

Abstract
Recent studies highlighted an emerging possibility of using Drosophila as a model system for investigating the mechanisms of human congenital muscular dystrophies, called dystroglycanopathies, resulting from the abnormal glycosylation of α-dystroglycan. Several of these diseases are associated with defects in O-mannosylation, one of the most prominent types of α-dystroglycan glycosylation mediated by two protein O-mannosyltransferases. Drosophila appears to possess homologs of all essential components of the mammalian dystroglycan-mediated pathway; however, the glycosylation of Drosophila Dystroglycan (DG) has not yet been explored. In this study, we characterized the glycosylation of Drosophila DG using a combination of glycosidase treatments, lectin blots, trypsin digestion, and mass spectrometry analyses. Our results demonstrated that DG extracellular domain is O-mannosylated in vivo. We found that the concurrent in vivo activity of the two Drosophila protein O-mannosyltransferases, Rotated Abdomen and Twisted, is required for O-mannosylation of DG. While our experiments unambiguously determined some O-mannose sites far outside of the mucin-type domain of DG, they also provided evidence that DG bears a significant amount of O-mannosylation within its central region including the mucin-type domain, and that O-mannose can compete with O-GalNAc glycosylation of DG. We found that Rotated Abdomen and Twisted could potentiate in vivo the dominant-negative effect of DG extracellular domain expression on crossvein development, which suggests that O-mannosylation can modulate the ligand-binding activity of DG. Taken together these results demonstrated that O-mannosylation of Dystroglycan is an evolutionarily ancient mechanism conserved between Drosophila and humans, suggesting that Drosophila can be a suitable model system for studying molecular and genetic mechanisms underlying human dystroglycanopathies.
Keywords: Drosophila, Dystroglycan, dystroglycanopathy, glycosylation, protein O-mannosylation
Introduction
Dystroglycan, a highly glycosylated protein of mammalian muscle cells, is a central component of the dystrophin–glycoprotein complex (DGC) that provides structural stability to the sarcolemma during muscle contraction. Mammalian dystroglycan undergoes posttranslational cleavage into separate α- and β-subunits (Barresi and Campbell 2006). Proper glycosylation of α-dystroglycan (α-DG) has been proven to be essential for interaction with the extracellular matrix (ECM) ligands, such as laminin, agrin, and perlecan, thus providing functionality for the DGC at the sarcolemma. α-DG has a complex pattern of abundant glycosylation, including the presence of both N- and O-linked glycans. While not all structures of these glycans have been fully characterized, the O-glycans are reported to be initiated by O-GalNAc and O-mannose, the latter of which is rarely observed on other mammalian proteins (Endo 1999; Barresi and Campbell 2006; Martin 2006). The presence of the O-mannose-linked glycans is thought to be particularly important for ligand-binding activity of α-DG (Barresi and Campbell 2006; Martin 2007). Several human congenital muscular dystrophies (CMDs) were found to be caused by genetic defects in glycosyltransferases involved in the biosynthesis of the O-mannose-linked carbohydrates. These CMDs are associated with hypoglycosylation of α-DG and classified as dystroglycanopathies; they include Walker–Warburg syndrome (WWS), which is caused by mutations in protein O-mannosyltransferase genes, POMT1 and POMT2, responsible for the addition of O-mannose onto the protein backbone, and muscle-eye-brain disease (MEB) which results from defects in POMGnT1, the glycosyltransferase that elongates O-linked mannose with GlcNAc (Yoshida et al. 2001; Beltran-Valero de Bernabe et al. 2002; van Reeuwijk et al. 2005). Although substantial progress has been made in understanding the molecular and genetic bases of O-mannosylation of α-DG ((Willer et al. 2004; Liu et al. 2006), reviewed in Barresi and Campbell (2006) and Martin (2007)), the complexity of mammalian glycosylation pathways along with limitations of genetic approaches indicate that a suitable experimentally amenable model system would be a useful tool for studying biological mechanisms of O-mannosylation and its involvement in human pathologies. Several recent studies have hinted at Drosophila as a potential model organism for such studies.
Drosophila genome encodes two protein O-mannosyl- transferases, Rotated Abdomen (RT) and Twisted (TW) (aka DmPOMT1 and DmPOMT2, respectively), along with counterparts of all essential components of the mammalian DGC, including Dystroglycan. However, in Drosophila, unlike in mammals, DG appears not to be cleaved into α- and β-subunits upon maturation (Greener and Roberts 2000), while alternative splicing is predicted to produce three different DG isoforms, DG-A, -B, and -C (Deng et al. 2003). Out of these three isoforms, only DG-C includes a predicted mucin-type domain with the potential for extensive O-glycosylation, sharing this feature with mammalian α-DG (Figure 1A, (Deng et al. 2003; Schneider et al. 2006)). Drosophila Dg is required for apicobasal polarity in epithelial cells and antero-posterior polarity in the oocyte, while the downregulation of Dg expression in larvae and adult flies causes neuromuscular junction synaptic defects, muscle defects and degeneration (Deng et al. 2003; Schneider et al. 2006; Haines et al. 2007; Shcherbata et al. 2007; Bogdanik et al. 2008; Wairkar et al. 2008). The similarity of defects caused by Dystroglycan abnormalities in Drosophila and mammals has led to the hypothesis that DG functions are evolutionary conserved between Drosophila and humans (Deng et al. 2003; Haines et al. 2007; Shcherbata et al. 2007, Bogdanik et al. 2008; Kucherenko et al. 2008; Wairkar et al. 2008). Potential O-mannosylation of Drosophila DG and its putative role in DG regulation were discussed in several studies. Similar to their mammalian counterparts, RT and TW proteins appear to function as a heterocomplex (Ichimiya et al. 2004; Lyalin et al. 2006), and they can also add O-mannose to a fragment of mammalian α-DG in vitro (Ichimiya et al. 2004). Mutations in rt and tw were found to cause defects in larval NMJ similar to the defects found in Dg mutants, and these genes showed genetic interactions (Haines et al. 2007, Wairkar et al. 2008). Moreover, Western blot detection revealed the absence of a high-molecular-mass band of DG in rt mutants, which was interpreted as an evidence of the involvement of RT in DG glycosylation (Wairkar et al. 2008). However, the glycosylation of Drosophila DG has not been examined, and the presence of O-mannose modifications on the DG protein remains obscure. In this work, we analyzed the glycosylation of Drosophila DG using both in vivo and in vitro approaches. We found that the DG protein is a target of O-mannosyltransferase activity and that both RT and TW are simultaneously required for the modification of DG with O-linked mannose in vivo. Our results demonstrate that O-mannosylation of DG is an evolutionarily conserved mechanism and suggest that it plays an important role in the regulation of DG function in Drosophila. These data also indicate that Drosophila can be used as a model organism to study molecular and genetic mechanisms of CMDs.
Fig. 1.
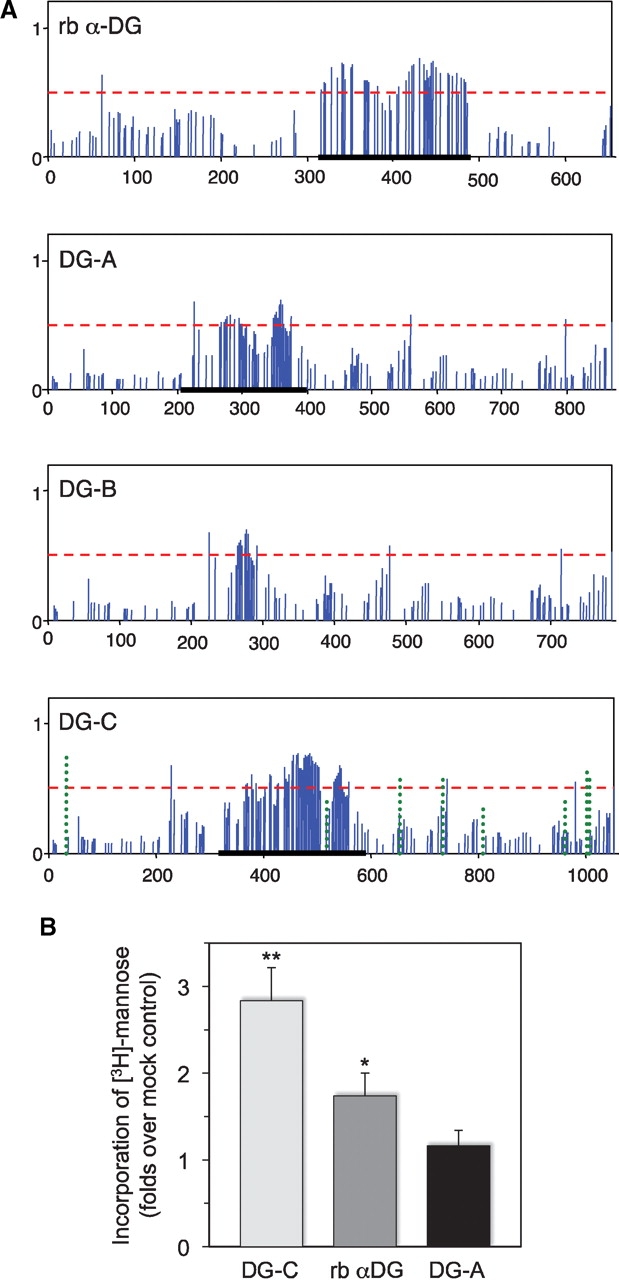
Predicted glycosylation and in vitro O-mannosylation of Drosophila Dystroglycan. (A) Prediction of mucin-type O-linked glycosylation of the extracellular domains of rabbit α-DG and Drosophila DG isoforms A, B, and C. Solid vertical bars show the G-score of corresponding S/T residues with respect to the glycosylation potential (Julenius et al. 2005); the predicted glycosylation sites have bars above the threshold (horizontal dashed line). DC-C panel also includes the prediction of N-linked glycosylation (dotted vertical bars). Regions of rabbit α-DG, DG-A, and DG-C proteins used in in vitro O-mannosylation assays are underlined. (B) O-Mannosylation assay of Drosophila DG-A and DG-C proteins. Purified fragments of extracellular domain of DG-A and DG-C isoforms were used as substrates in in vitro O-mannosylation assays with microsomal fraction from Drosophila larvae as a source of RT-TW mannosyltransferase activity and [3H]-mannosyl phosphoryl dolicol as a sugar donor. Incorporation of mannose is shown as the ratio of incorporated radioactivity for a substrate to that for BSA as a mock control. Error bars indicate SEM calculated from six independent assays. ** and * – indicate significant differences from the mock control with t-test P < 0.01 and P < 0.05, respectively. Note that the results for DG-C and rabbit α-DG were also significantly different from each other (t-test, P < 0.05), while incorporation of O-mannose in DG-A was not significantly different from the background control (t-test, P > 0.4).
