

Summary
Almost four decades of research into the role of human leukocyte antigen-B27 (HLA-B27) in susceptibility to spondyloarthritis has yet to yield a convincing answer. New results from an HLA-B27 transgenic rat model now demonstrate quite convincingly that CD8+ T cells are not required for the inflammatory phenotype. Discoveries that the HLA-B27 heavy chain has a tendency to misfold during the assembly of class I complexes in the endoplasmic reticulum (ER) and to form aberrant disulfide-linked dimers after transport to the cell surface have forced the generation of new ideas about its role in disease pathogenesis. In transgenic rats, HLA-B27 misfolding generates ER stress and leads to activation of the unfolded protein response (UPR), which dramatically enhances the production of interleukin-23 (IL-23) in response to pattern recognition receptor agonists. These findings have led to the discovery of striking T-helper 17 cell activation and expansion in this animal model, consistent with results emerging from humans with spondyloarthritis and the discovery of IL23R as an additional susceptibility gene for ankylosing spondylitis. Together, these results suggest a novel link between HLA-B27 and the T-helper 17 axis through the consequences of protein misfolding, and open new avenues of investigation as well as identifying new targets for therapeutic intervention in this group of diseases.
Keywords: antigen presentation/processing, monocytes/macrophages, ankylosing spondylitis, major histocompatibility complex, Toll-like receptors/pattern recognition receptors, unfolded protein response
Introduction
Spondyloarthritides are a relatively common group of immune-mediated inflammatory diseases occurring in an estimated 0.5 to 1% of the population (1). The prototypic form of disease, ankylosing spondylitis, represents approximately one third to one half of cases and exhibits a phenotype that can include axial and peripheral arthritis, gastrointestinal inflammation, inflammation where tendons and joint capsules insert on bone (enthesitis), and abnormal bone formation in the sacroiliac joints and ascending along the spine leading to ankylosis (2). In 1973, two groups working independently reported a striking association between what we now know as human leukocyte antigen-B27 (HLA-B27) and ankylosing spondylitis. In a four-paragraph letter to Nature, Caffrey and James (3) wrote, ‘A study of human lymphocyte phenotypes in unrelated Caucasian individuals with ankylosing spondylitis has revealed a striking similarity in their antigenic pattern. The lymphocytes of fifty such patients, when tested against a panel of twenty-six different specific typing sera, using a two stage lymphocytotoxicity micro-method, were shown to have in common either the antigen HL-A27 (96%) or the antigen W5 (4%). This remarkably high frequency of the antigen HL-A27 compares with the incidence of 5–6% of this antigen in random Caucasian populations’. This report and two additional reports appearing a month later in The Lancet (4) and The New England Journal of Medicine (5) marked the beginning of an odyssey that has lasted for over three and a half decades as investigators have sought to explain how HLA-B27 contributes to disease. Peaks, valleys, and occasional detours have marked this journey, and it is not yet clear when it will end. Paralleling discoveries of the heterogeneity of HLA class I molecules and their function displaying peptides, it was first proposed that HLA-B27 was the target of autoreactive antibodies and then autoreactive T cells. More recently, considerable attention has been directed toward unusual features of this allele, including a tendency of the HLA-B27 heavy chain to misfold in the ER prior to assembly into complexes with peptide and β2 microglobulin (β2m) (6, 7) and to form disulfide-linked homodimers after reaching the cell surface (8, 9). There is evidence that abnormal forms of HLA-B27 may be recognized by major histocompatibility complex (MHC) class I receptors on various leukocyte populations, which could play a role in disease (10, 11). Thus, these features evoke very different ideas about underlying pathogenic mechanisms, including the heretical concept that immunological recognition of HLA-B27 might not be necessary to cause disease if intracellular effects are paramount.
While HLA-B27 constitutes a large proportion of genetic susceptibility to ankylosing spondylitis (20–40%), it is now well recognized that this is a complex genetic disease, and HLA-B27 is only one of several genes determining predisposition and phenotype (12). Technological advances that enable high throughput genotyping of single nucleotide polymorphisms (SNPs) have revolutionized the field of complex genetics and led to the identification of multiple risk alleles in diseases such as rheumatoid arthritis, lupus, and now ankylosing spondylitis (12, 13). These genome-wide association studies not only provide a more complete picture of genetic susceptibility to ankylosing spondylitis but should also begin to inform hypotheses about pathogenesis including where HLA-B27 fits in the puzzle. Despite this optimism, experience tells us that understanding the steps in the journey from genotype to phenotype will continue to be a major challenge in this and other complex genetic diseases.
The purpose of this review is to discuss unusual properties of HLA-B27 documented in the last decade, why they occur, and how they may be relevant to its role in disease in light of results from animal models as well as humans. Our focus will be on the concept of HLA-B27 misfolding and its consequences, including new data from transgenic rats suggesting how it may be linked to activation of the interleukin-23 (IL-23)/IL-17 axis and T-helper 17 (Th17) cells. We also identify, from our viewpoint, some of the key questions that need to be addressed.
HLA-B27 transgenic animal models
Transgenic rats
Typical reductionist approaches to address the question of how HLA-B27 causes disease such as the production of transgenic animals have met with mixed success. The first breakthrough occurred in 1990 when Taurog and colleagues (14) showed that HLA-B27/human β2m (hβ2m) transgenic rats developed a spontaneous spondyloarthritis-like phenotype that included colitis and arthritis. This study provided the first direct evidence that HLA-B27 itself could cause disease and dispelled the notion that ankylosing spondylitis might be caused by an HLA-B27-linked gene, a finding that is now consistent with accumulated evidence that HLA-B27 is associated with disease in multiple genetically diverse populations around the globe. In the years since HLA-B27/hβ2m transgenic rats were first described, this model has taught us a great deal about the cellular immunology of disease (15). One important observation, now confirmed by two independent approaches, is that CD8α/β T cells are not necessary for the development of arthritis or colitis. Both antibody-mediated CD8+ T-cell depletion (16) and a chemically induced mutation in the CD8a gene that eliminates CD8α protein expression (17) fail to prevent the spondyloarthritis-like phenotype from occurring in rats. These results are consistent with earlier observations, including the finding that CD4+ T cells efficiently transfer colitis to athymic nude rats, as long as they are expressing high levels of HLA-B27/hβ2m in the bone marrow (15). Interestingly, expression of HLA-B27 in non-hematopoietic cells including the thymus is not necessary for the inflammatory phenotype. Transgenic rat lines with variable copy number have demonstrated a ‘threshold’ effect, where a certain level of HLA-B27/hβ2m expression must be achieved in order to obtain the phenotype. However, spondyloarthritis-like disease is not merely a consequence of MHC class I overexpression, in so far as comparable expression of HLA-B7 with hβ2m does not result in inflammation. Taken together, the picture that has emerged from HLA-B27/hβ2m transgenic rats is that spondyloarthritis-like disease is due to some consequence of high expression of HLA-B27 in the hematopoietic compartment. However, the canonical function of HLA-B27, presentation of antigenic peptides to CD8+ T cells, is not involved in disease causation. CD4+ T cells are strongly implicated in pathogenesis (18), while natural killer (NK) and NKT cells have not been specifically examined. To the extent that HLA-B27/hβ2m transgenic nude (rnu/rnu) rats have functional NK cells (19) (lack B and T cells) and remain healthy (15), this observation would imply that they not capable of causing disease at least in the absence of B, T, or NKT cells.
