucosylated. Given the inherent limitations of overexpression studies—especially when it comes to temperature-sensitive phenotypes of dosage-sensitive genes like Notch and Delta—the proof for the above notions will await in vivo structure–function analyses on Notch and Delta at endogenous levels.
Potential molecular mechanism for temperature-sensitive phenotypes of rumi mutants
How does the lack of glucose on Notch result in a temperature-sensitive loss of signaling? In order for canonical Notch signaling to occur, Notch needs to reach the plasma membrane, bind the ligands and undergo proteolytic cleavages, so that the Notch intracellular domain is released from the membrane to enter the nucleus (Figure 1). Cell surface staining of Drosophila imaginal discs harboring rumi mutant clones raised at 28–30°C with an antibody against the extracellular domain of Notch indicates that high levels of Notch accumulate at the surface of rumi-null clones, ruling out lack of cell surface expression as the cause of rumi mutant phenotype (Acar et al. 2008). Also, several lines of evidence suggest that a defect in ligand binding cannot explain the temperature-sensitive loss of Notch signaling in rumi clones: (1) a secreted hybrid protein comprising the Notch extracellular domain and Alkaline phosphatase (N-AP) made in Rumi knock-down cells can bind Delta as strongly as N-AP made in control cells; (2) overexpression of Delta or Serrate in cells adjacent to rumi mutant cells in Drosophila tissues fails to induce signaling at high temperatures; (3) the ligand-independent gain of Notch signaling observed in mutant clones of the Drosophila tumor suppressor gene lethal giant discs (lgd) can be fully suppressed by simultaneous loss of rumi (Acar et al. 2008). However, Western blots on protein extracts from Drosophila larval brains and imaginal discs indicate a severe decrease in the Notch cleavage products in rumi animals raised at 28–30°C (Acar et al. 2008). Importantly, rumi mutations cannot suppress the overexpression phenotypes of NEXT, a membrane-bound form of Notch which is independent of the S2 cleavage but still requires the S3 cleavage (Figure 1). Therefore, based on the above data, we propose that the best explanation for the temperature-sensitive phenotypes of rumi is that a critical step between the ligand binding and the S2 cleavage of Notch requires the function of Rumi.
A series of reports from the Blacklow lab has provided significant insights into the role of the so-called negative regulatory region (NRR) of Notch in preventing its proteolytic S2 cleavage in the absence of ligands (Sanchez-Irizarry et al. 2004; Malecki et al. 2006; Gordon et al. 2007; Gordon, Roy, et al. 2009). The NRR consists of three LNR motifs and a hetero dimerization (HD) domain and resides between the EGF-like repeats and the transmembrane domain of Notch (Figure 2). The S2 cleavage site is normally buried in a hydrophobic interface between the HD domain and the LNR motifs (Figure 5). Therefore, in the absence of ligand, Notch is protease-resistant because the S2 cleavage site is not exposed (Gordon et al. 2008). Ligand binding induces conformational changes in the NRR region and results in disengagement of LNR motifs and relaxation of the interaction between the N-terminal and C-terminal parts of the HD domain (Gordon, Roy, et al. 2009). Thereby, the S2 cleavage site becomes accessible to a disintegrin and metalloproteinase (ADAM) proteases, rendering Notch protease-sensitive (Figure 5). Ligands bind to the EGF-like repeat region of the Notch extracellular domain (Xu et al. 2005). Therefore, in order for Notch to switch from protease-resistant to protease-sensitive, the effects of ligand binding should somehow be transmitted to the NRR. Genetic and cell biological studies in Drosophila and mammalian cell culture systems suggest that endocytosis of the Notch-bound ligand into the signal-sending cell exerts a pulling force on the Notch extracellular domain and thereby allows the protease cleavage and activation of Notch to occur (Figure 5) (Parks et al. 2000; Nichols et al. 2007; Windler and Bilder 2010). Since Rumi glycosylates the EGF-like repeats of Notch, one way to explain the rumi phenotype is that when rumi mutants are raised at high temperature, the “unglucosylated” extracellular domain of Notch undergoes conformational changes which are still compatible with ligand binding but do not allow the transmission of the pulling force from the ligand binding domain to the NRR. We should also note that even though the above-mentioned data argue against a defect in Notch–ligand binding as the underlying mechanism for temperature-sensitive phenotypes of rumi (Acar et al. 2008), it is still possible that at endogenous levels of these proteins in vivo, the Notch–ligand binding is not strong enough to ensure the transmission of the endocytosis-generated pulling force to the NRR.
Fig. 5.

Ligand endocytosis makes Notch sensitive to the S2 proteolytic cleavage. The negative regulatory region (NRR) consists of the LNR motifs (green ovals) and the N-terminal and C-terminal parts of the heterodimerization domain (HD). At the cell membrane, the S2 cleavage site (orange circle) is buried in a cleft in the C-terminal part of the HD domain (yellow). A loop between the first two LNR motifs functions as a plug to ensure that the ADAM protease cannot gain access to the S2 cleavage site (Gordon et al. 2007). Binding of ligand from the neighboring signal-sending cell to the Notch receptor and endocytosis of the Notch-bound ligand into the signal-sending cell lead to the disengagement of the LNR motifs from the HD domain and relaxation of the interaction between the C-terminal and N-terminal parts of the HD domain. As a result, the S2 cleavage site is exposed, and Notch becomes a substrate for cleavage by the ADAM protease. The force generated by ligand endocytosis is thought to cause the above-mentioned conformational changes in the extracellular domain of Notch, but it is also possible that allosteric changes—independent of force—are involved (Gordon et al. 2008). The folds drawn in the Notch ECD are based on the reports that the linkage between non-Ca+//0-binding EGF-like repeats and their neighboring EGF-like repeats can be flexible, whereas the linkage between two Ca+//0-binding EGF-like repeats is rigid (Downing et al. 1996; Hambleton et al. 2004; Irvine 2008). See Figure 3 for the distribution of Ca+//0-binding EGF-like repeats in Notch receptors. Although not shown in this schematic, the three-dimensional structure of the Notch ECD might also change upon ligand binding and endocytosis
An alternative, but not mutually exclusive, explanation for the temperature-sensitive phenotypes of rumi comes from the possible role of inter- or intramolecular interactions of the Notch receptors in regulating the signaling (Xu et al. 2005; Pei and Baker 2008). A recent study by the Baker group showed that a fragment of Notch containing EGF-like repeats 11–20 bound another fragment of Notch containing EGF-like repeats 21–30 with a strength comparable to that between EGF-like repeats 11–20 and Delta (Pei and Baker 2008). Work from the Downing and Handford groups indicates that when neighboring EGF-like repeats are both Ca+//0-binding, they form a rigid, rod-like structure and that non-Ca+//0-binding EGF-like repeats can form flexible links with their neighboring EGF-like repeats (Downing et al. 1996; Hambleton et al. 2004). The distribution of the non-Ca+//0 binding EGF-like repeats of Notch suggests that the Ca+//0-binding EGF-like repeats 11–21 might form a rigid structure flanked by flexible links with non-Ca+//0-binding EGF-like repeats 10 and 22 (Figure 3) (Xu et al. 2005). Accordingly, it is possible that interaction between regions containing EGF11–20 and EGF21–30 of the same Notch molecule or other Notch molecules contributes to the stability of Notch in a protease-resistant conformation and/or its proper response to ligand binding. Given that the majority of EGF-like repeats in the 11–21 region are Rumi targets (Figure 3), loss of Rumi might affect the intra- or intermolecular Notch–Notch interactions and therefore result in a temperature-sensitive phenotype. Although speculative in nature, this model could potentially explain how ligand-independent activation of Notch in lgd mutants can be suppressed by rumi mutations (Acar et al. 2008).
As mentioned previously, Fringe proteins add a GlcNAc residue to the O-linked fucose and thereby regulate Notch signaling in some contexts. It is therefore reasonable to hypothesize that addition of xylose residues to O-linked glucose might also have a modulatory effect on Notch signaling. The Bakker group recently reported the identification and biochemical characterization of two human glucoside xylosyltransferases (GXylT1 and GXylT2) capable of adding a xylose residue to O-glucose (Figure 2) (Sethi et al. 2010). It would be interesting to examine whether these enzymes or their Drosophila homolog play a role in Notch signaling.
Is the function of Rumi conserved?