Results
Drosophila DG is a substrate for protein O-mannosylation activity of RT and TW in vitro
We tested if Drosophila DG can be used as a substrate for O-mannosylation by RT and TW in vitro. Previous experiments revealed that the mucin-type domain of mammalian α-DG is a target of O-mannose modification (Manya et al. 2007; Breloy et al. 2008). Thus, we focused our analysis on Drosophila DG-C isoform that, similarly to its mammalian counterpart, also includes a mucin-type domain, a potential target of O-mannosylation. A region of mucin-type domain of the DG-C isoform (Figure 1A) was expressed in Escherichia coli, purified and tested as a substrate in an in vitro O-mannosylation assay using microsomal membrane fraction prepared from Drosophila larvae as a source of RT-TW activity (see Material and methods). We also tested a corresponding fragment of DG-A extracellular domain that spans the predicted region of mucin-type O-glycosylation (Figure 1A). As a positive control, we used the region of mucin-type domain of rabbit α-DG that was previously shown to be a substrate for RT-TW in vitro (Ichimiya et al. 2004). Our results indicated that, similarly to mammalian α-DG, DG-C could serve as a substrate for in vitro O-mannosylation, with the DG-C fragment being apparently a better substrate for RT-TW activity in vitro than the region of mucin-type domain of rabbit α-DG. Incorporation of mannose into DG-A was not significantly above the background (Figure 1B).
Extracellular domain of DG is properly folded and trafficked in Drosophila cells with or without RT-TW activity
We expressed the entire extracellular part of DG-C tagged with 3xFLAG epitope (designated below as ExDG) as a transgenic construct in Drosophila. To confirm that ExDG is properly folded and trafficked in Drosophila cells, we verified by Western blot that the ExDG protein was efficiently secreted in a diffusible form into cell medium when expressed in Drosophila S2 cultured cells (Figure 2A). Next, we expressed ExDG in vivo within wing imaginal disk epithelium using a UAS-GAL4 expression system. We tested three different genetic backgrounds: rt mutant, wildtype, and rt-tw coexpression. The immunofluorescent detection of in vivo expressed ExDG revealed that it was similarly delivered to the cell surface and no difference in subcellular localization of ExDG could be detected between these backgrounds (Figure 2B). Thus, we concluded that the trafficking and subcellular localization of ExDG was not affected by RT-TW activity in vivo.
Fig. 2.

Secretion and subcellular localization of the ExDG protein. (A) Anti-FLAG Western blot of tissue culture media from S2 cells (control) or from S2 cells transfected with ExDG-expressing construct (with or without induction of expression). The results show that ExDG is efficiently secreted in a diffusible form outside of the cell. (B) Expression of ExDG in the third instar larval wing imaginal disks with a patched-GAL4 driver using the UAS-GAL4 system. Genotype of the disks: left disk (ptc::ExDg rt−) – ptc-GAL4 UAS-GFP/UAS-ExDg; rtP/rt2; middle disk (ptc::ExDg) – ptc-GAL4 UAS-GFP/+; UAS-ExDg/+; right disk (ptc::ExDg+rt+tw) – ptc-GAL4 UAS-GFP/UAS-rt UAS-tw; UAS-ExDg/+. ExDG expression is detected by immunofluorescent staining (red), while GFP signal (green) highlights the pattern of the ptc driver. Yellow dashed line indicates the position of Z cross-sections reconstructed for each disk in panel Z. Z cross-sections: no accumulation of the ExDG protein can be detected inside the columnar epithelium cells (asterisks), while the protein is efficiently delivered to the basal (arrows) or apical (arrowheads) surfaces of the disk epithelium. There is no significant difference in the subcellular localization of ExDG between the three genotypes. For all panels: dorsal is to the top; for the frontal-view sections: anterior is to the left; for Z cross-sections: basal is to the left.
The concurrent in vivo activity of RT and TW is necessary and sufficient to generate the high-molecular-mass form of DG
The ExDG protein was expressed in Drosophila with different genetic backgrounds corresponding to varied levels of RT and TW, and then it was analyzed by Western blotting using an anti-FLAG antibody. We found a drastic difference in the pattern of high-molecular-mass bands of ExDG expressed in rt and/or tw mutant, wildtype, and RT-TW coexpression backgrounds (Figure 3A and B). In wild-type flies, we observed two major bands of estimated sizes of 175 kDa (designated as S (small) band) and 215 kDa (designated as L (large) band) present at approximately equal amounts. The L band was undetectable in rt mutants, tw mutants, as well as in rt and tw double mutants. We also found that the relative amount of the L band was significantly increased in flies coexpressing both RT and TW. At the same time, the overexpression of one of the O-mannosyltransferases, RT or TW, resulted in no significant increase of the relative amount of the L band, as compared to a wild-type background. These results indicated that RT and TW are simultaneously required in vivo for producing the high-molecular-mass form of ExDG.
Fig. 3.

Western and lectin blot analyses of in vivo expressed ExDG. (A) Western blot detection of ExDG expressed in rt-tw double mutants (rt− tw−), rt mutants (rt−), tw mutants (tw−), wild-type background (WT), and backgrounds with ubiquitous ectopic expression of RT (rt+), TW (tw+), or RT-TW co-expression (rt+ tw+). The L band is the top band present in wild-type background and backgrounds with RT and TW expression, but absent in rt and/or tw mutants; the S band is present in all genetic backgrounds analyzed. (B) The ratio between the intensities of L and S bands was quantified for rt mutant, tw mutant, wild-type, and RT-TW co-expression backgrounds. Error bars represent SD.
Interestingly, we did not observe a band corresponding to the ∼110 kDa band that was detected for endogenously expressed DG with antibodies raised against its intracellular part, but not with the antibody specific for DG-C (Schneider et al. 2006; Wairkar et al. 2008), which confirmed previous conclusion that this smaller band represents only DG-A/B isoforms (Schneider et al. 2006).
Glycosidase treatments and lectin blots revealed O-mannosylation of high-molecular-mass form of DG
We analyzed the glycosylation of DG-C isoforms represented by L and S bands using treatments with specific glycosidases. In agreement with N-linked site prediction for DG-C (Figure 1A), the removal of N-linked glycans with PNGaseF resulted in a gel shift of ExDG, with the shift being similar for both L and S bands (Figure 4A). The S band was similarly decreased in size for both rt mutant and RT-TW coexpression backgrounds. The shift of ExDG bands after PNGaseF treatment, estimated as ∼10 kDa, corresponds to approximately six oligomannose-type N-glycans (typical N-linked structures present on Drosophila glycoproteins (North et al. 2006; Aoki et al. 2007; Koles et al. 2007)). This result is consistent with the presence of total eight putative N-linked sites within the extracellular part of DG-C, with five of them predicted to have a significant potential for N-glycosylation (Figure 1A). We also treated purified ExDG with α-mannosidase that cleaves off α-linked mannose residues. This treatment should both remove O-linked mannose (Ichimiya et al. 2004; Manya et al. 2004) and trim oligomannose N-linked glycans down to the 4-β-mannosyl chitobiose (Manβ1–4GlcNAcβ1–4GlcNAc…) of the core structure. α-Mannosidase treatment alone resulted in a significantly greater shift of the L band as compared to the S band, which suggested that the high-molecular-mass form is extensively modified with O-linked mannose. This conclusion was confirmed by sequential digestion with PNGaseF and α-mannosidase, which showed an additional decrease in the mass of the L band as compared to PNGaseF digestion alone. A similar comparison between PNGaseF-treated and double PNGaseF /α-mannosidase-treated S bands did not reveal a significant difference, thus suggesting that the S band represents a glycoform without extensive O-mannosylation.
Fig. 4.