Transgenic mice
For reasons that remain unclear, spondyloarthritis-like disease has not been observed with HLA-B27 expression in ‘normal’ mice (20–22). Given the experience with rats, the explanation for this could be as simple as insufficient copy number or due to more complicated immunological differences between species. However, when HLA-B27 was expressed on a β2m-deficient (β2m−/−) background, Khare et al. (23) reported a high frequency of spontaneous peripheral arthritis and ankylosis without colitis, when animals were housed in conventional rooms outside of specific pathogen-free conditions. The phenotype persisted when hβ2m was introduced into HLA-B27/β2m−/− mice (24) and in the absence of MHC class II molecules (25), and it correlated with the presence of free HLA-B27 heavy chains on the cell surface. In contrast, we found only a low frequency of spontaneous arthritis in HLA-B27/β2m−/− mice on the C57BL/6J (B6) background, but more concerning was that we observed a similar disease phenotype and frequency in β2m−/− B6 mice in the absence of HLA-B27 (26). Further investigation revealed that a deficiency in class I expression either due to a lack of β2m (β2m−/−) or peptide (TAP1−/−) was sufficient to cause this phenotype, and that the frequency of disease was highly dependent on the mouse strain (26). A mixed B6/129 background was most permissive, which may explain the earlier results of Khare et al., since the mice used to introduce the β2m null gene had a similar mixed B6/129 background (personal communication). It is tempting to draw parallels between results from these mice and HLA-B27/hβ2m transgenic rats such as the lack of a role for CD8+ T cells that are virtually absent in β2m−/− and TAP1−/− mice. It could also be that β2m deficiency creates a similar situation to HLA-B27 overexpression, a concept that is clarified in subsequent sections. However, definitive interpretation of these results in terms of a molecular and cellular mechanism of disease remains difficult because of the potential for confounding genetic background effects.
Alternatives to arthritogenic peptides: HLA-B27 misfolding and dimerization
In the early 1990s, with strong evidence for a causal relationship between HLA-B27 and spondyloarthritis and an animal model available, it seemed like the HLA-B27 question would be relatively straightforward to sort out. It was clear that MHC class I molecules were displaying peptides for recognition by CD8+ T cells, and Benjamin and Parham (27) had synthesized emerging data to propose the arthritogenic peptide hypothesis. They argued that the apparently unique peptide binding specificity of HLA-B27, due in large part to its unusual B pocket, could be responsible for selecting self-peptides resembling pathogen-derived peptides that might evoke an autoreactive T-cell response. To test this idea, we set out to create a mutant of HLA-B27 that was minimally altered and fully functional, but with a completely different peptide binding specificity. Six B pocket amino acids in HLA-B27 that differed between HLA-B27 and HLA-A2 were replaced with residues from HLA-A2. The result was a ‘hybrid’ class I molecule (referred to as B27.A2B) that could select and present a unique set of peptides. B27.A2B did not present naturally occurring HLA-B27-restricted peptides unless they were modified at position 2 so that the amino acid side chain would fit properly into an HLA-A2-like B pocket (6, 28). This provided one of the first functional demonstrations of the allelic specificity of pocket-peptide side chain interactions and suggested that it might be possible to address the role of HLA-B27-specific peptides in disease by creating a transgenic rat with this molecule. Indeed, our collaborators were able to produce animals expressing B27.A2B/hβ2m that remained free from disease over at least two years of observation (15, and personal communication). Unfortunately, the line was unintentionally sacrificed precluding further studies (personal communication). We initially considered the absence of an inflammatory phenotype to be consistent with the idea that HLA-B27-restricted peptides were involved in the pathogenesis of spondyloarthritis-like disease. However, in the course of further characterizing B27.A2B, we found a very striking difference in its assembly characteristics that changed our direction in the pursuit of a pathogenic mechanism.
Slow folding and ER-associated degradation
Armed with no specific hypothesis but as part of a comprehensive comparison, we found that the assembly characteristics of HLA-B27 and B27.A2B differed dramatically. The HLA-A2-like B pocket caused the HLA-B27 heavy chain (B27.A2B) to fold, bind β2m, and exit the ER much more rapidly than ‘wild type’ HLA-B27 and more like other A and B alleles (6). From the perspective of most alleles, it appeared that HLA-B27 was unusually slow to fold and that the B pocket ‘transplant’ essentially corrected the problem. We also noted that delayed folding was associated with a dramatic difference in peptide loading efficiency, with HLA-B27 requiring an approximately 30-fold greater concentration of peptide to achieve comparable binding. At the time of these observations, Cresswell and colleagues (29) had just shown that HLA class I heavy chains misfolded when expressed in the absence of β2m or peptide and were then eliminated from the ER by a mechanism known as ER-associated degradation (ERAD). ERAD was already recognized as part of an elaborate quality control process whereby proteins that misfolded or failed to properly assemble into larger oligomeric complexes were disposed of after dislocation from the ER into the cytosol (30). A careful examination of HLA-B27 revealed that a small proportion of newly synthesized heavy chains were undergoing ERAD, while this could not be detected for B27.A2B or other HLA class I alleles (6). It should be emphasized that ERAD of HLA-B27 was seen in normal cells without imposing any peptide or β2m deficiency, including Epstein-Barr virus (EBV)-transformed B cells with a single copy of the HLA-B27 gene. This provided the first evidence for HLA-B27 misfolding (6) and prompted us to examine additional properties of the heavy chain and eventually explore the consequences of misfolding.