As mentioned above, mouse Notch1 is decorated with many O-linked glucose residues (Shao et al. 2002). As shown in Figure 3, all four human Notch receptors contain numerous predicted O-glucosylation sites. Moreover, several C1–X–S–X–P–C2 motifs are found in each human ligand, except for Delta-like3 (Figure 3). Therefore, given the evolutionary conservation of many aspects of Notch pathway regulation between flies and mammals (Fortini 2009; Kopan and Ilagan 2009), it is possible that Rumi homologs also regulate mammalian Notch signaling. Database searches indicate that mouse and human genomes contain three genes predicted to encode soluble, ER-recycling proteins with a CAP10 domain: CAP10-like protein 46 kDa (CLP46; also called KTELC1), KDEL-containing 1 (KDELC1; also called ER protein 58 or EP58) and KDELC2. CLP46 cDNA was originally isolated from CD34 hematopoietic stem/progenitor cells obtained from myelodysplastic syndrome patients who had entered the leukemic phase of the disease (Teng et al. 2006) and was recently reported to be overexpressed in peripheral blood cells of acute myeloblastic leukemia and T-cell acute lymphoblastic leukemia patients and in several leukemic cell lines (Wang et al. 2010). KDELC1 and CLP46 are broadly expressed in mammalian tissues (Kimata et al. 2000; Teng et al. 2006). Tagged versions of both proteins were shown to be localized to the ER upon overexpression in COS-7 cells (Kimata et al. 2000; Teng et al. 2006). However, biochemical and physiological functions of these proteins have not been reported yet. Preliminary data from our group and the Haltiwanger lab suggest that the function of Rumi might indeed be conserved. Further biochemical, cell culture and in vivo studies are needed to establish whether O-glucosylation is required for mammalian Notch signaling.
Nucleotide-sugar transporters and the regulation of Notch pathway
Glycosylation of Notch in ER and Golgi requires nucleotide sugars as substrates for the glycosyltransferases. Most nucleotide sugars are synthesized in the cytosol and transported into the lumen of ER and the Golgi apparatus by nucleotide-sugar transporters (NSTs) (Kawakita et al. 1998; Liu et al. 2010). Given the crucial roles that glycosylation plays in various aspects of Notch pathway regulation, at least some NSTs should be required for Notch signaling. Indeed, genetic studies in Drosophila have led to the identification of three NSTs involved in Notch signaling: the Golgi GDP-fucose transporter (Gfr) (Ishikawa et al. 2005), the ER GDP-fucose transporter (Efr) (Ishikawa et al. 2010) and fringe connection (frc), which encodes an NST involved in the regulation of several important signaling pathways, including the Notch pathway (Goto et al. 2001; Selva et al. 2001).
Fringe connection is an NST with broad substrate specificity and biological roles
Mutations in frc were identified in two independent genetic screens in flies (Goto et al. 2001; Selva et al. 2001). Removal of the maternal and zygotic components of frc in Drosophila embryos results in phenotypes compatible with loss of fibroblast growth factor (FGF) signaling and also an impairment in the positive feedback loop between Wingless and Hedgehog signaling pathways (Selva et al. 2001). These embryos also show an overproliferation of neural tissue at the expense of epidermal cells (Goto et al. 2001), a phenotype usually observed in Notch pathway mutants. Furthermore, loss of frc results in impaired Notch signaling in a variety of postembryonic developmental contexts, including wing margin and eye and leg development (Goto et al. 2001; Selva et al. 2001). Interestingly, many—but not all—of the Notch-related frc phenotypes resemble the fringe mutant phenotypes. Moreover, in the context of external mechanosensory organ formation in adult flies, removing one copy of frc strongly enhances the mild haploinsufficient phenotype of fringe (Goto et al. 2001; Selva et al. 2001). These observations strongly suggest that the function of Frc is required for proper glycosylation of Notch by Fringe.
Transport assays in a yeast cell-free system indicate that Frc has rather broad substrate specificity toward UDP-sugars and is able to mediate the transport of UDP-GalNAc, UDP-GlcA, UDP-Gal, UDP-Glc and UDP-GlcNAc into the lumen of microsomes (Table II) (Goto et al. 2001). A similar transport assay using Leishmania microsomal vesicles shows that Frc can serve as a transporter for UDP-GlcNAc, UDP-GlcA and UDP-Xyl (but not for UDP-Glc and UDP-Gal) (Selva et al. 2001). Homologs of Frc in human (hFrc1 and UGTrel7/SLC35D1) and C. elegans (Sqv-7) are also able to function as a transporter for various UDP-sugars (Suda et al. 2004; Hiraoka et al. 2007), although the substrate specificities of these homologous proteins are not identical.
Table II.
Substrate specificity of the Drosophila NSTs involved in the Notch pathway
Several of the UDP-sugars transported by Frc (UDP-Xyl, UDP-GlcA and UDP-GlcNAc) are required for the formation and elongation of the heparan sulfate proteoglycans (HSPGs). Moreover, genetic experiments in Drosophila have demonstrated a critical role for HSPGs in Wingless, Hedgehog and FGF signaling (Hacker et al. 2005). Finally, altering the level of Frc in Drosophila embryos or the level of its human homolog hFrc1 in a human cell line affects the expression of HSPGs (Selva et al. 2001; Suda et al. 2004). These observations strongly suggest that defects in HSPG biosynthesis underlie the impairments of FGF and Wingless/Hedgehog signaling pathways in frc mutants. As mentioned above, Fringe proteins add GlcNAc to the O-linked fucose on some Notch EGF-like repeats and thereby regulate both Drosophila and mammalian Notch signaling in some contexts (Rampal et al. 2007). The Golgi localization of Frc (Goto et al. 2001), its ability to transport UDP-GlcNAc into the secretory pathway (Goto et al. 2001; Selva et al. 2001), the similarity between many of the frc phenotypes and the fringe phenotypes, and the synergistic, dosage-sensitive interaction between frc and fringe mutants strongly suggest that Frc regulates Notch signaling primarily by providing the UDP-GlcNAc substrate required for the modification of the Notch receptor by Fringe. Interestingly, a recessive mutation in the bovine gene Slc35a3 results in a cattle disease called Complex Vertebral Malformation, which is characterized by multiple axial skeleton defects and other deformities in calves (Thomsen et al. 2006). Since this gene encodes a Golgi-resident UDP-GlcNAc transporter and because mutations in some Notch pathway components (including Lunatic fringe) result in axial skeletal defects in mice and human (Table I) (Evrard et al. 1998; Zhang and Gridley 1998), it is possible that defects in Notch glycosylation due to decreased levels of UDP-GlcNAc in the Golgi play a causative role in the pathophysiology of this disease (Thomsen et al. 2006).
It is important to note that ∼30% of the embryos lacking both maternal and zygotic frc exhibit a classic neurogenic phenotype, which indicates an impairment in the Notch-mediated lateral inhibition process in the embryonic nervous system. fringe mutants do not affect the lateral inhibition process (Haines and Irvine 2003). Therefore, another glycosylation event involved in the regulation of Notch signaling must rely on the transport of substrate sugar-nucleotides by Frc (Table II). A review of the literature suggests at least five potential mechanisms to explain the neurogenic phenotype of frc mutants: (1) Even though Frc is localized to the Golgi membrane, it might also provide UDP-Glc or UDP-Xyl for the ER glycosyltransferases via retrograde transport from Golgi, similar to what has recently been shown for GDP-fucose (Ishikawa et al. 2010). Therefore, Frc might be required for the addition of O-glucose to Notch by Rumi or its elongation by GxylT. We note, however, that unlike frc mutants, rumi mutant cells show a strong accumulation of the Notch receptor both intracellularly and at the cell surface. (2) RNAi-mediated knock-down of heparan sulfate 3-O sulfotransferase-B in flies results in loss of Notch signaling in various developmental contexts (Kamimura et al. 2004), suggesting that HSPGs might regulate Notch signaling. As mentioned above, the level of HSPGs is controlled by Frc. Accordingly, the Fringe-independent phenotypes of frc mutants might result from defects in HSPG biosynthesis. (3) Mutations in the Golgi glycosyltransferases brainiac and egghead result in neurogenic phenotypes in Drosophila embryos (Goode et al. 1992, 1996). Brainiac is a β3GlcNAc-transferase, which uses UDP-GlcNAc to catalyze an early step in glycosphingolipid (GSL) synthesis and is essential for GSL biosynthesis in flies (Schwientek et al. 2002; Wandall et al. 2005). Accordingly, it has been proposed that GSLs might be involved in Notch pathway regulation. Schweisguth and colleagues have recently reported that Drosophila Notch signaling is sensitive to the level of the N5 GSL at the membrane (Hamel et al. 2010). Therefore, defects in GSL synthesis due to a decrease in UDP-GlcNAc pool in the Golgi might contribute to the neurogenic phenotype observed in frc mutants. (4) Even though embryos lacking complex and hybrid N-glycans do not appear similar to embryos with impaired Notch signaling (Stanley 2007), milder Notch pathway defects have not been ruled out in these mutants. Biochemical and cell culture studies indicate that the mammalian Nicastrin protein needs to be decorated with mature N-linked glycans in order to participate in the active γ-secretase complex (Kimberly et al. 2002; Tomita et al. 2002). Therefore, a decrease in the ER/Golgi levels of UDP-GlcNAc, UDP-Glc and UDP-Gal in frc mutants could potentially affect Notch signaling by altering the N-glycosylation of the Notch pathway components. (5) Okajima and colleagues recently reported that several EGF-like repeats of the Drosophila Notch and at least one EGF-like repeat of Delta contain an O-linked GlcNAc on conserved serine/threonines between cysteines C5 and C6 (Figure 2) (Matsuura et al. 2008). If future work establishes functional importance for this unusual glycosylation, a loss or decrease in the level of O-GlcNAc on Notch can provide an alternative explanation for Fringe-independent phenotypes of frc. Of note, one of the Notch1 missense mutations identified in patients with bicuspid aortic valve (T596A) alters a threonine in EGF-like repeat 15 predicted to be O-GlcNAcylated (Mohamed et al. 2006).