Analysis of ExDG glycosylation by glycosidase treatments. (A) left panel: ExDG purified from rt mutants was treated with PNGaseF or α-mannosidase, or with both glycosidases sequentially. No additional shift of the ExDG band (S band) is detected in double PNGaseF/α-mannosidase treatment as compared to PNGaseF treatment alone, suggesting that ExDG has no O-mannose modification. Right panel: glycosidase treatments of ExDG purified from RT-TW coexpression background. Top (L) band shows significant loss of mass (≥10 kDa) when PNGaseF-treated ExDG was digested with α-mannosidase, which suggests the presence of abundant O-mannose modifications. No such loss of mass was detected for the lower (S) band, suggesting that O-mannosylation of ExDG in this band is not significant. (B) Con A reactivity of purified ExDG after treatments with PNGaseF and α-mannosidase. The S glycoform purified from rt mutant background loses Con A reactivity either after the removal of N-linked glycans by PNGaseF or after treatment with α-mannosidase removing α-linked mannose residues (top panel, left), suggesting the absence of O-mannose modification and efficient removal of oligomannose structures either by trimming N-linked branches with α-mannosidase or by complete elimination of N-linked glycans. At the same time, the L glycoform purified from RT-TW co-expression background retains Con A reactivity after treatment with PNGaseF, α-mannosidase, or both glycosidases (top panel, right), suggesting that L glycoform is O-mannosylated, and that α-mannosidase does not remove O-mannose completely. The bottom panel shows anti-FLAG western control corresponding to the lectin blot. Red dashed line outlines the region of the L glycoform on the blots. Asterisk “*” indicates an additional minor band sometimes detected by FLAG Western blots that probably represents ExDG proteolytic degradation.
The presence of α-linked mannose on ExDG was examined by concanavalin A (Con A) lectin blot (Figure 4B). Con A strongly reacted with the L band, while showing weaker reactivity with the S band from rt mutant and RT-TW coexpression backgrounds. The Con A reactivity of the S band from rt mutants was eliminated by PNGaseF treatment, indicating that this reactivity was solely due to mannose structures of N-linked glycans and providing further evidence that ExDG was not modified with O-mannose in rt mutants. At the same time, PNGaseF treatment did not eliminate the reactivity of the L band, suggesting that O-mannose is present on this glycoform. Interestingly, the S glycoform from RT-TW coexpression also showed Con A reactivity after PNGaseF treatment, suggesting the presence of some O-linked mannose on this isoform as well. We also found that double PNGaseF/α-mannosidase-treated ExDG from RT-TW coexpression retains some residual reactivity for Con A, which suggest the presence of mannosidase-resistant O-mannose on ExDG (Figure 4B). This conclusion is consistent with the fact that O-linked mannose is a relatively poor substrate for α-mannosidase (Manya et al. 2004), while there is also a possibility that some modification present on O-mannose may inhibit α-mannosidase.
We also examined the glycosylation of ExDG using several other lectins. The presence of O-linked GalNAc on purified ExDG was analyzed using Vicia villosa (VVA) lectin (Fig- ure 5A). Only S band showed strong VVA staining, while the reactivity of the L band was barely above the background. Interestingly, the S band from rt mutants had a significantly stronger VVA reactivity than that from RT-TW coexpression, suggesting more extensive O-GalNAc modification of ExDG in the absence of O-linked mannose. O-GalNAc can be extended with β1–3 linked galactose, which produces the core 1 structure, one of the most abundant O-linked modification in Drosophila (Aoki et al. 2008; Breloy et al. 2008). Thus, we tested a possibility that O-GalNAc of the L band was masked from VVA recognition because of β1–3 galactose extension by lectin blot using Peanut agglutinin (PNA) that specifically recognizes this disaccharide. However, neither L nor S band showed PNA reactivity (data not shown), indicating the absence of core 1 structure on ExDG.
Fig. 5.

Analysis of ExDG glycosylation by lectin blots. ExDG was purified from rt mutant or RT-TW coexpression backgrounds. (A) VVA lectin blot (left two lanes) and anti-FLAG western control for protein amount (right two lanes). The S band from RT-TW overexpression has significantly weaker VVA reactivity than that from rt mutants, while the reactivity of the L band is even further reduced to a nearly undetectable level. (B) Anti-FLAG western control (left two lanes), WFA and HHL lectin blots (middle and right two lanes, respectively). While the WFA reactivity of ExDG bands shows similar pattern to VVA staining in (A), the reactivity to HHL is complementary to VVA and WFA stainings. Only the L band reveals strong HHL staining, with the S band from RT-TW coexpression showing much weaker staining, and the S band from rt mutants having no HHL reactivity at all. ExDG was treated with PNGaseF (de-N-glycosylated) in (B). Asterisk indicates an additional minor band representing some proteolytic degradation.
We also performed analysis of ExDG glycosylation by Wisteria floribunda agglutinin (WFA) and Hippeastrum hybrid lectin (HHL) that recognize terminal GalNAc and mannose residues, respectively. To exclude possible reactivity with N-linked glycans, purified ExDG was first treated with PNGaseF in these experiments. Similar to VVA, WFA strongly recognized the S band from rt mutants, had a weaker reactivity with the S band from RT-TW coexpression, and could barely detect the L band (Figure 5B). On the other hand, HHL staining had a pattern complementary to that of WFA and VVA: HHL strongly recognized the L band, showed weak reactivity with the S band from RT-TW coexpression, and did not react at all with the S band from rt mutants (Figure 5B). Together, these results further supported our conclusion that ExDG from RT-TW coexpression has O-mannose modification, with the L band representing more extensively O-mannosylated glycoform and the S band probably including only limited O-mannose modification. At the same time, the extent of O-linked GalNAc modification appears to be complementary to that of O-mannosylation, with the S band from rt mutants being most prominently modified with O-GalNAc, the amount of O-GalNAc being significantly decreased on the S band from RT-TW coexpression and even further reduced on the L glycoform.
Mass spectrometry of purified extracellular domain of DG identified O-mannose and O-GalNAc modifications
To further elucidate the glycosylation of ExDG, we performed mass spectrometry analyses of the L and S bands of ExDG from RT-TW coexpression, and the S band from rt mutants. To this end, samples of purified and gel-separated L and S bands were subjected to digestion either by trypsin or by a combination of trypsin and elastase. The peptide coverage was very similar for all three analyzed samples (see supplementary Table), revealing no difference in polypeptide sequences. The central region of ExDG including mucin-type domain (a.a. 309–552) was not amenable to the mass spectrometry analysis, which was likely because of its resistance to protease digestion due to a peculiar amino acid sequence (including only 4 trypsin sites) and putative extensive glycosylation. The rest of the protein was almost completely covered by mass spectrometry analysis. Glycopeptides were analyzed by a combination of BEMAD and a pseudo-neutral loss method on a linear ion trap (see Material and methods and Wells et al. (2002)), which allowed us to not only map the site of modification but also determine the identity of the O-linked glycan structure (Figure 6A and B). In total, we identified nine O-linked modifications on the L glycoform, five modifications on the S glycoform from RT-TW coexpression background, and two O-linked sites on the S glycoform from rt mutants (Table I, Figure 7, supplementary Figure 1). We found that the L and S glycoforms from RT-TW coexpression have six and three O-mannose sites outside of the mucin region, respectively. No O-mannose was found on ExDG purified from rt mutants, even though we specifically looked for the presence of glycopeptides observed in RT-TW coexpression background. Two O-GalNAc sites were identified on each of the analyzed glycoforms, with some of them overlapping with O-linked mannose sites on ExDG coexpressed with RT and TW. These results are consistent with our conclusions from glycosidase treatment and lectin blot analyses about the absence of O-mannose in rt mutants and the reciprocal distribution of O-GalNAc and O-mannose on the L and S glycoforms. These data also suggested that the extensive O-mannose modification predicted by glycosidase and lectin analyses is confined to the central region of ExDG including mucin-type domain.
Fig. 6.
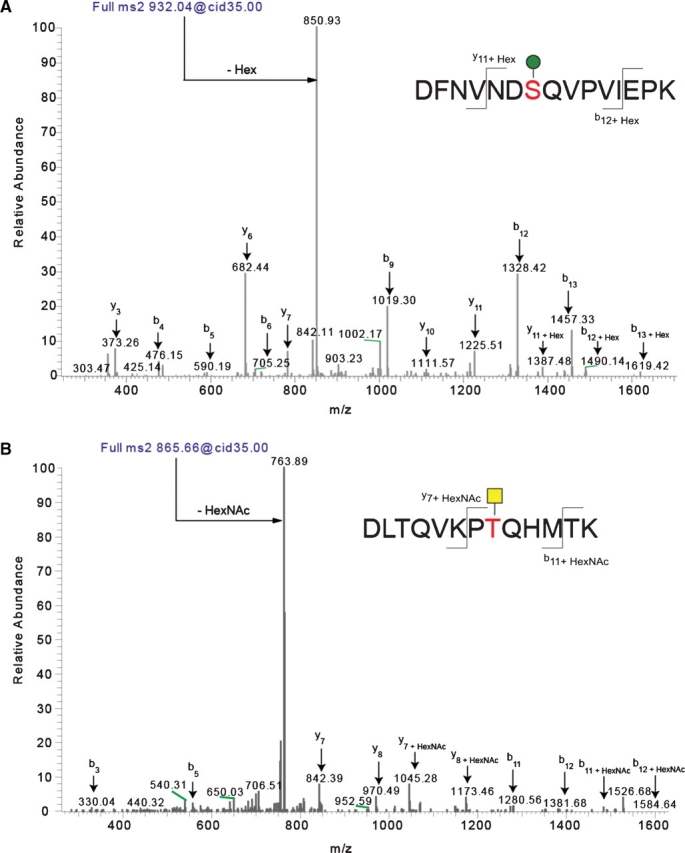
Identification of O-linked sites on ExDG glycopeptides using mass spectrometry. The pseudo-neutral loss method was applied using LC-MS/MS allowing for data-dependent MS3 fragmentation upon witnessing a neutral loss of a glycan mass in the MS2 spectra. Through the application of the pseudo-neutral loss method, it was possible to confirm the assignment of the glycosylated peptide that was identified using TurboSequest. Panel A shows the MS2 of an O-mannose-modified peptide, while panel B shows the MS2 of an O-GalNAc-modified peptide. Note that in both cases the major fragment is the loss of the glycan. Using the MS2 profile (shown) and the MS3 profile of the neutral-loss species (the unmodified peptide, not shown) allowed us to assign the peptide, the glycan, and the site of modification in an automated fashion using TurboSequest followed by manual validation. Sites of modification were manually validated by looking for the addition of Hex and HexNAc to the theoretical m/z of b- and y-ions for the glycopeptide.
