

Abstract
An ongoing research and multiple clinical trials involve new targeted therapies and less aggressive treatment regimens that improve survival in patients with lung cancer. Targeted therapeutic agents are based on the concept of discovering genetic alterations and the signaling pathways altered in cancer and have added significantly to our armamentarium in order to prolong patient survival and minimizing drug toxicity. Among 34 molecularly targeted drugs approved by U.S. Food and Drug Administration (FDA) for treatment of various cancers since 1998 three targeted therapies have been approved for treatment of lung cancer (gefitinib in 2002, erlotinib in 2003, and bevacizumab in 2006).
This review focuses on the targeted therapies in lung cancer, the molecular biomarkers that help identify patients that will benefit for these targeted therapies, describes the basic molecular biology principles and selected molecular diagnostic techniques and the pathological features correlated with molecular abnormalities in lung cancer. Lastly, new molecular abnormalities described in lung cancer that are predictive to novel promising targeted agents in various phases of clinical trials are discussed.
Keywords: Lung cancer, adenocarcinoma, squamous, tyrosine kinase inhibitors, molecular pathogenesis
OVERVIEW
Approximately 219 000 cases of lung cancer were diagnosed in the United States in 2009, accounting for about 15% of all new cancers.[1] Lung cancer is the second most common cancer in men, after prostate cancer and in women, after breast cancer. In 2009, lung cancer caused an estimated 159 000 deaths (89 000 among men and 70 000 among women). The overall lifetime risk of lung cancer is about 1 in 13 for men and 1 in 16 for women. The risk is significantly higher for smokers and lower for nonsmokers.[2,3] Unfortunately, the 5-year overall survival rate did not change substantially in the past 30 years and is 15% despite current therapeutic approaches.[4] Lung cancer is one of the most challenging cancers to treat and the standard therapy includes surgical resection, platinum-based chemotherapy, and radiation therapy alone or in combination. New research and ongoing clinical trials involve new targeted therapies and milder treatment regimens that improve survival. Targeted therapeutic agents are based on the concept of discovering genetic alterations and the signaling pathways altered in cancer and have added significantly to our armamentarium in order to prolong patient survival and minimizing drug toxicity. Although improvements seen in the trials are modest, the hope is that an increased number of biomarkers will be available in the near future to help oncologists and treating physicians predict which patients are most likely to benefit from such therapies. Since 1998, when Trastuzumab was the first molecularly targeted drug approved by U.S. Food and Drug Administration (FDA), 34 more drugs were introduced in the U.S.A. for treatment of various cancers until the end of 2009. However, the clinical response obtained from targeted therapies is extremely variable. [5]
Development of targeted therapies in lung cancer changed our pathology practice. Historically, the distinction between small cell and non-small cell lung carcinoma (NSCLC) was used to guide treatment decision. Three major subtypes of NSCLC including adenocarcinoma, squamous cell carcinoma and large cell carcinoma were grouped together as a single entity for therapy. With the recent development of targeted therapies, the discrimination between specific NSCLC subtypes has a significant impact on therapy selection. Therefore, if distinction cannot be made on routine H&E sections, ancillary studies such as a small panel of immunoperoxidase studies (e.g. TTF-1 and surfactant apoproteins favor adenocarcinoma; p63 and cytokeratin 5/6 favor squamous cell carcinoma) should be performed. After the conventional pathologic analysis, tissue samples are subjected to analysis of molecular abnormalities that may be able to predict sensitivity and resistance to various chemotherapeutic agents and targeted therapies.
MOLECULAR ALTERATIONS AND TARGETED THERAPIES IN LUNG CANCER
In the past years, given the development of new targeted therapies, tremendous efforts are directed towards identifying potentially drugable molecular alterations, especially against known activating mutations. Although numerous mutations have been described in lung adenocarcinoma the mutation status remains unknown in more than 50% of cases (Figure 1).[6–7] Despite these efforts, so far we can identify at the present moment therapeutic targets in only 20% of lung cancers.
Figure 1.

Clinically important oncogenic mutations of the lung adenocarcinoma.
Targeted therapies in lung cancers with EGFR abnormalities
EGFR
The epidermal growth factor receptor (EGFR, HER-1/ErbB1) is a member of the ErbB family of tyrosine kinase receptors (TK), which includes HER-1/ErbB1, HER-2/neu/ErbB2, HER-3/ErbB3 and HER-4/ErbB4. It is composed of extracellular (ligand binding), transmembrane and intracellular (tyrosine-kinase) domain. Upon ligand binding and receptor homo-or hetero-dimerization and activation, activated EGFR signals downstream to the PI3K/AKT and RAS/RAF/MAPK pathways. These intracellular signaling pathways regulate key processes such as apoptosis and proliferation.