ER- and Golgi-resident GDP-fucose transporters play a partially redundant role in Notch signaling
Further insight into the role of NSTs in the regulation of Notch pathway comes from biochemical and genetic analyses of two Drosophila GDP-fucose transporters by the Matsuno lab: Gfr (localized to the Golgi) (Ishikawa et al. 2005) and Efr (localized to the ER) (Ishikawa et al. 2010). Mutations in the human homolog of Gfr (SLC35C1) cause congenital disorders of glycosylation (CDG) Type IIc, which is characterized by slowed growth, mild dysmorphism, mental retardation and immunodeficiency (Lubke et al. 2001; Luhn et al. 2001). Loss of Slc35c1 in mice phenocopies some of the phenotypes observed in CDG IIc patients (Hellbusch et al. 2007). Flies homozygous for null alleles of Gfr are viable but show a cold-sensitive loss of wing margin (Ishikawa et al. 2005). Moreover, Gfr mutants exhibit genetic interaction with several genes involved in the Notch pathway. Gfr transports GDP-fucose into the lumen of Golgi (Ishikawa et al. 2005). Therefore, decreased Gfr function could affect O-linked fucosylation of proteins like Notch and/or terminal fucosylation of N-linked glycans. In Drosophila, loss of Gfr resulted in a severe decrease in terminal fucosylation of N-linked glycans on larval proteins, including the Notch receptor. Moreover, at restrictive temperature, Gfr mutants suppressed the gain-of-function phenotype resulting from Fringe overexpression, strongly suggesting a decrease in the level of O-linked fucose on Notch (Ishikawa et al. 2005). These observations indicate that loss of Gfr decreases the activity of the Notch pathway by reducing the supply of GDP-fucose in the secretory pathway.
As mentioned above, Gfr-null mutant flies are viable. Since Ofut1 is essential for Notch signaling and animal viability, another GDP-fucose transporter must compensate for the loss of Gfr in flies. Given that Pofut1/Ofut1 is a soluble ER protein, the alternative GDP-fucose transporter is predicted to reside in the ER membrane (Luo and Haltiwanger 2005; Okajima et al. 2005). Indeed, the Matsuno group has recently reported the identification and characterization of an ER GDP-fucose transporter, Efr (Figure 4) (Ishikawa et al. 2010). Unlike Gfr, which is fairly selective to GDP-fucose, biochemical assays indicate that Efr is able to transport GDP-fucose, UDP-GlcNAc and UDP-xylose (but not CMP-mannose or UDP-glucose) (Table II). Null alleles of Efr are homozygous lethal. Genetic experiments indicate that Efr and Gfr together provide the GDP-fucose required for the O-fucosylation of Notch by Ofut1 and that the weak phenotypes of Gfr are due to compensation by Efr. Indeed, even at 25°C—which is permissive for Gfr mutations—Efr; Gfr double-mutant animals completely lose the expression of the Notch downstream target Wingless in the wing margin (Ishikawa et al. 2010). Furthermore, the gain-of-function phenotype caused by Fringe overexpression is fully suppressed in Efr; Gfr double-mutant imaginal discs (Ishikawa et al. 2010). Since the function of Fringe is strictly dependent on the presence of O-linked fucose on Notch (Sasamura et al. 2003), these observations indicate a lack of Notch O-fucosylation in the absence of both Efr and Gfr. Moreover, in these assays, Efr; Gfr double mutants behave identically to animals mutant for gmd, which is essential for GDP-fucose synthesis in flies (Okajima et al. 2005; Sasamura et al. 2007). Altogether, these results indicate that Efr and Gfr play a redundant role in the transport of GDP-fucose into the secretory pathway and in the regulation of those aspects of the Notch signaling pathway which depend on the presence of O-linked fucose on Notch.
Efr and Gfr only show 10% sequence identity. Interestingly, analysis of null mutations in another Drosophila NST with significantly higher sequence similarity to Gfr, CG14971, does not show any role for this protein in GDP-fucose transport (Ishikawa et al. 2010). This provides further evidence for the notion that the primary sequence of NSTs has a limited predictive value for their substrate specificities (Berninsone and Hirschberg 2000). The observation that a Golgi NST (Gfr) is required for the function of an ER glycosyltransferase (Ofut1) suggests that retrograde vesicular transport from Golgi to the ER might be involved in the transport of sugar-nucleotides (Figure 4).
The Gfr phenotypes in flies can be rescued by transgenic expression of the human SLC35C1 (Ishikawa et al. 2005). Also, shRNA-mediated knock-down of mouse Gfr results in a significant decrease in Notch pathway activation in C2C12 mouse myoblast cells (Ishikawa et al. 2005). These observations indicate functional conservation between Gfr and its mammalian homologs and suggest that a reduction in Notch signaling might underlie some of the abnormalities observed in CDG IIc patients. It is worth mentioning though that biochemical experiments on a fibroblast cell line established from a CDG IIc patient showed a severe decrease in the bulk addition of terminal fucose to N-glycans but no changes in the level of O-linked saccharides containing fucose (Sturla et al. 2003). Specifically, the level of Notch1 O-fucosylation seemed similar in CDG and control fibroblasts. Accordingly, it was suggested that at least in some CDG IIc patients, a decrease in terminal fucosylation of N-glycans but not a decrease in O-linked fucosylation of proteins is probably responsible for the disease phenotypes (Sturla et al. 2003).
There are at least 17 human proteins in the solute carrier family SLC35—which comprises NSTs—with homologs in other vertebrate and invertebrate species (Ishida and Kawakita 2004). Even though Gfr, Efr and Frc are so far the only NSTs for which an important role in the Notch pathway has been reported, it is possible that other NSTs are also involved in the regulation of this pathway. As suggested by the analysis of Gfr and Efr mutants, redundancy between various NSTs might complicate the analysis of single gene NST mutants in the regulation of the Notch pathway. Nevertheless, the observation that loss of one copy of frc results in a significant enhancement of the heterozygous phenotypes of several Notch pathway components strongly suggests that at least in Drosophila, Notch signaling is sensitive to the level of UDP-sugars in the secretory pathway and that altering the level of these sugar donors in the Golgi (and possibly ER) can change the efficiency of Notch glycosylation and signaling.