Table I.
Summary of ExDG glycopeptides with O-linked mannose and GalNAc modifications identified by mass spectrometry
| Glycoform S | Glycoform S | Glycoform L |
|---|---|---|
| rt mutants | RT-TW coexpression | RT-TW coexpression |
| Glycopeptides identified | Glycopeptides identified | Glycopeptides identified |
| With O-mannose: | With O-mannose: | With O-mannose: |
| 30 DFNVND SQVPVIEPK 44 | ||
| 238 HHHGALEVSEK 248 | 238 HHHGALEVSEK 248 | |
| — | 277 LESVISK 283 | 277 LESVISK 283 |
| 785 DLTQVKPTQHMTK 797 | 785 DLTQVKPTQHMTK 797 | |
| 785 DLTQVKPTQHMTK 797 | ||
| With O-GalNAc: | With O-GalNAc: | With O-GalNAc: |
| 238 HHHGALEVSEK 248 | 238 HHHGALEVSEK 248 | 238 HHHGALEVSEK 248 |
| 785 DLTQVKPTQHMTK 797 | 785 DLTQVKPTQHMTK 797 | 785 DLTQVKPTQHMTK 797 |
| Other/ambiguous: | Other/ambiguous: | Other/ambiguous: |
| — | — | 200 RQSEGSGADEDDYDYGDDDEEVA 222 |
Fig. 7.

Glycosylation of the DG-C extracellular domain. Proposed glycosylation profiles of L and S glycoforms of the DG-C extracellular domain based on mass spectrometry analyses, glycosidase treatments, and lectin reactivity. Orange box indicates the mucin-type domain of DG-C; hatched region shows the part of the full-length DG-C protein that was truncated in ExDG (including the transmembrane domain shown in blue). N–C termini orientation is from left to right.
ExDG modified with O-mannose is resistant to protease digestion
We further characterized properties of purified ExDG from RT-TW coexpression by analyzing its sensitivity to trypsin. Our mass spectrometry analysis suggested a possibility that the central region of ExDG may be resistant to proteolytic cleavage because of extensive glycosylation. We tested this hypothesis by analyzing products of trypsin digestion of denatured PNGaseF-treated ExDG. Since it has been reported that Coomassie and silver staining methods often fail to efficiently detect highly glycosylated peptides, we used PAS and PAS-silver staining of SDS–PAGE-separated tryptic digestion to detect glycopeptides (Zacharius et al. 1969; Gradilone et al. 1998). A ∼120 kDa band was detected on the gel by PAS staining indicating the presence of a glycosylated peptide resistant to proteolysis (Figure 8). We analyzed this glycopeptide by binding to lectin affinity agarose beads. The glycopeptide was quantitatively bound by Con A beads; however, it did not bind to WFA or VVA agarose (Figure 8). This result suggested that the glycopeptide has significant O-mannose modification, while revealing no evidence for the presence of GalNAc. O-Mannosylation of this glycopeptide was further corroborated by treatment with α-mannosidase which resulted in a significant shift of the glycopeptide on the gel (Figure 8). The glycopeptide band also became less intensely stained after α-mannosidase treatment, which is consistent with lower sensitivity of PAS-silver staining for proteins with reduced amount of glycosylation (Gradilone et al. 1998). Taken together, these results provided further evidence that ExDG contains a trypsin-resistant region with extensive O-mannosylation, suggesting that this region corresponds to the central part of ExDG not amenable to mass spectrometry analysis and including mucin-type domain.
Fig. 8.

Analysis of trypsin-digested ExDG. Trypsin digestion of ExDG purified from RT-TW coexpression generates a ∼120 kDa trypsin-resistant glycopeptide that can be detected with PAS or PAS-silver staining. The glycopeptide quantitatively binds to Con A agarose and changes its gel mobility upon α-mannosidase treatment. PAS, PAS-staining of ExDG after trypsin digestion. Other lanes show PAS-silver staining: Input, amount control for the ExDG tryptic glycopeptide used for binding to lectin agarose beads; ConA, WFA, VVA, the glycopeptide bound to Con A, WFA, and VVA beads, respectively; α-Mannase “-” and “+”, α-mannosidase treatment of the glycopeptide with heat-inactivated (control) and active α-mannosidase, respectively. Note that PNGaseF-treated ExDG was used.
Function of ExDG is modulated by RT-TW activity in vivo
In order to test the functional influence of O-mannosylation on Drosophila DG, we developed an in vivo assay for ExDG activity. Intracellular truncation of membrane proteins involved in cell adhesion or signaling processes is known to often result in dominant-negative forms that could antagonize functions of their endogenous wild-type forms by participating in non-productive molecular interactions with ligands (Rebay et al. 1993; Sun and Artavanis-Tsakonas 1996). Since ExDG construct includes only the extracellular domain of DG and thus should be mostly expressed as a secreted soluble protein (Fig- ure 2A), we expected that it could function as a dominant-negative form by outcompeting endogenous DG-ligand interactions on the cell surface. Indeed, we found that the ubiquitous expression of ExDG in developing wing imaginal disks results in defective crossveins, causing the loss of crossvein tissue (Fig- ure 9A), with the phenotype penetrance of ∼12% (Figure 9B). This phenotype is characteristic for loss-of-function mutations of Dg and detached, the Drosophila ortholog of vertebrate dystrophin, which indicates the involvement of the DGC in crossvein development (Christoforou et al. 2008). Thus, ExDG can function as a dominant-negative form antagonizing endogenous DG activity. We found that the penetrance of the ExDG phenotype was significantly increased (∼2.5 times) by coexpression with RT and TW (Figure 9B), which suggests that the activity of ExDG is potentiated by O-mannosylation in vivo.
Fig. 9.
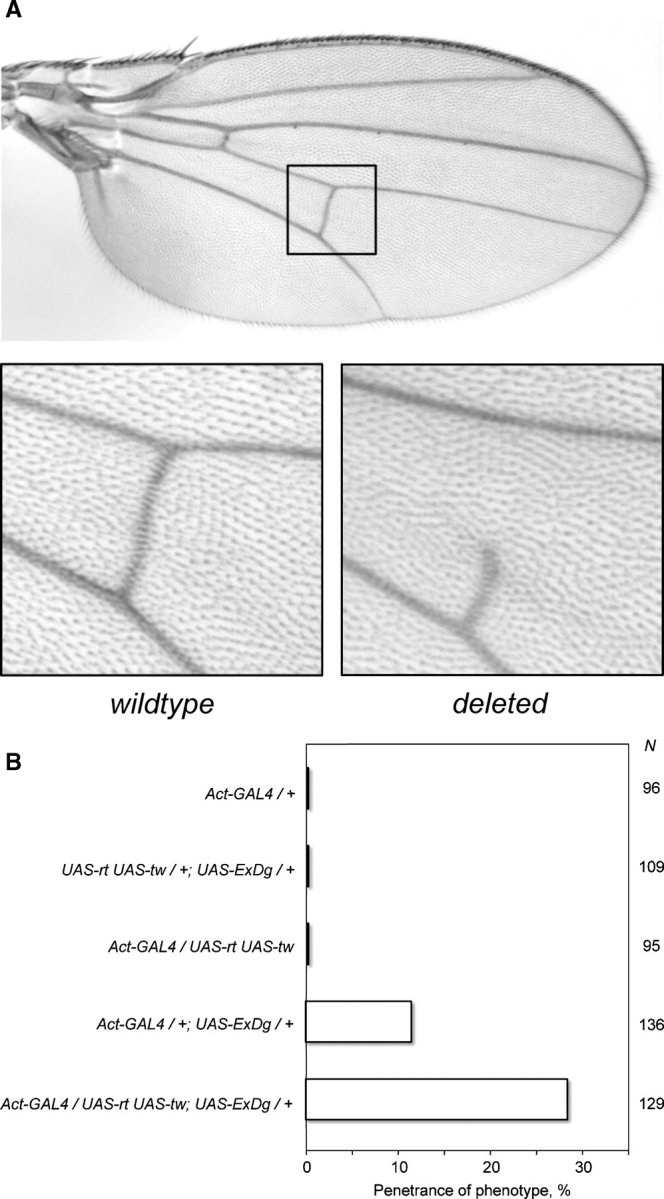
Effect of ExDG ectopic expression on the posterior crossvein development. (A) Top panel, wild-type Drosophila wing with the region of posterior crossvein shown by a rectangle. Bottom panels show enlarged examples of a wild-type and a defective crossvein (with deleted anterior part) as a result of ExDG ectopic expression using an Act-GAL4 driver. The phenotype varied from a severe ‘deletion’ of the crossvein (shown) to a less extreme but always obvious loss of crossvein tissue. (B) Penetrance of the crossvein defect is enhanced by RT-TW activity. N indicates the number of flies scored for each genotype. ExDG encompasses the complete extracellular domain of DG-C (a.a. 1–1048).