Aberrant disulfide bond formation and unusual forms of HLA-B27
Protein misfolding can result in, or be a consequence of, the formation of inappropriate disulfide bonds. Bowness and colleagues (8) had observed that HLA-B27 heavy chains tended to dimerize when re-folded in vitro in the absence of β2m and in TAP1/TAP2-deficient cells. where peptides cannot be transported from the cytosol into the ER. Dimerization depended on the Cys67 residue located at the edge of the B pocket, which had been shown previously to be reactive in vivo, in part because of the nearby Lys70 residue (31). This finding suggested that in the absence of their usual ‘cargo’ (i.e. β2m and peptide), HLA-B27 heavy chains might be particularly susceptible to aggregation because of the unpaired Cys67. Looking for additional evidence of misfolding in vivo, we found that up to 25% of newly synthesized HLA-B27 heavy chains formed disulfide-linked dimers in the ER immediately after synthesis. Again, this behavior was unusual in that it occurred in cells with an intact class I assembly pathway and an appropriate supply of β2m and peptide. About two thirds of the pool of newly made heavy chain was unfolded (or misfolded) in the ER (with almost 40% as dimers and 60% monomers), whereas approximately one third was represented by properly folded monomers (32). The ability to distinguish between unfolded (or pre-folded) and folded heavy chains is based on the conformation specific monoclonal antibodies (mAbs) HC10 and W6/32. HC10 recognizes HLA-B and C heavy chains before they have completed the folding process and acquired β2m or after losing β2m and unfolding. In contrast, W6/32 is a pan HLA class I mAb that recognizes folded heavy chain associated with β2m and peptide. About half of the unfolded ER dimers were gone in three hours, but they were not appearing as folded dimers. Instead, folded dimers were detected only after prolonged periods of chase and thus appeared to derive primarily from folded monomers. Consistent with this observation, Bowness and colleagues (33) demonstrated that cell surface dimers form during endosomal recycling of HLA-B27 heavy chains. However, it remains possible that some unfolded dimers arise on the cell surface without endosomal recycling, after heavy chains have lost β2m and/or peptide (32). Also, we cannot rule out the possibility that some of the dimers initially formed in the ER escape to the cell surface. However, this pool does not appear to be the primary source of cell surface dimers.
Heterogeneity of HLA-B27 dimers
Several forms of HLA-B27 heavy chain dimers distinguished by different mAbs exhibit heterogeneity on non-reducing SDS-polyacrylamide gel electrophoresis (PAGE). W6/32 immunoprecipitates a single ~90 kDa band referred to as a folded dimer (32). In contrast, HC10 immunoprecipitates unfolded dimers, which are heterogeneous, migrating as a series of heavy chain bands between ~80 and ~100 kDa on non-reducing gels. Additional evidence for distinct pools of dimers comes from sequential immunoprecipitations, where several rounds with W6/32 does not diminish the pool of HC10-reactive dimers, and similarly, multiple rounds of immunoprecipitation with HC10 does not remove W6/32-reactive dimers (32). A third mAb (5H7) that recognizes a conformational epitope in the α3 domain of HLA class I heavy chains immunoprecipitates only the ~90 kDa folded form of HLA-B27 dimers. This observation is interesting, as it suggests that the α3 domain in HC10-reactive dimers is either unfolded/misfolded or perhaps otherwise inaccessible due to steric issues. MARB4, which has been shown to react with a unique pool of HLA-B27 heavy chains that contain peptides longer than the canonical 9–11-mers (34) and may be devoid of β2m (35), also recognizes some ~90 kDa dimers but not the heterogeneous forms. The ratio of monomers to dimers recognized by MARB4 is close to one, while for the other mAbs monomers predominate at a ratio of approximately 10:1, which could indicate a link between forms of HLA-B27 that bind long peptides and dimerization.
While the 90 kDa band appears to be a homodimer of two HLA-B27 heavy chains, the precise nature of the heterogeneous (80–100 kDa) complexes has yet to be determined. On two-dimensional non-reducing/reducing separations of radiolabeled proteins from HLA-B27/hβ2m transgenic rats, the heterogeneous bands resolve to 44 kDa bands, identical to monomeric HLA-B27 heavy chain (36). This finding suggests they are homodimers of heavy chain and that altered migration under non-reducing conditions might result from intra- and inter-chain disulfides (although pairing with other ~44 kDa proteins cannot be ruled out). If the α3 domain is disrupted as suggested above (32), this would suggest a more fundamental folding abnormality, since theα3 domain is thought to form very rapidly or even co-translationally with heavy chain synthesis.
Fussell and colleagues (37) have recently examined the presence of unpaired and/or abnormally paired Cys residues in HLA-B27. They used the method of rapid acidification to trap cysteine residues in their native (oxidized or reduced) state in the cell, followed by covalent modification under non-reducing conditions with alkylating agents. When visualized after gel electrophoresis, this provides a picture of the in vivo redox status of the protein. This approach reveals more heterogeneity in HLA-B27 than appreciated previously, and stronger evidence for the involvement of residues beyond Cys67 in misfolding. Their results are consistent with the existence of multiple species of HLA-B27 heavy chains with aberrant disulfide bonds, including both intra- and inter-molecular species.
BiP binding to HLA-B27
BiP (GRP78/Hspa5) is an ER chaperone that interacts transiently with a number of newly synthesized proteins and forms a stable association with proteins that misfold. It binds to hydrophobic regions of unfolded (pre-folded) proteins as part of a large multi-protein complex (38) and prevents their oligomerization. It is also a key regulator of the cellular response to stress that emanates from the ER when protein folding and/or secretion are compromised (discussed below). BiP had been shown to bind transiently to newly synthesized MHC class I heavy chains (39). We decided to immunopurify HLA-B27 from C1R.B27 cells using W6/32 and HC10 and found substantial amounts of BiP associated with unfolded/misfolded heavy chains reactive with HC10 but not W6/32 (Fig. 1). Additional proteins co-purifying with unfolded/misfolded HLA-B27 included calnexin, calreticulin, and protein disulfide isomerase (PDI), whereas only small amounts of calreticulin were associated with W6/32-reactive species. PDI is an oxidoreductase that has been shown to associate with complexes containing BiP and misfolded proteins in the ER lumen that are being prepared for dislocation into the cytosol for degradation (40). These results, together with the finding of aberrant disulfide-linked complexes, provided a strong rationale for us to pursue the mechanism and consequences of HLA-B27 misfolding.
Fig. 1. ER chaperones associated with purified HLA-B27 heavy chains.