Conclusions
“Look what a grain of sugar does to my intoxicated soul!”—Rumi (translated from Persian)
Starting with two landmark reports on the glycosyltransferase activity of Fringe (Bruckner et al. 2000; Moloney, Panin, et al. 2000), the body of evidence published during the last decade on the glycobiology of Notch leaves no doubt that glycosylation of the Notch receptors is a critical mechanism for regulation of this pathway in animals. Genetic studies in model organisms combined with biochemical experiments have shown that lack of O-linked glycans can have profound effects on the activity of the Notch pathway and the development of organisms. However, analysis of the effects of abolishing O-fucosylation sites and loss of Pofut1 in several animal and cellular models has started to show that, although the role of O-fucose glycans in Notch pathway is evolutionarily conserved, the way these carbohydrates regulate Notch at molecular and cellular levels might not always be similar between invertebrates and vertebrates (Lei et al. 2003; Okajima et al. 2005; Rampal, Arboleda-Velasquez, et al. 2005; Ge and Stanley 2008; Stahl et al. 2008). Many important questions remain unanswered. Controversy in the cell biological mechanisms of Notch modulation by O-fucose and its elongated forms still exists, even between reports studying the same model organism (Hicks et al. 2000; Shimizu et al. 2001; Okajima et al. 2005; Sasaki et al. 2007; Sasamura et al. 2007; Okajima, Reddy, et al. 2008; Okamura and Saga 2008; Stahl et al. 2008). Most of the analysis on the profile of carbohydrates on Notch has been performed on Notch fragments overexpressed in cell lines. Therefore, the glycosylation pattern of the full-length Notch expressed at endogenous levels needs to be analyzed. What’s more, the level of occupancy of various predicted O-fucosylation and O-glucosylation sites on different Notch receptors has not been explored in vivo, and little is known about tissue- or cell type-specific variations in the distribution of O-linked glycans on Notch proteins. The effects of addition and elongation of O-linked carbohydrates on the three-dimensional structure of Notch EGF-like repeats in isolation or in the context of the full-length Notch is not clear. The recent identification of several novel glycosyltransferases that modify Notch provides new avenues for studying the glycobiology of this pathway (Acar et al. 2008; Sethi et al. 2010). It is not known how O-linked glucose regulates fly Notch signaling, whether it is evolutionarily conserved and whether its elongation plays a context-specific role in the pathway (like the addition of GlcNAc to O-fucose by Fringe). These and other questions will keep those of us interested in studying the glycobiology of Notch busy for the next few years. We anticipate that collaboration among geneticists, biochemists and structural biologists will continue to push this exciting field forward.
Acknowledgments
We thank Robert Haltiwanger, Tom Van Lee and Hideyuki Takeuchi for comments on the manuscript. We are also grateful to Robert Haltiwanger for sharing unpublished observations with us. We wish to apologize to those colleagues whose work on the glycobiology of Notch signaling has not been covered here.
Glossary
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- α2,3SA-T
α2,3sialyltransferase
- β4GalT-1
β4galactosyltransferase-1
- CBF1
C promoter binding factor 1
- CDG
congenital disorder of glycosylation
- CG14971
computed gene 14971
- CLP46
CAP10-like protein 46 kDa
- CHO
Chinese hamster ovary
- CR
cysteine-rich
- DSL
Delta-Serrate-Lag2
- Efr
ER GDP-fucose transporter
- EGF
epidermal growth factor
- ER
Endoplasmic reticulum
- ES cells
Embryonic stem cells
- FGF
Fibroblast growth factor
- FK
fucose kinase
- frc
fringe connection
- Fuc
fucose
- Fx
GDP-4-keto-6-deoxymannose 3,5-epimerase-4-reductase
- Gal
galactose
- GalNAc
N-acetylgalactosamine
- GDP
guanosine diphosphate
- Gfr
Golgi GDP-fucose transporter
- Glc
glucose
- GlcA
glucoronic acid
- GlcNAc
N-acetylglucosamine
- GMD
GDP-d-mannose dehydratase
- GPP
GDP-fucose pyrophosphorylase
- GSL
glycosphingolipid
- GXylT
glucoside xylosyltransferase
- HD
heterodimerization
- Hes
Hairy-Enhancer of Split
- Hey
Hairy-Enhancer of Split related with YRPW motif
- HSPG
heparan sulfate proteoglycans
- Jak-STAT
Janus kinase/signal transducers and activators of transcription
- KDELC1
KDEL-containing 1
- KDELC2
KDEL-containing 2
- lgd
lethal giant discs
- LNR
Lin12-Notch repeat
- N-AP
Notch-alkaline phosphatase
- NECD
Notch extracellular domain
- NEXT
Notch extracellular truncation
- NICD
Notch intracellular domain
- NRR
negative regulatory region
- NST
nucleotide-sugar transporter
- Ofut1
the Drosophila protein O-fucosyltransferase
- Pofut1
the mammalian protein O-fucosyltransferase 1
- RAM
RBP-J association module
- RBP-J
Recombination signal binding protein for immunoglobulin kappa J region
- S2 cells
Drosophila Schneider 2 cells
- SA
sialic acid
- SLC35
solute carrier 35
- Su(H)
Suppressor of Hairless
- Xyl
xylose
Funding
Funding was provided by the National Institute of Health (R01GM084135 to H.J.-N.) and the March of Dimes Foundation (Basil O’Connor Starter Scholar Research Award No. 5-FY07-654 to H.J.-N.).
References
- Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, Pan H, Haltiwanger RS, Bellen HJ. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247–258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker H, Oka T, Ashikov A, Yadav A, Berger M, Rana NA, Bai X, Jigami Y, Haltiwanger RS, Esko JD, et al. Functional UDP-xylose transport across the endoplasmic reticulum/Golgi membrane in a Chinese hamster ovary cell mutant defective in UDP-xylose synthase. J Biol Chem. 2009;284:2576–2583. doi: 10.1074/jbc.M804394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barolo S, Posakony JW. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 2002;16:1167–1181. doi: 10.1101/gad.976502. [DOI] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Berninsone PM, Hirschberg CB. Nucleotide sugar transporters of the Golgi apparatus. Curr Opin Struct Biol. 2000;10:542–547. doi: 10.1016/s0959-440x(00)00128-7. [DOI] [PubMed] [Google Scholar]
- Bjoern S, Foster DC, Thim L, Wiberg FC, Christensen M, Komiyama Y, Pedersen AH, Kisiel W. Human plasma and recombinant factor VII. Characterization of O-glycosylations at serine residues 52 and 60 and effects of site-directed mutagenesis of serine 52 to alanine. J Biol Chem. 1991;266:11051–11057. [PubMed] [Google Scholar]
- Bolt G, Steenstrup TD, Kristensen C. All post-translational modifications except propeptide cleavage are required for optimal secretion of coagulation factor VII. Thromb Haemost. 2007;98:988–997. doi: 10.1160/th07-05-0332. [DOI] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- Bulman MP, Kusumi K, Frayling TM, McKeown C, Garrett C, Lander ES, Krumlauf R, Hattersley AT, Ellard S, Turnpenny PD. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;24:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- Cagavi Bozkulak E, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29:5679–5695. doi: 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Conboy IM. Regulating the Notch pathway in embryonic, adult and old stem cells. Curr Opin Pharmacol. 2007;7:303–309. doi: 10.1016/j.coph.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Chang YC, Kwon-Chung KJ. Isolation, characterization, and localization of a capsule-associated gene, CAP10, of Cryptococcus neoformans. J Bacteriol. 1999;181:5636–5643. doi: 10.1128/jb.181.18.5636-5643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, et al. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci U S A. 2009;106:13838–13843. doi: 10.1073/pnas.0907008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lu L, Shi S, Stanley P. Expression of Notch signaling pathway genes in mouse embryos lacking beta4galactosyltransferase-1. Gene Expr Patterns. 2006;6:376–382. doi: 10.1016/j.modgep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Chen J, Moloney DJ, Stanley P. Fringe modulation of Jagged1-induced Notch signaling requires the action of beta 4galactosyltransferase-1. Proc Natl Acad Sci U S A. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- Cho KO, Choi KW. Fringe is essential for mirror symmetry and morphogenesis in the Drosophila eye. Nature. 1998;396:272–276. doi: 10.1038/24394. [DOI] [PubMed] [Google Scholar]