Discussion
Here we demonstrated for the first time, to our knowledge, that Drosophila Dystroglycan is O-mannosylated and serves as a substrate for two Drosophila protein O-mannosyltransferases, RT and TW, in vitro and in vivo. Our study was focused on the extracellular part of the DG-C isoform that includes a mucin-type domain and most closely resembles the structure of mammalian α-DG. For in vivo analyses, the entire extracellular domain of DG-C, ExDG, was expressed in flies with genetically varied levels of RT and TW. When characterized by Western blots, a characteristic pattern of two major bands (L and S) of ExDG was observed for the genetic backgrounds with wild-type or elevated levels of RT and TW, while only the lower-molecular-mass band (S) was detected when the activity of either rt or tw genes was compromised, as well as in double rt/tw mutants. These results are consistent with the recently reported absence of high-molecular-mass band of endogenous DG in rt mutants (Wairkar et al. 2008). Most importantly, we found that the relative amount of the L form was significantly elevated only by a concurrent increase of RT and TW, while increasing the level of either RT or TW alone had no significant effect on the amount of the L band (Figure 3 A, B). Together, these results indicate that RT and TW concurrent activity is required for generating the high-molecular-mass form of ExDG in vivo, suggesting that RT and TW work as a heterocomplex in vivo and providing further support for similar conclusions of in vitro (Ichimiya et al. 2004) and genetic (Lyalin et al. 2006) experiments. Since the expression of constructs was driven ubiquitously in these experiments, the fact that the elevated level of either RT or TW alone does not increase the amount of the L glycoform also suggests that the levels and patterns of endogenous RT and TW expression largely coincide throughout larval and pupal stages. This conclusion is consistent with in situ hybridization data that revealed the overlapping expression of rt and tw at embryonic and larval stages ((Lyalin et al. 2006); Lavrova and Panin, unpublished data).
To elucidate the nature of the L and S forms of ExDG, we investigated their glycosylation using a combination of approaches, including specific glycosidase treatments, lectins, tryptic digestion, and mass spectrometry. Treatment with PNGaseF suggested that there is no difference in N-linked glycosylation between ExDG species represented by L and S bands from rt mutant and RT-TW coexpression backgrounds, which indicates that RT-TW activity has no effect on N-linked glycosylation of ExDG. At the same time, α-mannosidase treatments of purified ExDG suggest that the L band represents a glycoform extensively modified with O-linked mannose, while the S band corresponds to the glycoform without significant O-mannosylation (Figure 4). This conclusion is consistent with the fact that α-mannosidase has similar effect on S bands from rt mutant and RT-TW coexpression backgrounds (Figure 3). Further evidence of O-mannosylation of the L glycoform was obtained by lectin blots with mannose-recognizing lectins, Con A and HHL (Figures 4B and 5B). The estimated decrease in the L glycoform molecular mass upon α-mannosidase digestion of PNGaseF-treated ExDG is ≥10 kDa (Figure 4A), which implies the presence of more than 50 O-mannose residues attached to ExDG. Interestingly, our mass spectrometry analysis identified only six O-linked mannoses outside of a central ∼250 a.a. region including a mucin-type domain (Table I, Figure 7, supplementary Figure 1), suggesting that this central region of ExDG bears most of the predicted O-mannosylation. Mass spectrometry results also suggested that this region may be resistant to proteases due to its extensive glycosylation. This possibility was further supported by the analysis of trypsin digestion of ExDG purified from RT-TW coexpression background that revealed the presence of ∼120 kDa trypsin-resistant glycopeptide. Moreover, the specific binding of this glycopeptide to Con A beads suggested that it is O-mannosylated (Figure 8). Digestion with α-mannosidase resulted in an estimated ∼8 kDa shift of this glycopeptide on the gel (Figure 8), which corresponds to the loss of ∼44 mannose residues. This result is consistent with the extensive α-mannosylation of the central region of ExDG suggested by mass spectrometry and glycosidase analyses of the full-length protein.
Our mass spectrometry analysis revealed a similar peptide coverage for purified L and S bands from both analyzed genotypes, rt mutant and RT-TW coexpression (supplementary Table 1 and Figure 1). Notably, the most-N-terminal peptide recovered for all three analyzed bands is the same, and it is located only 29 amino acids downstream of the ExDG N-terminus and two residues downstream of the predicted cleavage of the signal peptide. Since the integrity of ExDG C-terminus was conferred by anti-FLAG reactivity and purification, and a hypothetical cleavage of 29 N-terminal amino acids (corresponding to ∼3.5 kDa) could not account for the remaining ∼10 kDa mass difference between the L and S bands after a combined PNGaseF/α-mannosidase treatment, we concluded that this difference reflects a posttranslational modification of ExDG. At least in part, this difference is explained by the presence of O-mannose residues resistant to α-mannosidase since the L glycoform retains residual Con A reactivity after combined PNGaseF/α-mannosidase treatment (Figure 4). Thus, the L and S bands indeed most likely represent distinct glycoforms of ExDG that are primarily different in O-mannosylation, which, however, does not exclude a possible contribution of other types of modifications triggered or influenced by O- mannosylation.
Staining with VVA lectin that recognizes terminal GalNAc was previously found to correlate with the level of DG expressed in Drosophila muscles and NMJ (Haines et al. 2007), suggesting that Drosophila DG may bear this modification. Thus, besides O-mannose, the L and S glycoforms may be modified with O-linked GalNAc. Interestingly, we found that the reactivity of ExDG bands to VVA and WFA lectins that recognize O-linked GalNAc was essentially complementary to that of Con A and HHL that bind O-mannose, with the S band from rt mutants being most prominently stained with both VVA and WFA, the S band from RT-TW coexpression showing significantly weaker reactivity, and the L band being barely detectable with these lectins (Figure 5A and B). Thus, O-GalNAc appears to be present on ExDG glycoforms in a reverse proportion to O-mannose. This result is consistent with our finding that two of the identified O-mannose sites could also be modified by O-linked GalNAc (Table I, Figure 7). Since RT and TW function in the ER (Lyalin et al. 2006), upstream in the secretory pathway to O-GalNAc-transferases, PGANTs, that function in the Golgi (Rottger et al. 1998), this complementary pattern of glycosylation suggest that O-mannosylation can compete with O-GalNAc modification of DG. Thus, these data provide further support for our conclusion that the majority of O-mannosylation occurs within the region of mucin-type domain, where O-mannose could directly compete with the bulk of predicted O-GalNAc modification. This possible competition may underlie a regulatory mechanism that modulates DG function, for instance, via changing the ligand-binding activity of DG.
Importantly, the mass spectrometry analysis of ExDG purified from RT-TW co-expression background unequivocally detected O-mannosylation sites outside of the mucin-type region of ExDG (Table I, Figure 7, supplementary Figure 1). To our knowledge, this is the first report of animal O-mannosylation outside of mucin-type domain. This result has implication for understanding the substrate specificity of animal protein O-mannosyltransferases. The identified O-mannose sites on Drosophila DG neither agree with a consensus sequence proposed for mammalian O-mannosylation based on in vitro assays (Manya et al. 2007), nor conform to the sequence preferences proposed for in vivo O-mannose modification of α-DG (Breloy et al. 2008). It is worth noting that previous studies were focused on the modification α-DG mucin domain and used relatively short peptides or fragments of α-DG for assaying the substrate specificity of O-mannosylation. In our experiments, we analyzed O-mannosylation of a complete extracellular domain of DG. Thus, the mentioned difference in sequences of identified O-mannose sites can suggest that the substrate recognition for O-mannosylation in vivo occurs differently in a context of a full-length protein as opposed to a peptide substrate, which is in general agreement with the previous conclusion that structural elements distant from modification sites can determine the substrate recognition for O-mannosylation in vivo (Breloy et al. 2008). Alternatively, this result can suggest that Drosophila and mammalian O-mannosylation may be controlled by different structural properties of substrates. Interestingly, three out of the six identified O-mannose sites are located within the region that is shared by all three Drosophila DG isoforms produced by alternative splicing (Figures 1 and 7), which suggests that all of them, not just DG-C, may be targeted by O-mannosylation.