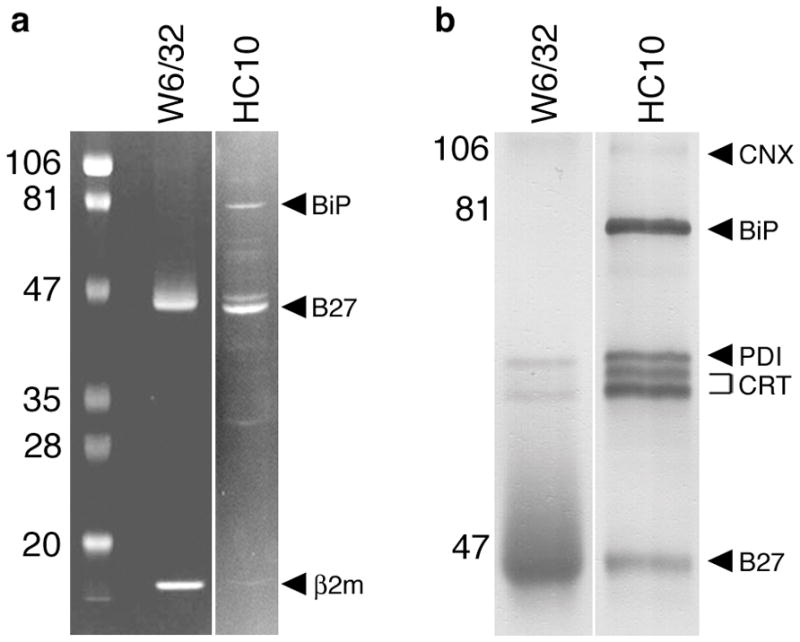
HLA-B27 was purified from C1R.B27 cell lysates using sequential W6/32 and HC10 immunoaffinity columns. (A) Coomasie blue stain of HLA-B27 purified on W6/32 and HC10 columns separated on SDS-PAGE under reducing conditions. A reverse image is shown to enhance visualization of faint bands. HLA-B27, β2m, and BiP are indicated by arrows (right) and pre-stained markers (left) by their MW. BiP was identified by mass spectroscopic (MALDI-TOF) analysis of in-gel digested protein (data not shown). (B) Immunoblots of purified HLA-B27 performed using a cocktail of antibodies including AF-8 (anti-calnexin; CNX), anti-BiP (BiP), anti-PDI (PDI), anti-calreticulin (CRT), and 3B10.7 (anti-class I HC; B27). Prior to preparing the cocktail chaperones were first identified by immunoblotting with single antibodies (not shown). Calreticulin migrates as two forms indicated by the bracket. Band intensity in (A) reflects relative amount of protein stained with Coomassie blue.
Multiple studies have reported BiP to be associated with HLA-B27 at steady state, implying a prolonged interaction that is consistent with heavy chain misfolding (32, 36, 41–43). Tran et al. (36) found that multimers of heavy chain, rather than monomers, were the predominant form of HLA-B27 bound to BiP in cells from HLA-B27/hβ2m transgenic rats. While it is unclear whether BiP binding occurs before or after multimers are formed, one possible scenario is that BiP binds to newly made HLA-B27 heavy chains and remains associated with the proportion that forms disulfide-linked complexes. Given the function of BiP, it might be expected to prevent self-association of these heavy chains if oligomerization were based on exposed hydrophobic regions. However, if oligomerization is based on reactive Cys residues forming intermolecular disulfides, then BiP might be powerless to prevent it and could become trapped and prevented from dissociating.
What is misfolding and why does HLA-B27 misfold?
Three characteristics of HLA-B27 support the contention that it misfolds (Fig. 2): first, heavy chains can be detected undergoing ERAD; second, they self-associate into complexes covalently linked by disulfide bonds in the ER; and third, they exhibit a prolonged association with the ER chaperone BiP (6, 32, 36, 41, 42). All of these events involve newly synthesized heavy chains and occur in the ER, prior to assembly into complexes with β2m and peptide, and parallel what has been described for other proteins that misfold. In addition, cell surface HLA-B27 complexes have a tendency to dimerize (Fig. 2), most likely after losing β2m and/or peptide, perhaps then acquiring a relaxed conformation (8, 33). There has been some confusion in the literature about what constitutes misfolding and whether it is synonymous with heavy chain dimerization. This has most likely arisen because dimerization occurs at two different times in the life of the heavy chain and in two different cellular locations. Because these have very different implications, we propose that misfolding be used in the context of aberrant events occurring in the ER, and that it should be distinguished from dimerization of heavy chains after they have reached the cell surface. Studies on misfolding and dimerization should delineate these events in the context of their temporal and spatial location.
Fig. 2. HLA-B27 misfolding in the endoplasmic reticulum and dimers on the cell surface.
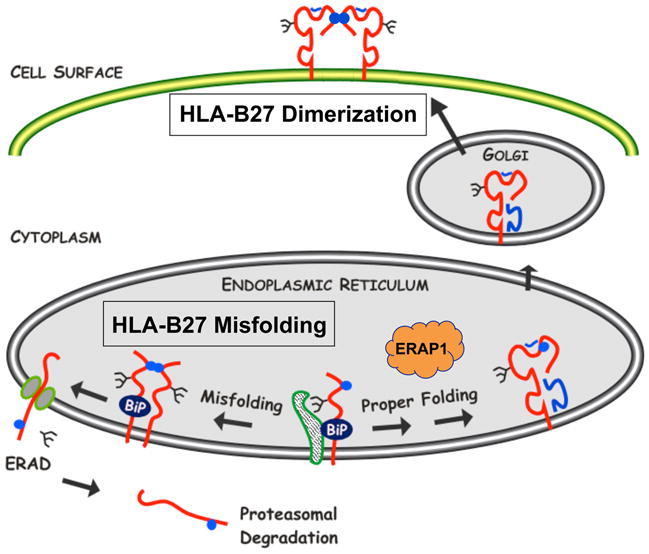
Newly synthesized HLA-B27 heavy chains exhibit three features of protein misfolding: (i) prolonged and stable association with the ER chaperone BiP, (ii) formation of disulfide-linked dimers and possibly oligomers, and (iii) enhanced ERAD. ERAD involves dislocation of heavy chains from the lumen of the ER into the cytosol via the Sec61 complex and involves heavy chain deglycosylation and proteasomal degradation. HLA-B27 also has a tendency to form homodimers via endosomal recycling that can be expressed on the cell surface. Cell surface dimers shown here are in a folded conformation. Unfolded dimers as well as unfolded monomers also exist on the cell surface in varying amounts. Heavy chains are shown in red, with β2m and peptide in blue. Blue circles depict the unpaired Cys67 residue. Other Cys residues have also been implicated in aberrant disulfide bond formation. The figure is reproduced in a modified form from ref. (7) with permission.