EGFR is expressed in 10–80% of NSCLCs depending on the methods of assessment This observation makes EGFR and its family members prime candidates for the development of targeted therapeutics (Figure 2)[8,9,10]. Status of the EGFR in lung cancer has been assessed by different methods including immunohistochemistry (protein expression), FISH/CISH (gene copy number changes) and mutational analysis. Somatic mutations in the exons 18–21 of the tyrosine-kinase (TK) domain of EGFR have been shown to correlate with a high likelihood of response to EGFR tyrosine kinase inhibitors (TKIs).[11,12,13] These mutations target four exons (18–21), which encode part of the tyrosine kinase domain (the entire kinase domain is encoded by exons 18–24) and are clustered around the ATP-binding pocket of the enzyme.[14] They occur in approximately 10% of Western and up to 50% of Asian patients. Several clinical features such as female gender, never smokers, Asian ethnicity and adenocarcinoma histology were found to be associated with increased response rate to EGFR TKIs and the presence of EGFR mutations. The most common are in-frame deletions in exon 19 (45%), followed by a point mutation (CTG to CGG) in exon 21 at nucleotide 2573 which results in substitution of leucine by arginine at codon 858 (L858R) (41%).
Figure 2.

Currently available therapies targeting EGFR signaling pathway.
Because EGFR gene mutations are the best predictor of patient’s response to EGFR TKI, various DNA mutational assays have been reported in literature. [15] Sangen DNA direct sequencing used to be the most common mutational technique used by many laboratories, but it may not detect mutations if tumor cells represent < 25% of the sample, which may be an issue with a small biopsy and cytology specimens. Therefore, more sensitive and comprehensive approaches have been developed.[16]
Genetic mutations in the gene encoding proteins involved in the EGFR signaling cascade (KRAS, HER2, BRAF, PI3K, LKB1, SHP2) seem to exist as mutually exclusive somatic mutations and are negative predictors of the tumor response to EGFR_TKI, with the possible exception of those in PI3K.[17] KRAS mutations which lead to substitutions of amino acids for glycines at positions 12 and 13 can be identified in up to 30% adenocarcinomas.[18,19,20] Secondary (acquired) resistance to EGFR TKI has been associated most frequently with mutation T790M in exon 20 of the TK domain, and less frequently with amplification of MET gene.[21, 22] Second generation EGFR TKIs are being develop to target T790M and other mechanisms of acquired resistance to gefitinib and erlotinib.[23] Therefore, clinical testing in lung adenocarcinomas goes beyond EGFR mutation status, and clinical laboratories are performing comprehensive mutational profiling of lung adenocarcinoma.
A subset of lung adenocarcinomas has activation of growth factor receptor (EGFR) by mutations and/or amplification, but the interaction between these is complex and unclear. EGFR gene amplification is detected in some EGFR-mutation positive patients as well, and is reported to be associated with disease progression.[24] However, this is an area of controversy, because different interpretation criteria and methods for EGFR amplification were used (FISH vs. qPCR). The group from University of Colorado has proposed a rather complex interpretation scheme for EGFR-FISH, which in addition to classical gene amplification also take into consideration a high polysomy as an indicator of FISH-positivity.[25] The importance of distinguishing increased copy number of chromosome 7 without EGFR gene amplification (high polysomy) and EGFR gene amplification is still uncertain. EGFR-FISH positivity is not limited to EGFR mutated group only. It is also present in tumors harboring KRAS mutations and in EGFR and KRAS wild type tumors.[26] This observation suggests that EGFR FISH cannot be solely used as a method to predict patient’s response to EGFR-TKI. Role of EGFR-FISH results in predicting the patient’s response to anti-EGFR antibodies has been suggested.[27] Recent reports suggest that chromogenic in-situ hybridization (CISH) can serve as a reliable alternative to FISH in determining EGFR copy number status in NSCLC. (Figure 3) [28]
Figure 3.