Intriguingly, the generation of the L glycoform by RT-TW activity appears to be an “all-or-nothing” process with the absence of intermediate forms. This intriguing observation suggests a possible mechanism of O-mannosylation that requires recognition of some determinants that results in quantitative modification of nearly all available sites. These determinants may belong to DG itself, such as some posttranslational modifications, or they may represent certain factors that regulate the function of the RT-TW complex within the cell. The first scenario includes an interesting possibility that recognition by RT-TW outside of the mucin domain may serve as a determinant that initiates processive O-mannosylation of DG within its mucin domain region. This possibility is consistent with the difference in O-mannose sites outside of the mucin region on the L and S glycoforms from RT-TW coexpression, as well as with the evidence that a region N-terminal to mucin-type domain plays a role in O-mannosylation initiation of human α-DG mucin domain (Breloy et al. 2008). In any case, these determinants, or factors that influence them, may provide an additional level of DG regulation and modulate the ratio of S and L glycoforms in a cell-specific manner. This hypothesis predicts that the glycoform ratio may vary between different cells independently from the level of RT-TW activity, and this interesting possibility will be addressed in future experiments.
Protein O-mannosylation in yeast has been implicated in secretory protein sorting, protein stability, and degradation via ERAD-mediated pathway (Lommel et al. 2004; Proszynski et al. 2004; Hirayama et al. 2008). However, in our experiments, the immunofluorescent detection and optical sectioning did not reveal significant differences in the level of expression or subcellular localization of ExDG within the wing imaginal disk epithelium in the presence or absence of RT-TW activity. At the same time, we found that the dominant-negative effect of ExDG on the crossvein formation is significantly potentiated by RT-TW activity (Figure 9), which suggests that the in vivo activity of ExDG is modulated by O-mannosylation and that the outcome of DG function depends on the balance between the amounts of L and S glycoforms. Thus, we think that O-mannosylation does not affect the stability or trafficking of Drosophila DG, but rather it changes its functional properties, e.g. by modifying interactions with extracellular ligands. These results suggest that O-mannosylation can function as a molecular regulator of the DGC-mediated pathway via changing the ligand-binding activity of DG.
In conclusion, the present study has unambiguously demonstrated that the extracellular domain of Drosophila DG-C is a target of O-mannosylation. We found that O-mannosylation is an abundant modification of ExDG that requires concurrent in vivo activity of two Drosophila protein O-mannosyltransferases, RT and TW. A combination of glycosidase treatments, lectin blotting, trypsin digestion, and mass spectrometry analyses provided strong evidence that the majority of O-mannose is located within a central 250 a.a. region of the extracellular part of DG that includes a mucin-type domain. Our experiments revealed that subcellular localization and trafficking of ExDG are not affected by RT and TW, while ExDG activity can be modulated by RT–TW activity in vivo ExDG, thus suggesting that O-mannosylation regulates ligand-binding activity of Drosophila DG. These results highlight the evolutionary conservation of mechanism of O-mannosylation between Drosophila and mammals and suggest that Drosophila can be a suitable model system to study molecular and genetic mechanisms of mammalian α-DG O-mannosylation. In addition, our experiments revealed an interplay between two O-glycosylation pathways that modify DG with O-mannose and O-GalNAc, which suggests that O-mannose can function as a molecular switch that regulates other types of glycosylation. In addition, we found that O-mannose sites can be located far outside of the mucin-type domain, which suggests that proteins without mucin-type domains may also be targets of O-mannosylation in vivo.
Material and methods
Drosophila strains and cDNA
The following Drosophila mutant alleles and transgenic insertions were obtained from the Bloomington Drosophila Stock Center, Indiana University: tw1, rtp, rt2, Df(1)su(s)83 (tw deficiency), Dp(1;Y)y2sc (tw duplication), Act5C-GAL4–25, tubP-GAL4 (LL7), ptc-GAL4. UAS-rt, and UAS-tw transgenes were previously described (Lyalin et al. 2006). Drosophila Dg-C cDNA was obtained from Dr. Ruohola-Baker (University of Washington, Seattle).
Constructs and proteins for in vitro O-mannosylation assays
Templates for engineering expression constructs were plasmids with cDNA sequences of Dg-A (obtained from DGRC, Indiana University), Dg-C (obtained from Hannele Ruohola-Baker, Seattle), and rabbit α-Dg (DGFc5, a gift from Michael Oldstone and Stephan Kunz from Scripps Institute, La Jolla, supported by NIH grant AI09484 (Kunz et al. 2001)). Fragments of Dg-A, Dg-C, and rabbit Dg cDNAs encoding protein regions with predicted abundant O-glycosylation (amino acids 204–399 for DG-A, 315–592 for DG-C, and 314–487 for rabbit α-DG) were PCR-amplified and cloned into the pET41a vector (Novagen, EMD USA, Gibbstown, NJ) between NcoI and EagI cloning sites and in frame with the N-terminal GST tag. The GST-fused proteins were expressed and purified from E. coli BL21(DE3) cells according to manufacturer's protocol (Novagen). Briefly, the protein expression was induced with 0.4 mM IPTG for 18 h, and then cells were harvested and lysed in the wash buffer (PBS, 0.1% NP-40, 0.1% PMSF) by sonication. The lysates were pre-cleared by centrifugation and 0.45 μm membrane filtering, and incubated with GST-beads on a nutator at 4°C overnight. Then, beads were extensively washed with the wash buffer and purified proteins were eluted with 100 mM glutathione in 0.5M Tris, pH 8.0. The eluted proteins were dialyzed against 2 mM EDTA, 0.1% PMSF, 20 mM Tris pH 8.0, concentrated by Millipore centrifugal filters (15 kDa cut-off), and used as substrates in the in vitro O-mannosylation assay.
In vitro O-mannosylation assays
The assays were performed essentially as described previously (Ichimiya et al. 2004). Briefly, a reaction mixture (20 μL total volume) contained 20 mM Tris, pH 8.0, 100 nM [3H]- mannosylphosphoryldolicol (ARC, Inc.), 2 mM 2-mercapto- ethanol, 10 mM EDTA, 0.5% n-octyl-β-d-thioglucoside, 10 μg GST-tagged acceptor protein, and 80 μg of microsomal membrane fraction as a source of O-mannosyltransferase activity. The microsomal membrane fraction was prepared from third instar Drosophila larvae using previously published protocol (Ichimiya et al. 2004). The concentration of proteins in microsomal fraction was determined by the Bradford assay. After 1 h incubation at 24°C, mannosyltransferase reactions were stopped by adding 200 μL PBS with 1% Triton X-100, the mixtures were centrifuged at 10,000 × g for 10 min at 4°C, and the supernatant was bound to pre-washed GST-beads on a nutator. Then, the beads were washed three times with PBS containing 0.5% Triton X-100, and the incorporation of radioactive mannose was measured in dpm using a liquid scintillation counter. Background control was determined by a mock assay following the same protocol but with 10 μg of BSA instead of a real acceptor. The results of the assay were presented as the ratio of radioactive mannose incorporation for Dystroglycan acceptors to that for BSA as a control.
ExDg construct
The ExDg construct was generated by in-frame fusion of cDNA region encoding the first 1048 amino acids of DG-C isoform (the entire predicted extracellular domain of DG-C) with the fragment encoding the 3xFLAG tag (Sigma) using standard molecular biology techniques. Details on molecular cloning of ExDg are available on request. The ExDG protein encoded by the construct lacks transmembrane domain and is predicted to be a secreted protein.
Expression of ExDG in Drosophila S2 cells
S2 tissue culture cells were maintained and transfected as previously described (Koles et al. 2004). For tissue culture expression, ExDg was subcloned into the pMK33 vector with metallothionein promoter using standard molecular biology techniques (details are available upon request). The expression of ExDG was induced in transiently transfected cells by 0.7 mM CuSO4.
In vivo expression and purification of ExDG
ExDg was cloned into the pUAST vector for in vivo expression, and transgenic Drosophila strains were obtained by P-element-mediated germline transformation. In vivo expression of the UAS-ExDg transgene was induced using the UAS-GAL4 system (Brand et al. 1994). The specificity of ExDG detection by Western blot was confirmed using ‘negative control’ samples without ExDG expression. ExDG was expressed in vivo in the following genetic backgrounds: rt−, rtP/rt2; tw−, tw1/ Df(1)su(s)83 (where Df(1)su(s)83 is a tw deficiency); rt+tw+, UAS-rt,UAS-tw/Act5C-GAL4; rt+, UAS-rt/Act5C-GAL4; tw+, UAS-tw/Act5C-GAL4. ExDG protein was purified from pupae with anti-FLAG M2 agarose (Sigma). For each purification experiment, 15–80 larvae or pupae were collected and lysed in 300 μL–1.5 mL of lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X100) including cocktail of protease inhibitors (Complete, Roche). Following centrifugation at 18,000 × g for 20 min at 4°C, the supernatant was added to 10–40 μL of FLAG agarose beads and incubated for 2 h at 4°C with nutation. The agarose beads were then washed four times with the lysis buffer. The purified ExDG bound to beads was directly used in later assays.
Glycosidase treatments
ExDG protein purified from 3–4 pupae was incubated with 500 units of PNGaseF (NEB) in 5 mM sodium phosphate buffer (pH 7.5) for 1 h at 37°C (mock control was without PNGaseF). It was later treated with 0.4 milliunits of α-mannosidase from Jack beans (Sigma) (or without α-mannosidase for control) in the 50 mM sodium citrate buffer (pH 4.5) for 3 h at 37°C. Reactions were stopped by adding 2× SDS loading buffer and used in Western or lectin blot analyses.