Our current understanding of HLA-B27 misfolding suggests it occurs as a consequence of slow heavy chain folding and aberrant disulfide bond formation, the latter being a result of prolonged exposure of unpaired Cys residues to the oxidizing environment of the ER (32, 41). We first suggested this based on results from a panel of HLA-B27 B pocket mutants that separated folding and dimerization properties. For example, replacing Glu45 with Met is sufficient to dramatically increase heavy chain folding efficiency, which also prevents ER dimerization despite the presence of Cys67. Alternatively, replacing Cys67 with Ala prevented dimerization in C1R cells, yet heavy chain folding was minimally enhanced (32). Although it is not known why Glu45 slows folding so dramatically, it is positioned deep within the peptide-binding groove at the base of the B pocket (44) and the need to neutralize its negative charge may be important to complete folding and achieve a compact stable structure (6).
In support of this model, Antoniou and colleagues (41) demonstrated that HLA-B27 heavy chain dimerization could be enhanced by reducing the rate of assembly of class I complexes by incubating cells at 26°C rather than 37°C. At low temperatures, homodimer formation and stable BiP binding could even be induced in a normal class I molecule (HLA-A2) when its assembly was slowed in this fashion (41). They also discovered that Cys164, which normally forms a disulfide with Cys101, participates in an aberrant disulfide bond in some HLA-B27 dimers. Together with the α3 domain disulfide, the Cys101–164 disulfide is important in overall HLA class I stability (45). This observation is consistent with the idea that some of the heterogeneity in dimeric HLA-B27 complexes is due to variable intrachain disulfides. This working model does not preclude effects of other polymorphic residues on the folding and assembly of HLA-B27. It is well recognized that residues outside the B pocket can dramatically influence HLA-A2 folding and/or chaperone association (46), and there are undoubtedly additional polymorphic amino acid residues that can alter important properties of HLA class I alleles.
Specificity of misfolding
No other naturally occurring HLA class I allele has been shown to misfold. However, the several alleles that have been studied represent only a handful of the >1,200 HLA-B alleles encoding >1,000 different proteins (as of July 2009; http://www.ebi.ac.uk/imgt/hla/). Residues in HLA-B27 that contribute to misfolding are polymorphic and unusual enough to predict that other alleles would be unlikely to exhibit the same behavior as HLA-B27 (7). While the aberrant behavior of HLA-B27 has been documented either in EBV-transformed or primary cells derived from individuals with single copies of HLA-B27, much of the work in this area has relied on transfected cells, cells from HLA-B27 transgenic rats, and/or cells that are lacking one or more components of the HLA class I assembly pathway. There are caveats associated with overexpression, expression in the absence of class I cargo or chaperones, and/or expression of human heavy chains in the presence of chaperones from other species that should be considered. While in most cases appropriate allelic controls have been included, this is not always the case. These variables could affect the quantity and or quality of aberrant HLA-B27 behavior.
The role of class I cargo and chaperones in HLA-B27 folding and misfolding
Class I cargo
The availability of cell lines lacking β2m (e.g. Daudi cells), peptide transporters (e.g. T2 cells), or tapasin (.220 cells) has been extremely useful in defining many critical features of MHC class I assembly and quality control. In cells that lack TAP1 and TAP2, cytosolic peptides generated by proteasomal degradation of proteins are not transported into the ER. As a consequence, class I complexes are expressed very poorly at the cell surface but can be partially rescued by adding exogenous peptides. Under these circumstances and in the absence of β2m, most class I heavy chains are eventually degraded via ERAD (29). We examined two TAP1/TAP2-deficient cell lines expressing HLA-B27 (either after DNA transfection or as a natural allele) and were unable to detect either folded (W6/32-reactive) or unfolded (HC10-reactive) disulfide-linked HLA-B27 complexes (32). It is tempting to speculate that peptides are necessary for HLA-B27 misfolding, perhaps by stabilizing unfolded HLA-B27 complexes in the ER and thus prolonging exposure of unpaired Cys residues. Consistent with this idea, in T2 cells where TAP1 and TAP2 have been re-expressed, there is greater accumulation of unfolded HLA-B27 heavy chains. Interestingly, the effect of peptide may be allele specific, as there is reduced accumulation of unfolded HLA-B5 heavy chains (32). Such a paradoxical effect could occur if the effect of peptide in the ER is to induce a conformational change in most alleles (e.g. HLA-B5), allowing them to complete the folding process and exit the ER. Incompletely folded forms of HLA-B27 might be stabilized by peptide but not complete the folding process, consistent with the requirement for higher concentrations of peptide to induce the folded conformation recognized by W6/32 (6). This raises the question of why unfolded forms of HLA-B27 would accumulate in the ER rather than be removed by ERAD. We have no good explanation but can postulate that dimerization might prevent recognition by ER degradation-enhancing mannosidase (EDEM), which has been shown to be important for ERAD of certain proteins (47–49) and can accelerate the degradation of misfolded species (50). We have overexpressed EDEM in cells expressing HLA-B27 and cannot detect any effect on reducing the accumulation of misfolded disulfide-linked heavy chains (unpublished observations).
Tapasin
Tapasin is an MHC class I-specific chaperone that facilitates the formation of peptide-heavy chain-β2m complexes in the ER (45) and thus promotes the expression of stable, peptide-loaded class I molecules on the cell surface. Tapasin interacts with ERp57 (a thiol oxidoreductase) through a disulfide bond between Cys95 on tapasin and Cys57 on ERp57 and is a key component of the peptide loading complex (PLC). The PLC contains heterodimers of class I heavy chain-β2m, tapasin-ERp57, and TAP1-TAP2, and also contains calreticulin bound to ERp57 and the N-linked carbohydrate side chain of the class I heavy chain. Formation of the PLC, including interactions between tapasin and ERp57 and tapasin and class I heavy chain, are necessary to support optimal peptide loading and editing. When the PLC cannot form, for example when tapasin is absent, class I complexes containing β2m and peptide can form and be expressed on the cell surface, but often only transiently, as they may contain suboptimal peptides that readily dissociate. As a result, cell surface expression of many class I alleles is reduced in the absence of tapasin (51).
Dong and colleagues (52) have recently proposed a model, based on the crystal structure of the tapasin-ERp57 heterodimer, where tapasin stabilizes the groove of the MHC class I heavy chain-β2m heterodimer in a peptide-receptive form. Calreticulin is envisioned as protecting the empty peptide-binding groove from reduction by oxidoreductases in the ER (e.g. free ERp57 or PDI), which might target it for degradation prior to optimal peptide loading. They further suggest that this might be the structure of the MHC class I molecule that binds N-terminally extended peptides that are then trimmed by the ER aminopeptidase ERAP1.