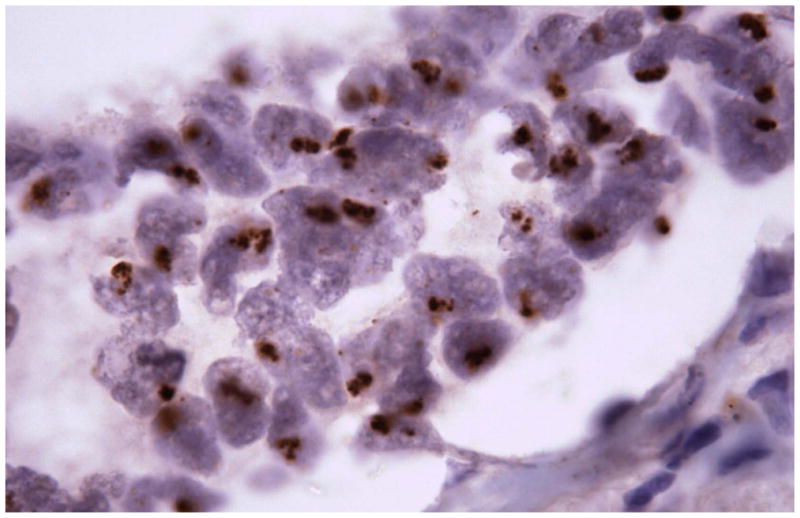
Figure 3a. Fluorescence in situ hybridization (FISH) with an EGFR probe (green) and 7q control probe (red) in NSCLC sections demonstrating gene amplification.
Figure 3b. The corresponding slides treated by chromogenic in situ hybridization (CISH) with the EGFR probe are illustrated in the same case of lung adenocarcinoma.
EGFR immunohistochemistry results highly depend on the antibody type procedure protocols, and interpretation criteria, and therefore the results with a wild type EGFR antibodies were contradictory.[29,30,31] Recently, mutation-specific rabbit monoclonal antibodies detecting two most common EGFR mutations (exon 19 deletions and exon 21 L858 mutation) have been reported [Figure 4].[32,33] This simple assay should be further validated, because it may provide a rapid and cost-effective clinical test to identify lung cancer patients responsive to EGFR-based therapies.
Figure 4.

Exon 19 deletion-specific antibody directly visualize the location of tumor cells with EGFR exon 19 deletion mutations and show heterogeneity in receptor overexpression among different tumor cells.
Mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) have prognostic significance because patients with EGFR-mutant NSCLC have prolonged survival compared with those with wild-type disease, regardless of the treatment received.[34,35] The other EGFR markers, such as protein expression determined by immunohistochemistry or gene amplification determined by FISH or CISH, are better predictors of survival. [36,37]
Targeted agents against lung cancer with EGFR mutations
The two TKI agents approved for use in lung cancer that target lung cancer with EGFR mutations are gefitinib (2002) and erlotinib (2003). EGFR mutation is a specific target for therapy by TKIs and is a validated biomarker of response. Its clinical utility is supported by prospective clinical trials that have demonstrated an increase in progression-free survival in EGFR mutant patients treated with TKI as first-line therapy.[38] Resistance to TKI therapy is associated with KRAS mutation and specific acquired EGFR mutations such as T790M. [39,40]These molecular events, as well as other genetic alterations in cMet (amplification), ERBB3 (over-expression) and epiregulin (autocrine loop activation), account for approximately 50% of cases of TKI-resistance. [22, 40,41]
Genotype-Phenotype Correlations
In patients with lung adenocarcinoma treated with erlotinib and gefitinib, favorable responses were associated with adenocarcinoma with bronchioloalveolar (BAC) and papillary patterns. [42,43]This finding led to trials of gefitinib and erlotinib in patients with BAC that showed that 20% of patients had a response to gefitinib.[44] The relationship of EGFR mutation status with adenocarcinoma subtype is a matter of intense debate.[26, 45–50] Genetic abnormalities can be seen in different histologies although with various frequency. One characteristic correlation is that mucinous adenocarcinoma (former mucinous BAC) is exclusively TTF-1 negative, EGFR mutation negative, but may have KRAS mutations. (Figure 5). [50]
Figure 5.
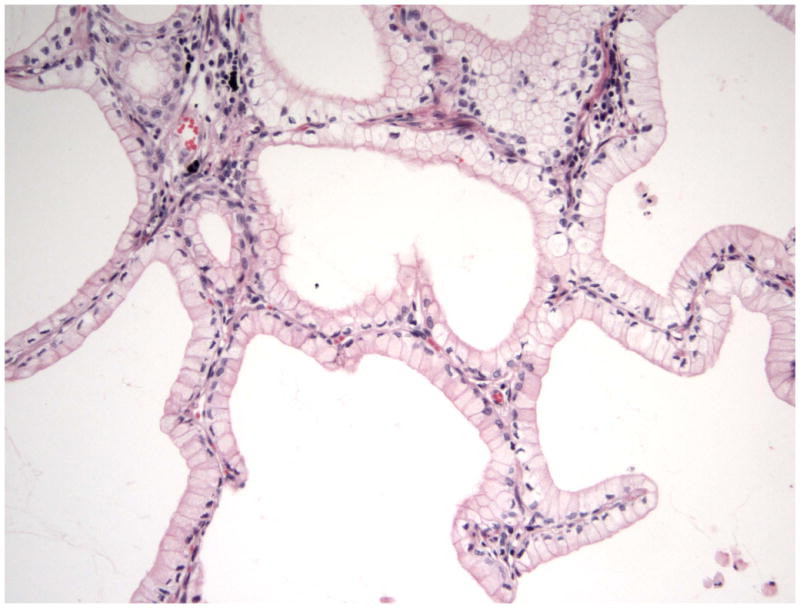
Bronchioloalveolar mucinous adenocarcinoma (mucinous BAC) is exclusively TTF-1 negative, EGFR mutation negative, but may have KRAS mutation.