Western and lectin blot analyses
Analyses were performed according to standard protocols. Briefly, the tissue lysates normalized by the Bradford method for protein amount, or purified ExDG from pupae, were run on 5% SDS–PAGE gel and then the separated proteins were transferred onto a nitrocellulose membrane. For Western blotting, mouse anti-FLAG M2 primary (Sigma) and rabbit anti-mouse HRP-conjugated secondary (Jackson Laboratories) antibodies were used for detection by chemiluminescence (SuperSignal, Thermo Scientific). For lectin blots, proteins on the membrane were blocked with 2% BSA (Roche) in TBST (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween 20) and then incubated with biotinylated lectins (2.5 μg/mL of Con A, or 10 μg/mL of VVA, WFA, HHL, and PNA, all from Vector) for 1 h at room temperature followed by detection with a Vectastain ABC kit (Vector). The specificity of lectin staining was confirmed by incubation with lectin in the presence of corresponding inhibiting sugars (i.e., 0.2 M GalNAc for VVA and WFA, 0.2 M galactose for PNA, 0.2 M methyl α-d-mannopyranoside for Con A and HHL). Blots were quantified using the ChemiDoc XRS system (Bio-Rad).
In-gel digestion of purified ExDG
ExDG was purified from ∼60 pupae and separated by 5% SDS–PAGE. Protein bands visualized by Coomassie G250 staining were cut from the gel. In-gel digestion was performed as follows: Coomassie G250 was removed from the selected bands by repeated swelling and shrinking with 40 mM ammonium bicarbonate and acetonitrile, respectively. After destaining, the proteins within the gel bands were reduced by swelling in 40 mM ammonium bicarbonate containing 10 mM DTT for 1 h at 56°C, followed by carbamidomethylation with 55 mM iodoacetomide (Sigma). Bands were swelled, dehydrated, and dried to dryness using SpeedVac. Both sequence grade trypsin (Promega) and elastase (Worthington Biochemical) were reconstituted using 40 mM ammonium bicarbonate. The dried gel pieces were swelled on ice by adding 5 μg of both trypsin and elastase. After swelling for a period of 45 min on ice, the bands were then subjected to overnight digestion at 37°C. The resulting glycopeptides were extracted three times for 15 min using 5% formic acid in 25%, 50%, and 75% acetonitrile. The extracted glycopeptides were dried down using a SpeedVac. Sequencing of the mucin region of DG proved to be difficult possibly because of the extensive glycosylation and/or the lack of suitable amino acids preventing enzymatic cleavage of the protein backbone.
Mass spectrometry analyses: BEMAD
β-Elimination through Michael addition with DTT (BEMAD) was carried out essentially as described previously (Wells et al. 2002). Briefly, lyophilized peptides were β-eliminated by resuspending them in 1% triethylamine and 0.1% sodium hydroxide. The peptides were then subjected to Michael addition with DTT. The reaction was incubated at 52°C for 2 h. After completion of incubation, the reaction was quenched with trifluoroacetic acid (final concentration ∼1%) and the peptides were dried in a Speed Vac. BEMAD allowed us to identify sites of glycosylation by detecting a mass increase of 136.2 Daltons due to DTT addition. However, the identity of the glycan present at those sites could not be distinguished. To reveal the identity of glycosylation, the BEMAD data were combined with the results of the pseudo-neutral loss method on a linear ion trap.
Nano-LC-MS3
Extracted ExDG glycopeptides were also analyzed using Nano-LC-MS3 on a linear ion trap mass spectrometer (LTQ; ThermoFisher). The glycopeptides were resuspended in 0.5 μL of solvent B (0.1% formic acid/80% acetonitrile) and 19.5 μL of solvent A (0.1% formic acid) and loaded on a 75 mm × 8.5 cm C18 reverse phase column (packed in-house, YMC GEL ODS-AQ120ÅS-5) using a nitrogen bomb. Peptides were eluted using a linear gradient increasing from 5% to 50% B over 85 min. Each full MS scan from 300 to 2000 m/z yielded five MS/MS scans of the top five most intense peaks with a dynamic exclusion of 2 for 30 s. Data-dependent MS3 scans were triggered if a neutral loss was observed equal to the singly or doubly charged mass of Hexose, HexNAc, Fucose, or Neu5Ac (sialic acid) within the top three peaks from the MS/MS scan.
Data analysis
The resulting data were searched against a nonredundant Drosophila database (Flybase), as well as a database only containing the Drosophila melanogaster protein sequences for α-DG (NCBI) added to the common contaminants database (Thermo Fisher), using the TurboSequest algorithm (Bio-Works 3.3, Thermo Fisher). For identifying glycopeptides, a mass increase of 162.1, 203.1, and 365.2 Daltons was allowed on serines and threonines looking for the addition of mannose, GalNAc, and Hex-HexNAc. Additionally, when searching the peptides that underwent the BEMAD procedure, a mass increase of 136.2 Daltons on both serines and threonines was allowed dynamically. All peptides indicating a posttranslational modification were confirmed through manual inspection after automatically deleting all peptides matching back to α-DG with Sf and P scores below 0.3 and 20.
Analysis of trypsin-resistant glycopeptides of ExDG
ExDG purified from ∼80 Drosophila pupae was treated with 1000 units of PNGaseF as described above. After incubation in 4 mM DTT /6.4 M urea at ∼100°C for 20 min, denatured ExDG was incubated with 0.5 μg of trypsin (TPCK treated, Sigma) in 0.9 M urea 43 mM Tris, pH 8.0, overnight. Trypsin was inactivated by boiling and the sample was dialyzed against 37.5 mM NaCl 12.5 mM Tris-HCl, pH 7.4, for binding to lectin affinity beads, or against 10 mM sodium citrate, pH 4.5, for α-mannosidase treatment (see below). The dialyzed ExDG peptides were incubated with ConA, WFA, and VVA agarose beads (Vector) in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1 mM CaCl2, 0.01 mM MnCl2, and 0.05% Tween-20 for 2 h at room temperature. The beads were washed 3 times with the incubation buffer, and the bound fraction was analyzed by SDS–PAGE and PAS-silver staining. Treatment with α-mannosidase was performed in 45 mM sodium citrate, pH 4.5, at 37°C overnight using 2.2 μg (0.05 milliunits) of α-mannosidase from jack beans (Sigma). In the control experiment, ExDG peptides were incubated using this protocol with the same amount of heat-inactivated α-mannosidase. The samples were analyzed by SDS–PAGE and PAS/PAS-silver staining.
PAS and PAS-silver staining
PAS (periodic acid-Schiff) and PAS-silver staining was performed essentially as described before with some modifications (Zacharius et al. 1969; Gradilone et al. 1998). Briefly, for PAS staining, gels were incubated in 1% periodic acid for 15 min at 4°C following rinse with 7.5% acetic acid for 5 min. After six washes with H2O (5 min each), gels were incubated in Schiff reagent for 15 min at 4°C and then washed with 0.5% sodium metabisulfite three times for 10 min followed by extensive washes with H2O. For PAS-silver staining, gels were PAS-stained and then incubated in 0.2% AgNO3/0.03% formaldehyde for 15 min. The staining was developed in 2.3% Na2CO3/0.01% formaldehyde/0.001% sodium thiosulfate for 5–10 min, and the development of staining was stopped by of 2.5% acetic acid.
Immunostaining and microscopy
Expression of the pUAST-ExDg construct was induced in wing imaginal disks using the ptc-GAL4 driver. Third instar imaginal disks were dissected, fixed, and stained as described previously (Panin et al. 1997). The following antibodies and corresponding dilutions were used for immunostaining: primary mouse anti-FLAG (1:2,000) (Sigma) and secondary donkey anti-mouse Cy3 (1:250) (Jacksons Laboratories). Digital images were obtained using a Zeiss Axioplan 2 fluorescent microscope with the ApoTome module for optical sectioning. Z-sections were reconstructed using Zeiss AxioVision software.
Bioinformatic analyses
Prediction of DG glycosylation and signal peptide cleavage was performed by NetNGlyc, NetOGlyc, and Signal IP software at the Center for Biological Sequence Analysis site, DTU, Denmark (http://www.cbs.dtu.dk/services/) (Bendtsen et al. 2004; Julenius et al. 2005).
Supplementary data
Supplementary data for this article is available online at http://glycob.oxfordjournals.org/.
Supplementary Material
Acknowledgments
We thank Kate Koles for help with generating pET-Dg-A construct, Dr. Ruohola-Baker for Dg-C cDNA, Dr. Kunz and Dr. Oldstone for rabbit -Dg construct. We are grateful to Michiko Nakamura for help with Drosophila experiments.
-Dg construct. We are grateful to Michiko Nakamura for help with Drosophila experiments.
Funding
NIGMS grant (GM069952 to V.M.P); a NCRR grant (P41RR018502, L.W. senior investigator); and the Muscular Dystrophy Association (MDA4074 to L.W.).