What does this mean for HLA-B27? HLA-B27 is unusual in that it is expressed relatively well on the cell surface in the absence of tapasin (51), although it interacts with tapasin when present, and tapasin influences its peptide repertoire (53, 54). We have speculated that this behavior of HLA-B27 might be the result of its tendency to fold slowly (7), perhaps allowing the peptide-binding groove remain peptide-receptive for longer periods of time enabling it to bind relatively high affinity peptides in the absence of editing and thus be more stable on the cell surface. As mentioned previously, a small proportion of cell surface HLA-B27 complexes (recognized by MARB4) contain long N-terminally extended peptides (34), consistent with some HLA-B27 complexes escaping editing in the ER by ERAP1.
Tapasin has a very interesting reciprocal effect on disulfide-linked dimers of HLA-B27. The HC10-reactive, heterogeneous disulfide-linked complexes that have been shown to form in the ER are very prominent when HLA-B27 is expressed in the absence of tapasin (.220 cells), while W6/32-reactive dimer complexes are not detectable (32). With tapasin re-expression there is a dramatic decrease in the accumulation of HC10-reactive disulfide-linked complexes, and the W6/32-reactive dimers re-appear. Tapasin also reduces the overall accumulation of HC10-reactive monomers and increases W6/32-reactive monomers. This finding is consistent with an overall effect of tapasin on HLA-B27 folding and provides further evidence for differences in the nature of the HC10 and W6/32-reactive disulfide-linked species. It also confirms that despite HLA-B27 being expressed well on the cell surface in the absence of tapasin, the chaperone has a dramatic effect on its immunobiology. To the extent that HC10-reactive disulfide-linked complexes are formed in the ER and are associated with misfolding as we have shown (32), tapasin could prevent their formation and/or reduce accumulation, perhaps by facilitating their removal via ERAD. This effect may not be a direct effect of tapasin but rather a consequence of having a fully functional PLC. For example, perhaps some of the disulfide bond heterogeneity that is prominent with HLA-B27 and exacerbated in the absence of tapasin is a result of the peptide-binding groove in HLA-B27 (particularly the Cys101-Cys164) being accessible and susceptible to reduction with subsequent oxidative formation of aberrant disulfides (e.g. with Cys67) by free ERp57 or other PDIs. This observation would be consistent with its slow folding features and the existing data on the effect of tapasin. The role of tapasin in influencing the immunobiology of HLA-B27, including misfolding, is worth further investigation.
Consequences of HLA-B27 misfolding
ER stress and the unfolded protein response
With strong biochemical evidence for misfolding (6, 32), we became interested in how it might affect cells expressing HLA-B27. For the interested reader, we suggest recent comprehensive reviews on the myriad consequences of protein misfolding in the pathogenesis of human disease (55, 56). While the consequences of protein misfolding depend on several factors, an important one is the site of synthesis of the protein. A large proportion of cellular proteins, including those that are membrane bound and those destined for secretion, are produced in the ER along with MHC class I complexes. The importance of maintaining ER homeostasis in the face of this large protein flux is underscored by the existence of cellular systems devoted to maintaining the fidelity of protein folding (protein quality control) and eliminating proteins that fail to achieve proper conformations (ERAD). When the balance is tipped and unfolded or misfolded proteins begin to accumulate, ER stress ensues, and the cell activates multiple signaling pathways to orchestrate what is known as the unfolded protein response (UPR) (57). In addition to protein misfolding, ER function can be perturbed by a number of extrinsic factors such as ischemia or depleting glucose and other nutrients or calcium. If successful, the UPR enhances the cell’s ability to fold, secrete, and degrade proteins, thus resolving the ER stress. However, depending on the severity and duration of the stress, the outcome of the UPR can be apoptosis. Chemical agents typically used to study UPR activation include tunicamycin, which blocks protein glycosylation, or thapsigargin, which inhibits the main ER Ca2+ ATPase (SERCA) causing Ca2+ depletion. Reducing agents can induce ER stress and activate the UPR by preventing oxidative protein folding. Exposure of cells to these agents results in global ER protein misfolding and robust UPR activation.
Sensing and responding to ER stress
Three proteins appear to be responsible for transducing signals to the nucleus when stress is sensed in the ER: (1) inositol requiring enzyme 1α (IRE1α), pancreatic ER eIF2α kinase (PERK), and activating transcription factor 6α (ATF6α) (58). IRE1α, PERK, and ATF6α are transmembrane proteins with cytosolic and ER lumenal domains that are normally bound to BiP. In addition to its role as a protein-folding chaperone, BiP also serves as a negative regulator of UPR activation by maintaining IRE1α, PERK, and ATF6α in their inactive state. BiP binding to misfolded ER proteins is thought to titrate it away from these stress sensors, allowing them to become activated. Freed from BiP, IRE1α and PERK form homodimers leading to trans self-autophosphorylation, while ATF6α is released from the ER to translocate to the Golgi where it is proteolytically cleaved by site-1 and site-2 intramembrane proteases, resulting in release of the active protein into the cytosol. Active IRE1α is a cytosolic endoribonuclease that acts on the constitutively expressed mRNA for X-box binding protein 1 (XBP1), removing a 26-nucleotide intron. As a consequence there is a translational frame shift, resulting in the loss of a stop codon (out of frame) and synthesis of the full-length XBP1 protein. Active PERK phosphorylates eIF2α resulting in the inhibition of protein synthesis initiation.
HLA-B27 expression and UPR activation
Since bone marrow (BM)-derived cells have been implicated in initiating spondyloarthritis-like disease in HLA-B27/hβ2m transgenic rats, we began a series of studies comparing BM macrophages from wildtype (WT) and transgenic animals before and during disease. In our colony in Cincinnati, 4-week-old rats were healthy with no clinical evidence of inflammation (pre-morbid), whereas by 10 weeks of age, colitis was readily apparent in all transgenic animals. We found striking evidence of UPR activation in BM macrophages derived from 10-week-old animals, whereas cells made from 4-week-old pre-morbid rats were minimally affected (42). UPR target genes BiP, CHOP (C/EBP homologous protein or CHOP10/GADD153), and XBP1 were upregulated several fold in cells from HLA-B27/hβ2m transgenic rats with inflammatory disease. A more comprehensive examination of gene expression differences with microarrays confirmed the presence of a robust UPR, and also revealed a strong interferon signature. Since no exogenous cytokines (except macrophage colony-stimulating factor) had been added during the 5–6 day macrophage derivation protocol, this finding suggested an autocrine effect of type I (or possibly type II) IFNs produced by developing BM macrophages. It also raised the possibility that IFN stimulation might be contributing to UPR activation. To better address the latter question, we derived BM macrophages from 4-week-old pre-morbid rats and treated them with IFN-γ. Robust UPR activation was seen in cells from HLA-B27/hβ2m transgenic but not WT rats. IFNs are potent at upregulating MHC class I expression, which we documented at the mRNA and protein levels (Fig. 3). Importantly, IFN-γ treatment caused the accumulation of disulfide-linked and BiP bound HLA-B27 heavy chains over the 24 h treatment period, indicating misfolding was exacerbated. In contrast, in macrophages derived from transgenic rats that overexpress HLA-B7 and hβ2m, no aberrant disulfide-linked or BiP bound HLA-B7 heavy chains could be found (Fig. 3), and the UPR was not activated (42). In a 24 h time course experiment, we established a temporal connection between HLA-B27 upregulation and UPR activation, consistent with a cause and effect relationship (43).