Targeted Therapies with Angiogenesis Inhibitors in non-squamous NSCLC
Recent studies show that NSCLC with histologies different than squamous cell carcinoma appear to be more associated with response to treatment with bevacizumab. Bevacizumab (Avastin®) is a monoclonal antibody with high affinity for VEGF. Despite the promise supporting a benefit with bevacizumab for some patients with previously untreated advanced NSCLC, [51–52] the appropriate clinical setting for the use of antiangiogenic agents should be stringent, due to safety issues raised in patients with lung squamous cell carcinoma (SCC), which requires an accurate diagnosis on the pretreatment biopsy specimens. The clinical activity of bevacizumab in inoperable locally advanced, metastatic or recurrent NSCLC was first shown in chemotherapy-naïve patients.[53] Patients with non-squamous NSCLC histology are the only patients who benefit from treatment with bevacizumab in combination with chemotherapy. [51]
Bevacizumab is currently contraindicated in patients with SCC on the basis of the results of a recently published phase II trial that 31% of patients with SCC histology developed a life-threatening or fatal hemoptysis associated with bevacizumab, although it is still not clear whether histology alone is the reason for increased bleeding risk.[53] Excluding patients with SCC appeared to markedly limit the risk of life-threatening bleeding complications associated with bevacizumab.
Targeted therapies in lung cancers with ALK abnormalities
Anaplastic large cell lymphoma kinase gene (ALK) was originally identified through cloning of the t(2;5)(p23;35) translocation found in a subset of anaplastic large cell lymphomas (ALCLs), a tumor of T-cell lineage.[54–55] ALK encodes a tyrosine kinase receptor that is normally expressed only in select neuronal cell types. In ALK-rearranged anaplastic large cell lymphomas (ALCLs), the intracytoplasmic portion of ALK is fused to the N-terminal portion of nucleophosmin (NPM) resulting in a chimeric protein with constitutive kinase activity. Several other balanced translocations involving ALK have been discovered in ALCLs and in inflammatory myofibroblastic tumors; however the various resulting chimeric proteins all retain the ALK kinase domain. The importance of the kinase activity is exemplified by ALK-rearranged ALCL cell lines which are dependent upon ALK enzymatic activity for growth and survival.
Recently, ALK rearrangements were identified in rare non-small cell lung cancer (NSCLC) cell lines and in isolated primary adenocarcinomas from Japanese and Chinese populations.(Figure 6) [56, 57] The majority of the ALK rearrangements within NSCLCs result from an interstitial deletion and inversion in chromosome 2p and result in the EML4-ALK fusion gene product (Figure 7). [56,57] Murine tumors and human cell lines expressing EML-ALK are sensitive to inhibitors of ALK kinase activity. [58, 59] Together these data indicate that, like EGFR, ALK is an important molecular target in lung carcinoma. Thus, it will be critical to efficiently and accurately identify those lung adenocarcinomas that harbor ALK rearrangements in routine practice in order to guide the appropriate clinical therapy.
Figure 6.

ALK translocations with multiple gene partners.
Figure 7.

ALK fusion products in lung adenocarcinoma.
None of ALK-rearranged adenocarcinomas showed coexistent mutations in EGFR. Recently published studies show that ALK-rearranged adenocarcinomas are more likely to present in younger patients with a history of never-smoking, and at higher stage relative to those without ALK rearrangements (ALK germline).[60] The majority of ALK-rearranged adenocarcinomas had a distinct histology represented by solid tumor growth and frequent signet-ring cells with abundant intracellular mucin in Western population, however this was not demonstrated in Asian population (Figure 8). [61,62] Identification of lung cancers with chromosomal translocations involving ALK requires fluorescence in situ hybridization (FISH) on formalin-fixed, paraffin-embedded tumor tissues using a break-apart probe to the ALK gene (Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe, Abbott Molecular) (Figure 9). In contrast to ALCL, immunohistochemical detection of ALK translocation fusion products in lung carcinoma does not necessarily correlate with the presence of translocation. However, recently developed monoclonal antibody shows increased sensitivity for ALK detection and promise for applicability in the clinical laboratory (Figure 10). [63] This novel IHC assay correctly identifies ALK-rearranged lung adenocarcinoma with high reproducibility.