- Dexter JS. The analysis of a case of continuous variation in Drosophila by a study of its linkage relations. The American Naturalist. 1914;48:712–758. [Google Scholar]
- Dominguez M, de Celis JF. A dorsal/ventral boundary established by Notch controls growth and polarity in the Drosophila eye. Nature. 1998;396:276–278. doi: 10.1038/24402. [DOI] [PubMed] [Google Scholar]
- Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID, Handford PA. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the Marfan syndrome and other genetic disorders. Cell. 1996;85:597–605. doi: 10.1016/s0092-8674(00)81259-3. [DOI] [PubMed] [Google Scholar]
- Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001;10:163–169. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- Evrard YA, Lun Y, Aulehla A, Gan L, Johnson RL. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 1998;394:377–381. doi: 10.1038/28632. [DOI] [PubMed] [Google Scholar]
- Fleming RJ, Gu Y, Hukriede NA. Serrate-mediated activation of Notch is specifically blocked by the product of the gene fringe in the dorsal compartment of the Drosophila wing imaginal disc. Development. 1997;124:2973–2981. doi: 10.1242/dev.124.15.2973. [DOI] [PubMed] [Google Scholar]
- Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–1411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- Gazave E, Lapebie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, Borchiellini C, Vervoort M, Renard E. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol Biol. 2009;9:249. doi: 10.1186/1471-2148-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Stanley P. The O-fucose glycan in the ligand-binding domain of Notch1 regulates embryogenesis and T cell development. Proc Natl Acad Sci U S A. 2008;105:1539–1544. doi: 10.1073/pnas.0702846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J. 1998 Warkany lecture: signaling pathways in development. Teratology. 1999;60:226–239. doi: 10.1002/(SICI)1096-9926(199910)60:4<226::AID-TERA7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Goode S, Melnick M, Chou TB, Perrimon N. The neurogenic genes egghead and brainiac define a novel signaling pathway essential for epithelial morphogenesis during Drosophila oogenesis. Development. 1996;122:3863–3879. doi: 10.1242/dev.122.12.3863. [DOI] [PubMed] [Google Scholar]
- Goode S, Wright D, Mahowald AP. The neurogenic locus brainiac cooperates with the Drosophila EGF receptor to establish the ovarian follicle and to determine its dorsal-ventral polarity. Development. 1992;116:177–192. doi: 10.1242/dev.116.1.177. [DOI] [PubMed] [Google Scholar]
- Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling—a structural and biochemical perspective. J Cell Sci. 2008;121:3109–3119. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, Blacklow SC. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood. 2009;113:4381–4390. doi: 10.1182/blood-2008-08-174748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007;14:295–300. doi: 10.1038/nsmb1227. [DOI] [PubMed] [Google Scholar]
- Gordon WR, Vardar-Ulu D, L'Heureux S, Ashworth T, Malecki MJ, Sanchez-Irizarry C, McArthur DG, Histen G, Mitchell JL, Aster JC, et al. Effects of S1 cleavage on the structure, surface export, and signaling activity of human Notch1 and Notch2. PLoS ONE. 2009;4:e6613. doi: 10.1371/journal.pone.0006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Taniguchi M, Muraoka M, Toyoda H, Sado Y, Kawakita M, Hayashi S. UDP-sugar transporter implicated in glycosylation and processing of Notch. Nat Cell Biol. 2001;3:816–822. doi: 10.1038/ncb0901-816. [DOI] [PubMed] [Google Scholar]
- Greenwald I. lin-12, a nematode homeotic gene, is homologous to a set of mammalian proteins that includes epidermal growth factor. Cell. 1985;43:583–590. doi: 10.1016/0092-8674(85)90230-2. [DOI] [PubMed] [Google Scholar]
- Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell. 1983;34:435–444. doi: 10.1016/0092-8674(83)90377-x. [DOI] [PubMed] [Google Scholar]
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol. 2003;4:786–797. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- Haines N, Irvine KD. Functional analysis of Drosophila beta1, 4-N-acetlygalactosaminyltransferases. Glycobiology. 2005;15:335–346. doi: 10.1093/glycob/cwi017. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS. Regulation of signal transduction pathways in development by glycosylation. Curr Opin Struct Biol. 2002;12:593–598. doi: 10.1016/s0959-440x(02)00371-8. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- Hambleton S, Valeyev NV, Muranyi A, Knott V, Werner JM, McMichael AJ, Handford PA, Downing AK. Structural and functional properties of the human notch-1 ligand binding region. Structure. 2004;12:2173–2183. doi: 10.1016/j.str.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Hamel S, Fantini J, Schweisguth F. Notch ligand activity is modulated by glycosphingolipid membrane composition in Drosophila melanogaster. J Cell Biol. 2010;188:581–594. doi: 10.1083/jcb.200907116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RJ, Spellman MW. O-linked fucose and other post-translational modifications unique to EGF modules. Glycobiology. 1993;3:219–224. doi: 10.1093/glycob/3.3.219. [DOI] [PubMed] [Google Scholar]
- Hase S, Kawabata S, Nishimura H, Takeya H, Sueyoshi T, Miyata T, Iwanaga S, Takao T, Shimonishi Y, Ikenaka T. A new trisaccharide sugar chain linked to a serine residue in bovine blood coagulation factors VII and IX. J Biochem (Tokyo) 1988;104:867–868. doi: 10.1093/oxfordjournals.jbchem.a122571. [DOI] [PubMed] [Google Scholar]
- Hase S, Nishimura H, Kawabata S, Iwanaga S, Ikenaka T. The structure of (xylose)2glucose-O-serine 53 found in the first epidermal growth factor-like domain of bovine blood clotting factor IX. J Biol Chem. 1990;265:1858–1861. [PubMed] [Google Scholar]
- Hellbusch CC, Sperandio M, Frommhold D, Yakubenia S, Wild MK, Popovici D, Vestweber D, Grone HJ, von Figura K, Lubke T, et al. Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation IIc/leukocyte adhesion deficiency II. J Biol Chem. 2007;282:10762–10772. doi: 10.1074/jbc.M700314200. [DOI] [PubMed] [Google Scholar]
- Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat Cell Biol. 2000;2:515–520. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- Hiraoka S, Furuichi T, Nishimura G, Shibata S, Yanagishita M, Rimoin DL, Superti-Furga A, Nikkels PG, Ogawa M, Katsuyama K, et al. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat Med. 2007;13:1363–1367. doi: 10.1038/nm1655. [DOI] [PubMed] [Google Scholar]
- Huang LH, Ke XH, Sweeney W, Tam JP. Calcium binding and putative activity of the epidermal growth factor domain of blood coagulation factor IX. Biochem Biophys Res Commun. 1989;160:133–139. doi: 10.1016/0006-291x(89)91631-8. [DOI] [PubMed] [Google Scholar]
- Irvine KD. A notch sweeter. Cell. 2008;132:177–179. doi: 10.1016/j.cell.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Irvine KD, Wieschaus E. fringe, a Boundary-specific signaling molecule, mediates interactions between dorsal and ventral cells during Drosophila wing development. Cell. 1994;79:595–606. doi: 10.1016/0092-8674(94)90545-2. [DOI] [PubMed] [Google Scholar]
- Ishida N, Kawakita M. Molecular physiology and pathology of the nucleotide sugar transporter family (SLC35) Pflugers Arch. 2004;447:768–775. doi: 10.1007/s00424-003-1093-0. [DOI] [PubMed] [Google Scholar]