Fig. 3. Upregulation of MHC class I heavy chains with IFN-γ exacerbates HLA-B27 misfolding in rat macrophages.

(A) Flow cytometric analysis of untreated (NS) and IFN-γ treated (IFN) (100 U/ml for 24 h) HLA-B27/hβ2m transgenic (B27) and HLA-B7/hβ2m transgenic (B7) rat macrophages stained with W6/32 or isotype (Iso) control antibody. (B and C) Immunoblots of HC10 (B) and HC10- and W6/32- (C) immunoprecipitated (IP) heavy chains from wildtype (WT), HLA-B27/hβ2m transgenic (B27), and HLA-B7/hβ2m transgenic (B7) macrophages treated without (−) and with (+) IFN-γ (IFN) for 24 h. (B) Non-reducing SDS-PAGE showing monomers (lower band) and disulfide-linked oligomers (upper bands) visualized with 3B10.7, a rat mAb that recognizes HLA class I heavy chains. (C) Reducing SDS-PAGE showing monomers (upper panel) visualized with 3B10.7, and co-precipitating BiP visualized with anti-KDEL (lower panel). W6/32 immunoprecipitates separated on non-reducing SDS-PAGE shows no evidence of disulfide-linked oligomers (unpublished observations). Some of the data in this figure were published previously in ref. (43) and are reprinted with permission.
The UPR and IFN-β production
To further explore the connection between UPR activation and IFNs, gene expression patterns in BM macrophages from 4-week-old healthy HLA-B27/hβ2m-expressing rats and WT controls were compared using microarrays. This analysis revealed low-level upregulation (~1.5–2-fold) of both IFN-response genes and UPR target genes in HLA-B27/hβ2m-expressing cells (42). Qualitatively, this result appeared similar to what we had observed in macrophages from 10-week-old rats, but the magnitude of both the UPR and the IFN signature was greatly reduced. To look for evidence that these cells were expressing type I and/or type II IFNs, we quantified IFN-β and IFN-γ mRNAs, which revealed an approximate two-fold increase in IFN-β mRNA (Fig. 4) with no difference in IFN-γ (unpublished observations). When treated with IFN-γ, HLA-B27-expressing cells upregulated IFN-β mRNA transcripts another ~3-fold along with UPR activation (Fig. 4), with no change in IFN-γ expression (42). To our knowledge, IFN-γ has not been reported to induce IFN-β, suggesting that this response was secondary to UPR activation. We found additional evidence of this activity in a previously published microarray experiment using pharmacologic agents to induce the UPR (59). Thus, results with HLA-B27-expressing macrophages are consistent with the idea that ER stress and UPR activation secondary to HLA-B27 misfolding causes low-level IFN-β production and that it has autocrine effects. We would anticipate paracrine effects in vivo, although it has not yet been documented. Interestingly, Ivashkiv and colleagues (60) have recently shown that TNF-α can initiate an IFN-β-mediated autocrine loop in macrophages that sustains inflammation and exacerbates pro-inflammatory TNF-α effects. Since TNF-α, either alone or together with IFNs, induces MHC class I expression and exacerbates UPR activation in HLA-B27-expressing macrophages (43), one might observe a more pronounced effect on IFN-β induction. This possibility suggests a mechanism by which an HLA-B27-induced UPR could promote pro-inflammatory effects of TNF-α, which is worth further consideration.
Fig. 4. IFN-β transcripts are upregulated in HLA-B27/hβ2m transgenic macrophages with UPR activation.

BM macrophages derived from 4-week-old HLA-B27/hβ2m transgenic rats remained untreated (−) or were treated with 100 U/ml IFN-γ (+) for 24 h. RNA was isolated and IFN-β transcripts quantified with quantitative real time RT-PCR (qPCR). Results are normalized to actin mRNA. Bars represent mean of triplicate cultures with standard deviation shown. P < 0.05 for B27 vs. WT, and for B27 (+) vs. B27 (−), but P > 0.05 for WT (−) vs. WT (+). Microarray results from this experiment are shown in Fig. 7 in ref. (42).
Since IFN-β production is dramatically increased in response to pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) [e.g. Toll-like receptors (TLRs)], we explored how the UPR influences IFN-β induction by reagents mimicking bacterial and viral infection. After inducing the UPR with pharmacologic agents (tunicamycin or thapsigargin), further stimulation with TLR4 or TLR3 agonists such as lipopolysaccharide (LPS) and double-stranded RNA (dsRNA) results in log-fold increases in IFN-β transcripts compared with TLR agonists alone and at least a sixfold increase in IFN-β production (61). A similar response is seen with intracellular dsRNA, which activates the IFNB gene via the MDA-5/IPS-1 pathway. The effects of tunicamycin and thapsigargin are rapid and only require a period of time sufficient to activate the UPR. This UPR-PRR synergy requires XBP1, as it is completely abolished in XBP1-deficient cells or when XBP1 mRNA expression is knocked down with short interfering RNA (siRNA). In contrast, UPR-PRR synergy is not affected by absence of PERK or inhibition of ATF6 activation. It appears that the spliced form of XBP1 can enhance LPS-induced IFN-β expression, yet optimal synergistic induction still required full UPR activation. This finding indicates that while spliced XBP1 is necessary for synergy, it is not sufficient.