Figure 8.
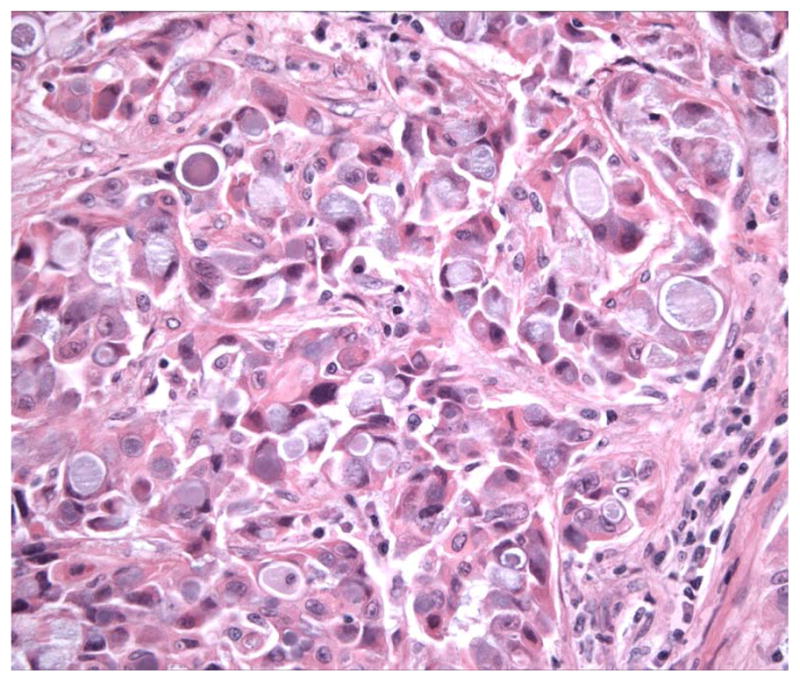
The majority of ALK-rearranged adenocarcinomas had a distinct histology represented by solid tumor growth and frequent signet-ring cells with abundant intracellular mucin.
Figure 9.

Identification of lung cancers with chromosomal translocations involving ALK requires fluorescence in situ hybridization (FISH) on formalin-fixed, paraffin-embedded tumor tissues using a break-apart probe to the ALK gene.
Figure 10.
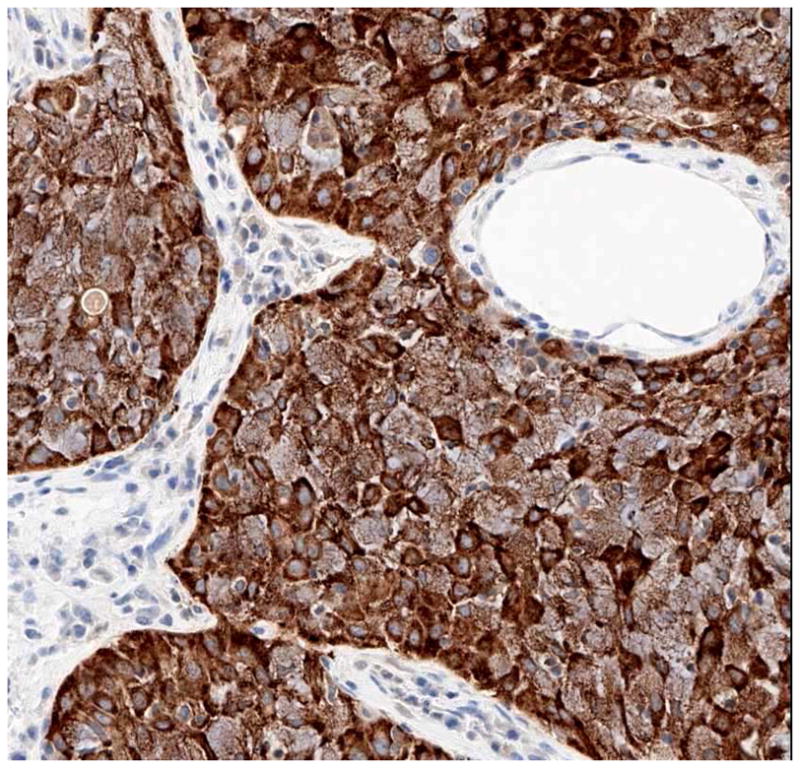
A novel immunoperoxidase assay that shows excellent sensitivity and specificity (100% and 99%, respectively) for the detection of ALK-rearranged lung adenocarcinomas in biopsy specimens with excellent interobserver agreement between pathologists.