Abbreviations
- BEMAD
β-elimination through Michael addition with DTT
- Con A
concanavalin A
- Dg
dystroglycan (gene)
- DG
Dystroglycan (protein)
- DGC
dystrophin-glycoprotein complex
- ECM
extracellular matrix
- GalNAc
N-acetyl-d-galactosamine
- GlcNAc
N-acetyl-d-glucosamine
- HHL
Hippeastrum hybrid lectin
- PAS
periodic acid-Schiff
- PGANT(s)
polypeptide GalNAc-transferase(s)
- PNA
Peanut agglutinin
- rt
rotated abdomen (gene)
- RT
rotated abdomen (protein)
- tw
twisted (gene)
- TW
twisted (protein); VVA, Vicia villosa; WFA, Wisteria floribunda agglutinin
References
- Aoki K, Perlman M, Lim JM, Cantu R, Wells L, Tiemeyer M. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J Biol Chem. 2007;282:9127–9142. doi: 10.1074/jbc.M606711200. [DOI] [PubMed] [Google Scholar]
- Aoki K, Porterfield M, Lee SS, Dong B, Nguyen K, McGlamry KH, Tiemeyer M. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J Biol Chem. 2008;283:30385–30400. doi: 10.1074/jbc.M804925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: From biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Bogdanik L, Framery B, Frolich A, Franco B, Mornet D, Bockaert J, Sigrist SJ, Grau Y, Parmentier ML. Muscle dystroglycan organizes the postsynapse and regulates presynaptic neurotransmitter release at the Drosophila neuromuscular junction. PLoS ONE. 2008;3:e2084. doi: 10.1371/journal.pone.0002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Manoukian AS, Perrimon N. Ectopic expression in Drosophila. Methods Cell Biol. 1994;44:635–654. doi: 10.1016/s0091-679x(08)60936-x. [DOI] [PubMed] [Google Scholar]
- Breloy I, Schwientek T, Gries B, Razawi H, Macht M, Albers C, Hanisch FG. Initiation of mammalian O-mannosylation in vivo is independent of a consensus sequence and controlled by peptide regions within and upstream of the alpha-dystroglycan mucin domain. J Biol Chem. 2008;283:18832–18840. doi: 10.1074/jbc.M802834200. [DOI] [PubMed] [Google Scholar]
- Christoforou CP, Greer CE, Challoner BR, Charizanos D, Ray RP. The detached locus encodes Drosophila Dystrophin, which acts with other components of the Dystrophin Associated Protein Complex to influence intercellular signalling in developing wing veins. Dev Biol. 2008;313:519–532. doi: 10.1016/j.ydbio.2007.09.044. [DOI] [PubMed] [Google Scholar]
- Deng WM, Schneider M, Frock R, Castillejo-Lopez C, Gaman EA, Baumgartner S, Ruohola-Baker H. Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development. 2003;130:173–184. doi: 10.1242/dev.00199. [DOI] [PubMed] [Google Scholar]
- Endo T. O-Mannosyl glycans in mammals. Biochim Biophys Acta. 1999;1473:237–246. doi: 10.1016/s0304-4165(99)00182-8. [DOI] [PubMed] [Google Scholar]
- Gradilone SA, Arranz SE, Cabada MO. Detection of highly glycosylated proteins in polyacrylamide gels. Anal Biochem. 1998;261:224–227. doi: 10.1006/abio.1998.2747. [DOI] [PubMed] [Google Scholar]
- Greener MJ, Roberts RG. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett. 2000;482:13–18. doi: 10.1016/s0014-5793(00)02018-4. [DOI] [PubMed] [Google Scholar]
- Haines N, Seabrooke S, Stewart BA. Dystroglycan and protein O-mannosyltransferases 1 and 2 are required to maintain integrity of Drosophila larval muscles. Mol Biol Cell. 2007;18:4721–4730. doi: 10.1091/mbc.E07-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama H, Fujita M, Yoko-o T, Jigami Y. O-Mannosylation is required for degradation of the endoplasmic reticulum-associated degradation substrate Gas1*p via the ubiquitin/proteasome pathway in Saccharomyces cerevisiae. J Biochem. 2008;143:555–567. doi: 10.1093/jb/mvm249. [DOI] [PubMed] [Google Scholar]
- Ichimiya T, Manya H, Ohmae Y, Yoshida H, Takahashi K, Ueda R, Endo T, Nishihara S. The twisted abdomen phenotype of Drosophila POMT1 and POMT2 mutants coincides with their heterophilic protein O-mannosyltransferase activity. J Biol Chem. 2004;279:42638–42647. doi: 10.1074/jbc.M404900200. [DOI] [PubMed] [Google Scholar]
- Julenius K, Molgaard A, Gupta R, Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology. 2005;15:153–164. doi: 10.1093/glycob/cwh151. [DOI] [PubMed] [Google Scholar]
- Koles K, Irvine KD, Panin VM. Functional characterization of Drosophila sialyltransferase. J Biol Chem. 2004;279:4346–4357. doi: 10.1074/jbc.M309912200. [DOI] [PubMed] [Google Scholar]
- Koles K, Lim JM, Aoki K, Porterfield M, Tiemeyer M, Wells L, Panin V. Identification of N-glycosylated proteins from the central nervous system of Drosophila melanogaster. Glycobiology. 2007;17:1388–1403. doi: 10.1093/glycob/cwm097. [DOI] [PubMed] [Google Scholar]
- Kucherenko MM, Pantoja M, Yatsenko AS, Shcherbata HR, Fischer KA, Maksymiv DV, Chernyk YI, Ruohola-Baker H. Genetic modifier screens reveal new components that interact with the Drosophila dystroglycan–dystrophin complex. PLoS ONE. 2008;3:e2418. doi: 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Sevilla N, McGavern DB, Campbell KP, Oldstone MB. Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]-dystroglycan. J Cell Biol. 2001;155:301–310. doi: 10.1083/jcb.200104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ball SL, Yang Y, Mei P, Zhang L, Shi H, Kaminski HJ, Lemmon VP, Hu H. A genetic model for muscle—eye–brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1) Mech Dev. 2006;123:228–240. doi: 10.1016/j.mod.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Lommel M, Bagnat M, Strahl S. Aberrant processing of the WSC family and Mid2p cell surface sensors results in cell death of Saccharomyces cerevisiae O-mannosylation mutants. Mol Cell Biol. 2004;24:46–57. doi: 10.1128/MCB.24.1.46-57.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyalin D, Koles K, Roosendaal SD, Repnikova E, Van Wechel L, Panin VM. The twisted gene encodes Drosophila protein O-mannosyltransferase 2 and genetically interacts with the rotated abdomen gene encoding Drosophila protein O-mannosyltransferase 1. Genetics. 2006;172:343–353. doi: 10.1534/genetics.105.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, Margolis RU, Endo T. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manya H, Suzuki T, Akasaka-Manya K, Ishida HK, Mizuno M, Suzuki Y, Inazu T, Dohmae N, Endo T. Regulation of mammalian protein O-mannosylation: Preferential amino acid sequence for O-mannose modification. J Biol Chem. 2007;282:20200–20206. doi: 10.1074/jbc.M702369200. [DOI] [PubMed] [Google Scholar]
- Martin PT. Mechanisms of disease: Congenital muscular dystrophies-glycosylation takes center stage. Nat Clin Pract Neurol. 2006;2:222–230. doi: 10.1038/ncpneuro0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med. 2007;7:417–425. doi: 10.2174/156652407780831601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North SJ, Koles K, Hembd C, Morris HR, Dell A, Panin VM, Haslam SM. Glycomic studies of Drosophila melanogaster embryos. Glycoconj J. 2006;23:345–354. doi: 10.1007/s10719-006-6693-4. [DOI] [PubMed] [Google Scholar]
- Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates Notch–ligand interactions. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- Proszynski TJ, Simons K, Bagnat M. O-Glycosylation as a sorting determinant for cell surface delivery in yeast. Mol Biol Cell. 2004;15:1533–1543. doi: 10.1091/mbc.E03-07-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebay I, Fehon RG, Artavanis-Tsakonas S. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell. 1993;74:319–329. doi: 10.1016/0092-8674(93)90423-n. [DOI] [PubMed] [Google Scholar]
- Rottger S, White J, Wandall HH, Olivo JC, Stark A, Bennett EP, Whitehouse C, Berger EG, Clausen H, Nilsson T. Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J Cell Sci. 1998;111(Pt 1):45–60. doi: 10.1242/jcs.111.1.45. [DOI] [PubMed] [Google Scholar]
- Schneider M, Khalil AA, Poulton J, Castillejo-Lopez C, Egger-Adam D, Wodarz A, Deng WM, Baumgartner S. Perlecan and Dystroglycan act at the basal side of the Drosophila follicular epithelium to maintain epithelial organization. Development. 2006;133:3805–3815. doi: 10.1242/dev.02549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, Baker D, Ruohola-Baker H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. Embo J. 2007;26:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Artavanis-Tsakonas S. The intracellular deletions of Delta and Serrate define dominant negative forms of the Drosophila Notch ligands. Development. 1996;122:2465–2474. doi: 10.1242/dev.122.8.2465. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker–Warburg syndrome. J Med Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wairkar YP, Fradkin LG, Noordermeer JN, DiAntonio A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci. 2008;28:3781–3789. doi: 10.1523/JNEUROSCI.0478-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics. 2002;1:791–804. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- Willer T, Prados B, Falcon-Perez JM, Renner-Muller I, Przemeck GK, Lommel M, Coloma A, Valero MC, de Angelis MH, Tanner W, et al. Targeted disruption of the Walker–Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc Natl Acad Sci USA. 2004;101:14126–14131. doi: 10.1073/pnas.0405899101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- Zacharius RM, Zell TE, Morrison JH, Woodlock JJ. Glycoprotein staining following electrophoresis on acrylamide gels. Anal Biochem. 1969;30:148–152. doi: 10.1016/0003-2697(69)90383-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.