- Ishikawa HO, Ayukawa T, Nakayama M, Higashi S, Kamiyama S, Nishihara S, Aoki K, Ishida N, Sanai Y, Matsuno K. Two pathways for importing GDP-fucose into the endoplasmic reticulum lumen function redundantly in the O-fucosylation of Notch in Drosophila. J Biol Chem. 2010;285:4122–4129. doi: 10.1074/jbc.M109.016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa HO, Higashi S, Ayukawa T, Sasamura T, Kitagawa M, Harigaya K, Aoki K, Ishida N, Sanai Y, Matsuno K. Notch deficiency implicated in the pathogenesis of congenital disorder of glycosylation IIc. Proc Natl Acad Sci U S A. 2005;102:18532–18537. doi: 10.1073/pnas.0504115102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimizu T, Sano K, Uchida T, Teshima H, Omichi K, Hojo H, Nakahara Y, Hase S. Purification and substrate specificity of UDP-D-xylose:beta-D-glucoside alpha-1, 3-D-xylosyltransferase involved in the biosynthesis of the Xyl alpha1–3Xyl alpha1–3Glc beta1-O-Ser on epidermal growth factor-like domains. J Biochem. 2007;141:593–600. doi: 10.1093/jb/mvm064. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Andrews HK, Acar M, Bayat V, Wirtz-Peitz F, Mehta SQ, Knoblich JA, Bellen HJ. Sec15, a component of the exocyst, promotes notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev Cell. 2005;9:351–363. doi: 10.1016/j.devcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Johansen KM, Fehon RG, Artavanis-Tsakonas S. The notch gene product is a glycoprotein expressed on the cell surface of both epidermal and neuronal precursor cells during Drosophila development. J Cell Biol. 1989;109:2427–2440. doi: 10.1083/jcb.109.5.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Kamimura K, Rhodes JM, Ueda R, McNeely M, Shukla D, Kimata K, Spear PG, Shworak NW, Nakato H. Regulation of Notch signaling by Drosophila heparan sulfate 3-O sulfotransferase. J Cell Biol. 2004;166:1069–1079. doi: 10.1083/jcb.200403077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakita M, Ishida N, Miura N, Sun-Wada GH, Yoshioka S. Nucleotide sugar transporters: elucidation of their molecular identity and its implication for future studies. J Biochem. 1998;123:777–785. doi: 10.1093/oxfordjournals.jbchem.a022004. [DOI] [PubMed] [Google Scholar]
- Kentzer EJ, Buko A, Menon G, Sarin VK. Carbohydrate composition and presence of a fucose-protein linkage in recombinant human pro-urokinase. Biochem Biophys Res Commun. 1990;171:401–406. doi: 10.1016/0006-291x(90)91407-j. [DOI] [PubMed] [Google Scholar]
- Kidd S, Kelley MR, Young MW. Sequence of the notch locus of Drosophila melanogaster: relationship of the encoded protein to mammalian clotting and growth factors. Mol Cell Biol. 1986;6:3094–3108. doi: 10.1128/mcb.6.9.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Irvine KD, Carroll SB. Cell recognition, signal induction, and symmetrical gene activation at the dorsal–ventral boundary of the developing Drosophila wing. Cell. 1995;82:795–802. doi: 10.1016/0092-8674(95)90476-x. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ooboki K, Nomura-Furuwatari C, Hosoda A, Tsuru A, Kohno K. Identification of a novel mammalian endoplasmic reticulum-resident KDEL protein using an EST database motif search. Gene. 2000;261:321–327. doi: 10.1016/s0378-1119(00)00499-6. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Complex N-linked glycosylated nicastrin associates with active gamma-secretase and undergoes tight cellular regulation. J Biol Chem. 2002;277:35113–35117. doi: 10.1074/jbc.M204446200. [DOI] [PubMed] [Google Scholar]
- Klutts JS, Levery SB, Doering TL. A beta-1, 2-xylosyltransferase from Cryptococcus neoformans defines a new family of glycosyltransferases. J Biol Chem. 2007;282:17890–17899. doi: 10.1074/jbc.M701941200. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld K, Reitman ML, Kornfeld R. The carbohydrate-binding specificity of pea and lentil lectins. Fucose is an important determinant. J Biol Chem. 1981;256:6633–6640. [PubMed] [Google Scholar]
- Lake RJ, Grimm LM, Veraksa A, Banos A, Artavanis-Tsakonas S. In vivo analysis of the Notch receptor S1 cleavage. PLoS ONE. 2009;4:e6728. doi: 10.1371/journal.pone.0006728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Caignec C, Lefevre M, Schott JJ, Chaventre A, Gayet M, Calais C, Moisan JP. Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal-growth-factor-like domain of jagged 1. Am J Hum Genet. 2002;71:180–186. doi: 10.1086/341327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecourtois M, Schweisguth F. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Genes Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100:920–926. doi: 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Xu A, Panin VM, Irvine KD. An O-fucose site in the ligand binding domain inhibits Notch activation. Development. 2003;130:6411–6421. doi: 10.1242/dev.00883. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- Li Y, Lei L, Irvine KD, Baker NE. Notch activity in neural cells triggered by a mutant allele with altered glycosylation. Development. 2003;130:2829–2840. doi: 10.1242/dev.00498. [DOI] [PubMed] [Google Scholar]
- Liu L, Xu YX, Hirschberg CB. The role of nucleotide sugar transporters in development of eukaryotes. Semin Cell Dev Biol. 2010 doi: 10.1016/j.semcdb.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Hu B, Choi YY, Chung M, Ullenbruch M, Yu H, Lowe JB, Phan SH. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol. 2009;174:1745–1755. doi: 10.2353/ajpath.2009.080618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A. 1998;95:8108–8112. doi: 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loriol C, Audfray A, Dupuy F, Germot A, Maftah A. The two N-glycans present on bovine Pofut1 are differently involved in its solubility and activity. FEBS J. 2007;274:1202–1211. doi: 10.1111/j.1742-4658.2007.05663.x. [DOI] [PubMed] [Google Scholar]
- Lubke T, Marquardt T, Etzioni A, Hartmann E, von Figura K, Korner C. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet. 2001;28:73–76. doi: 10.1038/ng0501-73. [DOI] [PubMed] [Google Scholar]
- Luhn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet. 2001;28:69–72. doi: 10.1038/ng0501-69. [DOI] [PubMed] [Google Scholar]
- Luo Y, Haltiwanger RS. O-fucosylation of notch occurs in the endoplasmic reticulum. J Biol Chem. 2005;280:11289–11294. doi: 10.1074/jbc.M414574200. [DOI] [PubMed] [Google Scholar]
- Luther KB, Haltiwanger RS. Role of unusual O-glycans in intercellular signaling. Int J Biochem Cell Biol. 2009;41:1011–1024. doi: 10.1016/j.biocel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26:4642–4651. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, Nadano D, Matsuda T, Okajima T. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem. 2008;283:35486–35495. doi: 10.1074/jbc.M806202200. [DOI] [PubMed] [Google Scholar]
- McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, Cole SE. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet. 2008;17:2886–2893. doi: 10.1093/hmg/ddn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew MJ, Dale JK, Fraboulet S, Pourquie O. The lunatic fringe gene is a target of the molecular clock linked to somite segmentation in avian embryos. Curr Biol. 1998;8:979–982. doi: 10.1016/s0960-9822(98)70401-4. [DOI] [PubMed] [Google Scholar]
- McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM., 3rd Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134:290–296. doi: 10.1016/j.jtcvs.2007.02.041. [DOI] [PubMed] [Google Scholar]
- Minamida S, Aoki K, Natsuka S, Omichi K, Fukase K, Kusumoto S, Hase S. Detection of UDP-d-xylose: alpha-d-xyloside alpha 1–>3xylosyltransferase activity in human hepatoma cell line HepG2. J Biochem. 1996;120:1002–1006. doi: 10.1093/oxfordjournals.jbchem.a021492. [DOI] [PubMed] [Google Scholar]
- Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH, Erdmann J. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. 2006;345:1460–1465. doi: 10.1016/j.bbrc.2006.05.046. [DOI] [PubMed] [Google Scholar]
- Mohr OL. Character changes caused by mutation of an entire region of a chromosome in Drosophila. Genetics. 1919;4:275–282. doi: 10.1093/genetics/4.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney DJ, Lin AI, Haltiwanger RS. The O-linked fucose glycosylation pathway. Evidence for protein-specific elongation of o-linked fucose in Chinese hamster ovary cells. J Biol Chem. 1997;272:19046–19050. doi: 10.1074/jbc.272.30.19046. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, et al. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem. 2000;275:9604–9611. doi: 10.1074/jbc.275.13.9604. [DOI] [PubMed] [Google Scholar]
- Moran JL, Shifley ET, Levorse JM, Mani S, Ostmann K, Perez-Balaguer A, Walker DM, Vogt TF, Cole SE. Manic fringe is not required for embryonic development, and fringe family members do not exhibit redundant functions in the axial skeleton, limb, or hindbrain. Dev Dyn. 2009;238:1803–1812. doi: 10.1002/dvdy.21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TH, Bridges CB. Sex-linked inheritance in Drosophila. Publs Carnegie Instn. 1916;237:1–88. [Google Scholar]