Other Molecular Abnormalities That Show Promise for Targeted Therapies in Lung Cancer
HER-2
Unlike the other members of the HER family, HER-2 is not strictly a receptor tyrosine kinase because no high-affinity endogenous ligand has been identified. HER-2 acts as a signaling network coordinator and amplifier when it heterodimerizes with other HER family members. HER-2 mutations occur in 2% of NSCLCs.[64,65] They are in-frame insertions in exon 20 and have targeted the corresponding TK domain region, as in EGFR-insertion mutations. These mutations occur in the same subpopulation as those with EGFR mutations (adenocarcinoma, never-smoker, East Asian, and women). HER-2 is frequently overexpressed in NSCLC and appears to be associated with drug resistance, increased metastatic potential, increased production of vascular endothelial growth factor (VEGF), and poor prognosis.[66] HER-2–mediated resistance to DNA-damaging agents requires the activation of Akt, which phosphorylates murine double minute 2 (MDM2) and therefore enhances MDM2-mediated ubiquitination and degradation of p53. Blocking the Akt pathway mediated by HER-2 increases the cytotoxic effect of DNA-damaging drugs in tumour cells with wild-type p53. Furthermore, recent studies have shown that the G/G genotype of the MDM2 polymorphism is associated with worse overall survival among early-stage NSCLC patients, particularly those with squamous cell histology.[67]
Trastuzumab (Herceptin) is a chimerized monoclonal antibody against HER-2. Combinations of trastuzumab and chemotherapy are well tolerated, with response rates of 21% to 40%.[66] One trial showed that patients whose tumors highly overexpressed HER-2 (3+) by immunohistochemistry or evidence of amplification by FISH showed a good response. It appears that highly overexpressing HER-2 cases of NSCLC (3+ by immunohistochemistry), although relatively infrequent (3% to 9%), may show benefit with treatment with trastuzumab. [67]
OTHER TARGETED MOLECULAR THERAPIES
There has been tremendous research and investment in the development of small molecules that target key proteins in cell signaling pathways that are aberrantly altered in disease, particularly in carcinogenesis. For instance, receptor tyrosine kinases (RTKs) serve as potential therapeutic targets in several solid tumors, including lung cancer. The RTK c-kit is highly expressed in SCLC (although it is not mutated), and this has led to clinical trials with the specific c-KIT inhibitor (STI-571, Gleevec, Novartis), alone and in combination therapy.
Summary
Surgical excision remains the only therapeutic modality that can cure selected lung cancer patients. Pathologists play an important role in the surgical management of patients with lung cancer, from preoperative diagnosis and staging, to intraoperative evaluation of the extent of distant disease and margin status, to postoperative assessment of tumor genetic alterations. They offer guidance in selecting appropriate surgical therapies and chemotherapeutic regimens, as well as in identifying morphologic and molecular prognostic markers and predictors of response to therapeutic agents. They are expected to provide accurate diagnosis and classification of the patient’s lung cancer. Most importantly, pathologists are required to provide pathologic staging information in lung resection specimens, surgical resection margin status, and information about lung cancer subtype, and more recently, to identify histological correlates that can uncover molecular abnormalities predictive of response targeted therapies.
Furthermore, classification algorithms based on the presence of genetic alterations found in lung cancer can be used to identify drugs or therapeutic agents targeting these alterations. The clinical application of molecular diagnostic techniques has allowed a more precise and rapid assessment of lung cancer. Molecular biology is increasingly being used to elucidate the various steps involved in tumor initiation, progression, and metastasis. In addition, technical advances in recombinant DNA technology have resulted in the identification of novel biomarkers that are of prognostic importance and may aid in early diagnosis and present potential molecular targets for novel therapeutic strategies. In this era of genomics and proteomics, there is an expectation of significant advances in early diagnosis of lung cancer, the elucidation of new relevant categories of disease, and the development of more targeted therapy. Specific disease profiles may be generated for individual patients to assist in the selection of therapy, and individualized therapy may be present in the future. Understanding the molecular events underlying pathogenic processes will hopefully translate into developing curative systemic therapy and improved patient survival.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lucian R. Chirieac, Email: lchirieac@partners.org, Harvard Medical School, Staff Pathologist, Department of Pathology, Brigham and Women’s Hospital, 75 Francis Street, Boston, MA 02465, Phone: 617-732-8126, Fax: 617-264-5118.
Sanja Dacic, Email: dacics@upmc.edu, University of Pittsburgh Medical Center, Department of Pathology – PUH C608, 200 Lothrop St. Pittsburgh, PA 15213, Phone: 412-647-8694, Fax: 412-647-3455.
References
- 1.Jemal A, et al. Cancer statistics. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Hampton T. Lung cancer trials probe effects of maintenance therapy, targeted agents. JAMA. 2009;302(6):616–618. doi: 10.1001/jama.2009.1118. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, et al. A translational view of the molecular pathogenesis of lung cancer. J Thor Oncol. 2007;2(4):327–43. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 4.Winer E, et al. Clinical cancer advances 2008: major research advances in cancer treatment, prevention, and screening--a report from the American Society of Clinical Oncology. J Clin Oncol. 2009;27(5):812–26. doi: 10.1200/JCO.2008.21.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin Cancer Res. 2009;15(24):7471–7478. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 6.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giaccone G, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol. 2004;22(5):777–84. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Herbst RS, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23(25):5892–9. doi: 10.1200/JCO.2005.02.840. [DOI] [PubMed] [Google Scholar]
- 10.Thatcher N, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366(9496):1527–37. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 11.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 12.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 13.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma SV, et al. Epidermal growth factor receptor mutations in lung cancer. Nature Rev Cancer. 2007;7(3):169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, Ladanyi M. Epidermal growth factor receptor mutation testing in lung cancer: searching for the ideal method. ClinCancer Res. 2007;13(17):4954–5. doi: 10.1158/1078-0432.CCR-07-1387. [DOI] [PubMed] [Google Scholar]
- 16.Pratilas CA, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68(22):9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linardou H, et al. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nature Rev Clin Oncol. 2009;6(6):352–66. doi: 10.1038/nrclinonc.2009.62. [DOI] [PubMed] [Google Scholar]
- 18.Ahrendt SA, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92(6):1525–30. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Eberhard DA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900–9. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 20.Riely GJ, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14(18):5731–4. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 23.Sequist LV. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Oncologist. 2007;12(3):325–30. doi: 10.1634/theoncologist.12-3-325. [DOI] [PubMed] [Google Scholar]
- 24.Dacic S. EGFR assays in lung cancer. Adv Anat Pathol. 2008;15(4):241–7. doi: 10.1097/PAP.0b013e31817bf5a9. [DOI] [PubMed] [Google Scholar]
- 25.Varella-Garcia M. Stratification of non-small cell lung cancer patients for therapy with epidermal growth factor receptor inhibitors: the EGFR fluorescence in situ hybridization assay. Diagn Pathol. 2006;1:19. doi: 10.1186/1746-1596-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dacic S, et al. Clinicopathological predictors of EGFR/KRAS mutational status in primary lung adenocarcinomas. Mod Pathol. 2010;23(2):159–68. doi: 10.1038/modpathol.2009.154. [DOI] [PubMed] [Google Scholar]
- 27.Hirsch FR, et al. Increased EGFR gene copy number detected by fluorescent in situ hybridization predicts outcome in non-small-cell lung cancer patients treated with cetuximab and chemotherapy. J Clin Oncol. 2008;26(20):3351–7. doi: 10.1200/JCO.2007.14.0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sholl LM, et al. Validation of chromogenic in situ hybridization for detection of EGFR copy number amplification in nonsmall cell lung carcinoma. Mod Pathol. 2007;20(10):1028–35. doi: 10.1038/modpathol.3800946. [DOI] [PubMed] [Google Scholar]
- 29.Clark GM, et al. Clinical utility of epidermal growth factor receptor expression for selecting patients with advanced non-small cell lung cancer for treatment with erlotinib. J Thor Oncol. 2006;1(8):837–46. [PubMed] [Google Scholar]
- 30.Hirsch FR, et al. Epidermal growth factor receptor immunohistochemistry: comparison of antibodies and cutoff points to predict benefit from gefitinib in a phase 3 placebo-controlled study in advanced nonsmall-cell lung cancer. Cancer. 2008;112(5):1114–21. doi: 10.1002/cncr.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon YK, et al. Clinicopathologic features and prognostic implications of epidermal growth factor receptor (EGFR) gene copy number and protein expression in non-small cell lung cancer. Lung Cancer. 2006;54(3):387–98. doi: 10.1016/j.lungcan.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 32.Brevet M, Arcila M, Ladanyi M. Assessment of EGFR mutation status in lung adenocarcinoma by immunohistochemistry using antibodies specific to the two major forms of mutant EGFR. J Mol Diagn. 2010;12(2):169–76. doi: 10.2353/jmoldx.2010.090140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J, et al. Mutation-specific antibodies for the detection of EGFR mutations in non-small-cell lung cancer. Clin Cancer Res. 2009;15(9):3023–8. doi: 10.1158/1078-0432.CCR-08-2739. [DOI] [PubMed] [Google Scholar]