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Nichols JT, Miyamoto A, Olsen SL, D'Souza B, Yao C, Weinmaster G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J Cell Biol. 2007;176:445–458. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Kawabata S, Kisiel W, Hase S, Ikenaka T, Takao T, Shimonishi Y, Iwanaga S. Identification of a disaccharide (Xyl-Glc) and a trisaccharide (Xyl2-Glc) O-glycosidically linked to a serine residue in the first epidermal growth factor-like domain of human factors VII and IX and protein Z and bovine protein Z. J Biol Chem. 1989;264:20320–20325. [PubMed] [Google Scholar]
- Nishimura H, Yamashita S, Zeng Z, Walz DA, Iwanaga S. Evidence for the existence of O-linked sugar chains consisting of glucose and xylose in bovine thrombospondin. J Biochem (Tokyo) 1992;111:460–464. doi: 10.1093/oxfordjournals.jbchem.a123780. [DOI] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- Okabayashi K, Hasegawa A, Watanabe T. Microreview: capsule-associated genes of Cryptococcus neoformans. Mycopathologia. 2007;163:1–8. doi: 10.1007/s11046-006-0083-0. [DOI] [PubMed] [Google Scholar]
- Okajima T, Irvine KD. Regulation of notch signaling by O-linked fucose. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- Okajima T, Matsuura A, Matsuda T. Biological functions of glycosyltransferase genes involved in O-fucose glycan synthesis. J Biochem. 2008;144:1–6. doi: 10.1093/jb/mvn016. [DOI] [PubMed] [Google Scholar]
- Okajima T, Reddy B, Matsuda T, Irvine KD. Contributions of chaperone and glycosyltransferase activities of O-fucosyltransferase 1 to Notch signaling. BMC Biol. 2008;6:1. doi: 10.1186/1741-7007-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okajima T, Xu A, Irvine KD. Modulation of notch-ligand binding by protein O-fucosyltransferase 1 and fringe. J Biol Chem. 2003;278:42340–42345. doi: 10.1074/jbc.M308687200. [DOI] [PubMed] [Google Scholar]
- Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science. 2005;307:1599–1603. doi: 10.1126/science.1108995. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Saga Y. Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech Dev. 2008;125:663–673. doi: 10.1016/j.mod.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Omichi K, Aoki K, Minamida S, Hase S. Presence of UDP-d-xylose: beta-d-glucoside alpha-1, 3-d-xylosyltransferase involved in the biosynthesis of the Xyl alpha 1–3Glc beta-Ser structure of glycoproteins in the human hepatoma cell line HepG2. Eur J Biochem. 1997;245:143–146. doi: 10.1111/j.1432-1033.1997.00143.x. [DOI] [PubMed] [Google Scholar]
- Overstreet E, Fitch E, Fischer JA. Fat facets and liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development. 2004;131:5355–5366. doi: 10.1242/dev.01434. [DOI] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates Notch-ligand interactions. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- Panin VM, Shao L, Lei L, Moloney DJ, Irvine KD, Haltiwanger RS. Notch ligands are substrates for protein O-fucosyltransferase-1 and Fringe. J Biol Chem. 2002;277:29945–29952. doi: 10.1074/jbc.M204445200. [DOI] [PubMed] [Google Scholar]
- Parks AL, Klueg KM, Stout JR, Muskavitch MA. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127:1373–1385. doi: 10.1242/dev.127.7.1373. [DOI] [PubMed] [Google Scholar]
- Pei Z, Baker NE. Competition between Delta and the Abruptex domain of Notch. BMC Dev Biol. 2008;8:4. doi: 10.1186/1471-213X-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petcherski AG, Kimble J. LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature. 2000;405:364–368. doi: 10.1038/35012645. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Mott R, Bork P, Copley RR. Novel protein domains and repeats in Drosophila melanogaster: insights into structure, function, and evolution. Genome Res. 2001;11:1996–2008. doi: 10.1101/gr.198701. [DOI] [PubMed] [Google Scholar]
- Poulson DF. The effects of certain X-chromosome deficiencies on the embryonic development of Drosophila melanogaster. J Exp Zool. 1940;83:271–325. [Google Scholar]
- Rampal R, Arboleda-Velasquez JF, Nita-Lazar A, Kosik KS, Haltiwanger RS. Highly conserved O-fucose sites have distinct effects on Notch1 function. J Biol Chem. 2005;280:32133–32140. doi: 10.1074/jbc.M506104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R, Li AS, Moloney DJ, Georgiou SA, Luther KB, Nita-Lazar A, Haltiwanger RS. Lunatic fringe, manic fringe, and radical fringe recognize similar specificity determinants in O-fucosylated epidermal growth factor-like repeats. J Biol Chem. 2005;280:42454–42463. doi: 10.1074/jbc.M509552200. [DOI] [PubMed] [Google Scholar]
- Rampal R, Luther KB, Haltiwanger RS. Notch signaling in normal and disease states: possible therapies related to glycosylation. Curr Mol Med. 2007;7:427–445. doi: 10.2174/156652407780831593. [DOI] [PubMed] [Google Scholar]
- Rauskolb C, Irvine KD. Notch-mediated segmentation and growth control of the Drosophila leg. Dev Biol. 1999;210:339–350. doi: 10.1006/dbio.1999.9273. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Bales C, Nelson A, Gonzalez DM, Underkoffler L, Segalov M, Wilson-Rawls J, Cole SE, Moran JL, Russo P, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology. 2008;48:1989–1997. doi: 10.1002/hep.22538. [DOI] [PubMed] [Google Scholar]
- Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol. 2004;24:9265–9273. doi: 10.1128/MCB.24.21.9265-9273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Sasamura T, Ishikawa HO, Kanai M, Ueda R, Saigo K, Matsuno K. Polarized exocytosis and transcytosis of Notch during its apical localization in Drosophila epithelial cells. Genes Cells. 2007;12:89–103. doi: 10.1111/j.1365-2443.2007.01037.x. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Ishikawa HO, Sasaki N, Higashi S, Kanai M, Nakao S, Ayukawa T, Aigaki T, Noda K, Miyoshi E, et al. The O-fucosyltransferase O-fut1 is an extracellular component that is essential for the constitutive endocytic trafficking of Notch in Drosophila. Development. 2007;134:1347–1356. doi: 10.1242/dev.02811. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Sasaki N, Miyashita F, Nakao S, Ishikawa HO, Ito M, Kitagawa M, Harigaya K, Spana E, Bilder D, et al. neurotic, a novel maternal neurogenic gene, encodes an O-fucosyltransferase that is essential for Notch-Delta interactions. Development. 2003;130:4785–4795. doi: 10.1242/dev.00679. [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Schuster-Gossler K, Harris B, Johnson KR, Serth J, Gossler A. Notch signalling in the paraxial mesoderm is most sensitive to reduced Pofut1 levels during early mouse development. BMC Dev Biol. 2009;9:6. doi: 10.1186/1471-213X-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwientek T, Keck B, Levery SB, Jensen MA, Pedersen JW, Wandall HH, Stroud M, Cohen SM, Amado M, Clausen H. The Drosophila gene brainiac encodes a glycosyltransferase putatively involved in glycosphingolipid synthesis. J Biol Chem. 2002;277:32421–32429. doi: 10.1074/jbc.M206213200. [DOI] [PubMed] [Google Scholar]
- Selva EM, Hong K, Baeg GH, Beverley SM, Turco SJ, Perrimon N, Hacker U. Dual role of the fringe connection gene in both heparan sulphate and fringe-dependent signalling events. Nat Cell Biol. 2001;3:809–815. doi: 10.1038/ncb0901-809. [DOI] [PubMed] [Google Scholar]
- Sethi MK, Buettner FF, Krylov VB, Takeuchi H, Nifantiev NE, Haltiwanger RS, Gerardy-Schahn R, Bakker H. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on O-glucosylated notch epidermal growth factor repeats. J Biol Chem. 2010;285:1582–1586. doi: 10.1074/jbc.C109.065409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Luo Y, Moloney DJ, Haltiwanger R. O-glycosylation of EGF repeats: identification and initial characterization of a UDP-glucose: protein O-glucosyltransferase. Glycobiology. 2002;12:763–770. doi: 10.1093/glycob/cwf085. [DOI] [PubMed] [Google Scholar]
- Shao L, Moloney DJ, Haltiwanger R. Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J Biol Chem. 2003;278:7775–7782. doi: 10.1074/jbc.M212221200. [DOI] [PubMed] [Google Scholar]
- Shi S, Stahl M, Lu L, Stanley P. Canonical Notch signaling is dispensable for early cell fate specifications in mammals. Mol Cell Biol. 2005;25:9503–9508. doi: 10.1128/MCB.25.21.9503-9508.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci U S A. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Chiba S, Saito T, Kumano K, Takahashi T, Hirai H. Manic fringe and lunatic fringe modify different sites of the Notch2 extracellular region, resulting in different signaling modulation. J Biol Chem. 2001;276:25753–25758. doi: 10.1074/jbc.M103473200. [DOI] [PubMed] [Google Scholar]
- Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, Homeister JW, Lowe JB. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer U, Liu H, Doering TL. An alpha-1, 3-mannosyltransferase of Cryptococcus neoformans. J Biol Chem. 2003;278:47724–47730. doi: 10.1074/jbc.M307223200. [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D, Dunwoodie SL. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78:28–37. doi: 10.1086/498879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Guillen-Navarro E, Fatkin D, Dunwoodie SL. Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum Mol Genet. 2008;17:3761–3766. doi: 10.1093/hmg/ddn272. [DOI] [PubMed] [Google Scholar]
- Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem. 2008;283:13638–13651. doi: 10.1074/jbc.M802027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. Regulation of Notch signaling by glycosylation. Curr Opin Struct Biol. 2007;17:530–535. doi: 10.1016/j.sbi.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Sturla L, Rampal R, Haltiwanger RS, Fruscione F, Etzioni A, Tonetti M. Differential terminal fucosylation of N-linked glycans versus protein O-fucosylation in leukocyte adhesion deficiency type II (CDG IIc) J Biol Chem. 2003;278:26727–26733. doi: 10.1074/jbc.M304068200. [DOI] [PubMed] [Google Scholar]
- Suda T, Kamiyama S, Suzuki M, Kikuchi N, Nakayama K, Narimatsu H, Jigami Y, Aoki T, Nishihara S. Molecular cloning and characterization of a human multisubstrate specific nucleotide-sugar transporter homologous to Drosophila fringe connection. J Biol Chem. 2004;279:26469–26474. doi: 10.1074/jbc.M311353200. [DOI] [PubMed] [Google Scholar]
- Surani MA. Glycoprotein synthesis and inhibition of glycosylation by tunicamycin in preimplantation mouse embryos: compaction and trophoblast adhesion. Cell. 1979;18:217–227. doi: 10.1016/0092-8674(79)90370-2. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Inoue T, Gossler A, Saga Y. Feedback loops comprising Dll1, Dll3 and Mesp2, and differential involvement of Psen1 are essential for rostrocaudal patterning of somites. Development. 2003;130:4259–4268. doi: 10.1242/dev.00629. [DOI] [PubMed] [Google Scholar]
- Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity. 2009;30:254–263. doi: 10.1016/j.immuni.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Teng Y, Liu Q, Ma J, Liu F, Han Z, Wang Y, Wang W. Cloning, expression and characterization of a novel human CAP10-like gene hCLP46 from CD34() stem/progenitor cells. Gene. 2006;371:7–15. doi: 10.1016/j.gene.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Thomsen B, Horn P, Panitz F, Bendixen E, Petersen AH, Holm LE, Nielsen VH, Agerholm JS, Arnbjerg J, Bendixen C. A missense mutation in the bovine SLC35A3 gene, encoding a UDP-N-acetylglucosamine transporter, causes complex vertebral malformation. Genome Res. 2006;16:97–105. doi: 10.1101/gr.3690506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Hansen D, Schedl T, Skeath JB. Epsin potentiates Notch pathway activity in Drosophila and C. elegans. Development. 2004;131:5807–5815. doi: 10.1242/dev.01459. [DOI] [PubMed] [Google Scholar]
- Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621–629. doi: 10.1083/jcb.200811141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T, Katayama R, Takikawa R, Iwatsubo T. Complex N-glycosylated form of nicastrin is stabilized and selectively bound to presenilin fragments. FEBS Lett. 2002;520:117–121. doi: 10.1016/s0014-5793(02)02802-8. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development. 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284:31018–31027. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Freeze HH, Vacquier VD. Glycans in development and systemic physiology. In: Varki A, Cummings R. D, et al., editors. Essenstial of Glycobiology. New York: Cold Spring Harbor Laboratory Press; 2009. pp. 531–536. [PubMed] [Google Scholar]
- Visan I, Tan JB, Yuan JS, Harper JA, Koch U, Guidos CJ. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nat Immunol. 2006;7:634–643. doi: 10.1038/ni1345. [DOI] [PubMed] [Google Scholar]
- Vodovar N, Schweisguth F. Functions of O-fucosyltransferase in Notch trafficking and signaling: towards the end of a controversy? J Biol. 2008;7:7. doi: 10.1186/jbiol68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandall HH, Pizette S, Pedersen JW, Eichert H, Levery SB, Mandel U, Cohen SM, Clausen H. Egghead and brainiac are essential for glycosphingolipid biosynthesis in vivo. J Biol Chem. 2005;280:4858–4863. doi: 10.1074/jbc.C400571200. [DOI] [PubMed] [Google Scholar]
- Wang W, Struhl G. Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development. 2004;131:5367–5380. doi: 10.1242/dev.01413. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang N, Zhang T, Liu H, Ma W, Chu Q, Lai Q, Liu L, Wang W. Overexpression of human CAP10-like protein 46 KD in T-acute lymphoblastic leukemia and acute myelogenous leukemia. Genet Test Mol Biomarkers. 2010;14:127–133. doi: 10.1089/gtmb.2009.0145. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shao L, Shi S, Harris RJ, Spellman MW, Stanley P, Haltiwanger RS. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem. 2001;276:40338–40345. doi: 10.1074/jbc.M107849200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez P, Piccoli DA, Krantz ID, Spinner NB. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006;27:436–443. doi: 10.1002/humu.20310. [DOI] [PubMed] [Google Scholar]
- Waterhouse CC, Johnson S, Phillipson M, Zbytnuik L, Petri B, Kelly M, Lowe JB, Kubes P. Secretory cell hyperplasia and defects in notch activity in a mouse model of leukocyte adhesion deficiency type II. Gastroenterology. 2010;138(1079–1090):e1071–1075. doi: 10.1053/j.gastro.2009.10.049. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, JPt Morris, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell. 1985;43:567–581. doi: 10.1016/0092-8674(85)90229-6. [DOI] [PubMed] [Google Scholar]
- Whittock NV, Sparrow DB, Wouters MA, Sillence D, Ellard S, Dunwoodie SL, Turnpenny PD. Mutated MESP2 causes spondylocostal dysostosis in humans. Am J Hum Genet. 2004;74:1249–1254. doi: 10.1086/421053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth GE, Zandberg WF, Clark T, Vocadlo DJ. Mammalian Notch is modified by D-Xyl-alpha1-3-D-Xyl-alpha1-3-D-Glc-beta1-O-Ser: implementation of a method to study O-glucosylation. Glycobiology. 2010;20:287–299. doi: 10.1093/glycob/cwp173. [DOI] [PubMed] [Google Scholar]
- Windler SL, Bilder D. Endocytic internalization routes required for Delta/Notch signaling. Curr Biol. 2010;20:538–543. doi: 10.1016/j.cub.2010.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26:484–489. doi: 10.1038/82644. [DOI] [PubMed] [Google Scholar]
- Xu A, Haines N, Dlugosz M, Rana NA, Takeuchi H, Haltiwanger RS, Irvine KD. In vitro reconstitution of the modulation of Drosophila Notch-ligand binding by Fringe. J Biol Chem. 2007;282:35153–35162. doi: 10.1074/jbc.M707040200. [DOI] [PubMed] [Google Scholar]
- Xu A, Lei L, Irvine KD. Regions of Drosophila Notch that contribute to ligand binding and the modulatory influence of Fringe. J Biol Chem. 2005;280:30158–30165. doi: 10.1074/jbc.M505569200. [DOI] [PubMed] [Google Scholar]
- Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell. 2005;16:927–942. doi: 10.1091/mbc.E04-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan YP, Schultz J, Mlodzik M, Bork P. Secreted fringe-like signaling molecules may be glycosyltransferases. Cell. 1997;88:9–11. doi: 10.1016/s0092-8674(00)81852-8. [DOI] [PubMed] [Google Scholar]
- Zakany J, Kmita M, Alarcon P, de la Pompa JL, Duboule D. Localized and transient transcription of Hox genes suggests a link between patterning and the segmentation clock. Cell. 2001;106:207–217. doi: 10.1016/s0092-8674(01)00436-6. [DOI] [PubMed] [Google Scholar]
- Zhang N, Gridley T. Defects in somite formation in lunatic fringe-deficient mice. Nature. 1998;394:374–377. doi: 10.1038/28625. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, Shim J, Chervin S, Lowe JB. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 2008;112:308–319. doi: 10.1182/blood-2007-11-115204. [DOI] [PMC free article] [PubMed] [Google Scholar]