- 34.Jackman DM, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(13):3908–14. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki H, et al. EGFR and erbB2 mutation status in Japanese lung cancer patients. Int J Cancer. 2006;118(1):180–4. doi: 10.1002/ijc.21301. [DOI] [PubMed] [Google Scholar]
- 36.Cappuzzo F, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97(9):643–55. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 37.Hirsch FR, et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol. 2005;23(28):6838–45. doi: 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 38.Mok TSK, et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2009;27(30):5080–7. doi: 10.1200/JCO.2008.21.5541. [DOI] [PubMed] [Google Scholar]
- 39.Zhu CQ, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26(26):4268–75. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 40.Cappuzzo F, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20(2):298–304. doi: 10.1093/annonc/mdn635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Z, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99(6):911–22. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller VA, et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J Clin Oncol. 2004;22(6):1103–9. doi: 10.1200/JCO.2004.08.158. [DOI] [PubMed] [Google Scholar]
- 43.Miller VA, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26(9):1472–8. doi: 10.1200/JCO.2007.13.0062. [DOI] [PubMed] [Google Scholar]
- 44.West HL, et al. Gefitinib therapy in advanced bronchioloalveolar carcinoma: Southwest Oncology Group Study S0126. J Clin Oncol. 2006;24(12):1807–13. doi: 10.1200/JCO.2005.04.9890. [DOI] [PubMed] [Google Scholar]
- 45.Marks JL, et al. Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thor Oncol. 2008;3(2):111–6. doi: 10.1097/JTO.0b013e318160c607. [DOI] [PubMed] [Google Scholar]
- 46.Motoi N, et al. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am J Surg Pathol. 2008;32(6):810–27. doi: 10.1097/PAS.0b013e31815cb162. [DOI] [PubMed] [Google Scholar]
- 47.Ninomiya H, et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas Significance of the micropapillary pattern and the hobnail cell type. Lung Cancer. 2009;63(2):235–40. doi: 10.1016/j.lungcan.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 48.Shigematsu H, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97(5):339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 49.Zakowski MF, et al. Morphologic features of adenocarcinoma of the lung predictive of response to the epidermal growth factor receptor kinase inhibitors erlotinib and gefitinib. Arch Path Lab Med. 2009;133(3):470–7. doi: 10.1043/1543-2165-133.3.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finberg KE, et al. Mucinous differentiation correlates with absence of EGFR mutation and presence of KRAS mutation in lung adenocarcinomas with bronchioloalveolar features. J Mol Diag. 2007;9(3):320–6. doi: 10.2353/jmoldx.2007.060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 52.West H, Harpole D, Travis W. Histologic considerations for individualized systemic therapy approaches for the management of non-small cell lung cancer. Chest. 2009;136(4):1112–8. doi: 10.1378/chest.08-2484. [DOI] [PubMed] [Google Scholar]
- 53.Johnson DH, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–91. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 54.Lamant L, et al. High incidence of the t(2;5)(p23;q35) translocation in anaplastic large cell lymphoma and its lack of detection in Hodgkin’s disease. Comparison of cytogenetic analysis, reverse transcriptase-polymerase chain reaction, and P-80 immunostaining. Blood. 1996;87(1):284–91. [PubMed] [Google Scholar]
- 55.Morris SW, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 56.Rikova K, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 57.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 58.Koivunen JP, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soda M, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–7. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaw AT, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodig SJ, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15(16):5216–23. doi: 10.1158/1078-0432.CCR-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Inamura K, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22(4):508–15. doi: 10.1038/modpathol.2009.2. [DOI] [PubMed] [Google Scholar]
- 63.Mino-Kenudson M, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16(5):1561–71. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shigematsu H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65(5):1642–6. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 65.Stephens P, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431(7008):525–6. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 66.Giaccone G. Clinical impact of novel treatment strategies. Oncogene. 2002;21(45):6970–81. doi: 10.1038/sj.onc.1205565. [DOI] [PubMed] [Google Scholar]
- 67.Heist RS, et al. MDM2 polymorphism, survival, and histology in early-stage non-small-cell lung cancer. J Clin Oncol. 2007;25(16):2243–7. doi: 10.1200/JCO.2006.08.8914. [DOI] [PubMed] [Google Scholar]
- 68.Azzoli CG, et al. Trastuzumab in the treatment of non-small cell lung cancer. Semin Oncol. 2002;29(1 Suppl 4):59–65. doi: 10.1053/sonc.2002.31526. [DOI] [PubMed] [Google Scholar]