Crosstalk between the UPR and cytokine regulation pathways has been noted for IL-6 and TNF-α. For example, cholesterol loading of macrophages activates the UPR and increases expression of these cytokines, probably secondary to NF-κB and mitogen-activated protein kinase activation (62). In addition, XBP1 is important for IL-6 production from plasma cells (63). However, both of these responses follow long lag-periods, in contrast to our studies, which involve short-term stimulation (e.g. thapsigargin for 1 h followed by 2–3 h of LPS). We have not found thapsigargin to have short-term effects on basal expression of either IL-6 or TNF-α or on LPS-induced IL-6, although TNF-α induction is augmented 3–4-fold (61). However, prolonged exposure of BM-derived macrophages to tunicamycin led to upregulation of IL-1α and IL-6 (unpublished observations). These observations underscore the fact that the UPR can influence many cellular pathways. Furthermore, differences between tunicamycin and thapsigargin that are unrelated to the UPR might be anticipated as these agents act through different mechanisms. For example, proteins that can be induced by changes in ER Ca2+ levels are influenced by thapsigargin but not tunicamycin (64). Along these same lines, the response to a single misfolded protein may represent a distinct category that differs qualitatively as well as quantitatively from both the chemical agents. Thus, the relevance of a pharmacologic UPR to a particular biological effect needs to be examined and established in each system under study, and there may be cell-and context-specific differences to consider.
The UPR and IL-23 production
To look for additional cytokines synergistically induced by the combination of UPR activation and TLR stimulation, we screened LPS-treated mouse macrophages that had been exposed to thapsigargin using microarrays. This revealed that the IL23A gene, which encodes the p19 subunit of the active IL-23 cytokine, was strongly upregulated (unpublished observations). This result has now been confirmed and extended to show that HLA-B27/hβ2m-expressing macrophages where the UPR has been activated by HLA-B27 upregulation, appear to be polarized to produce more IL-23 (65). Although we are still fully characterizing this response, it is seen with TLR3 and TLR2 agonists but is most robust with the TLR4 agonist LPS (Fig. 5). Since active IL-23 has a second subunit (IL-12/23p40) that also combines with IL-12p35 to form active IL-12 (also known as IL-12p70), we determined the effect of ER stress on IL-12p35 and IL-12/23p40 in addition to IL-23p19 mRNAs. In cells experiencing ER stress induced by thapsigargin, the LPS-mediated induction of IL-12p40 mRNA is somewhat attenuated, while IL-12p35 is increased up to fivefold over LPS alone (Fig. 6C, D). This observation is in contrast to the more than 100-fold greater induction of IL-23p19 mRNA by LPS during UPR activation (Fig. 6A). We documented BiP induction by thapsigargin and found it to be further induced by LPS, whereas LPS alone has no effect (Fig. 6B). We have documented exacerbation of ER stress and UPR activation by LPS previously, including the more modest stress induced by HLA-B27 misfolding (61, 65). The mRNA inductions translate into a large increase in IL-23 production in ER-stressed macrophages treated with LPS, whereas increases in IL-12p70 are much smaller (65). In rat macrophages experiencing ER stress due to HLA-B27 misfolding, there is little to no change in IL-12/23p40 and IL-12p35 mRNAs, while IL-23p19 induction is robust (65), consistent with a less robust UPR. Our data strongly suggest that ER stress and UPR activation polarize macrophages to produce much more IL-23 and perhaps more IL-12p70, with the effect on the latter cytokine depending on the magnitude of the UPR. IL-23 could be particularly important as it acts on Th17 T cells to enhance their survival and stimulate IL-17 production (66, 67).
Fig. 5. Multiple TLR ligands synergistically upregulate IL-23p19 mRNA in macrophages experiencing ER stress.

BM-derived macrophages from B6 mice remained untreated (−) or were pre-treated with thapsigargin (Tpg) (+) at a concentration of 1 μM for 1 h, then a TLR4 ligand (LPS, 10 ng/ml) was added for an additional 3 h, a TLR3 ligand (Poly I:C, 10 μg/ml) for 5 h, or a TLR2 ligand (Pam3cys, 1 μg/ml) for 3 h. RNA was isolated and IL-23p19 mRNA quantified with qPCR. All values have been normalized to β-actin mRNA. Bars represent mean of triplicate cultures with standard deviation shown. ND, not detected. Asterisks indicate P < 0.05 compared to respective (−) Tpg controls.
Fig. 6. Effects of ER stress on IL-12/23 subunit mRNA induction by LPS.

BM-derived macrophages from B6 mice were untreated or pre-treated with varying concentrations of thapsigargin (Tpg) (0 – 1 μM) for 1 hr, followed by LPS (10 ng/ml) for an additional 2 h. RNA was isolated and mRNA quantified with qPCR. All values have been normalized to GAPDH mRNA. Each data point is the mean of triplicate cultures with standard deviation shown. Effects of LPS alone are shown at ‘0’ Tpg. Asterisks indicate P < 0.05 compared to respective controls without LPS.
Activation of the IL-23/IL-17 axis in HLA-B27 transgenic rats
Recognizing that HLA-B27 expression might alter cellular responsiveness to TLR agonists and promote IL-23 production, we looked for evidence in the inflamed colon of HLA-B27/hβ2m transgenic rats. Colitis in this model has long been thought to be a Th1-mediated process, in part because of documented overexpression of IFN-γ, IL-1α/β, TNF-α, and IL-6 (15). However, we found an equally dramatic overexpression of IL-17 (IL-17A) that coincides precisely with the onset of colitis at ~6 weeks of age, as determined histologically, and preceding clinical manifestations such as changes in stool consistency that typically begin at 8–9 weeks of age (65). Spikes in IL-23p19 and IL-12/23p40 expression also accompany the IL-17 increase, while IL-12p35 is unchanged at this time. UPR activation is evident in CD11b/c+ antigen-presenting cells (APCs) isolated from the lamina propria, while purified CD4+ T cells overexpress IL-17. In addition, the fraction of CD4+ T cells expressing IL-17 is about 25% of the total, which represents (on average) about a sixfold increase over WT animals. Interestingly, CD4+ T cells expressing IFN-γ are expanded about threefold, and there is some overlap with double positives expressing both IL-17 and IFN-γ (unpublished observations). The APCs isolated from the colon of animals with established disease are strongly polarized to IL-23p19 expression, with smaller increases in IL-12/23p40 and IL-12p35 mRNAs. The increase in IL-12p35 expression appears in isolated APCs from older animals but was not readily apparent in whole tissue from younger rats, perhaps because of timing differences. In summary, there is a striking Th17 activation and expansion in the colon of HLA-B27/hβ2m transgenic rats that is likely to contribute to colitis. Th1 activation and expansion is also present, and the source of cytokines such as TNF-α needs to be addressed. It will be important to determine whether biologics that target IL-23 or both IL-12 and IL-23 (anti-IL-12/23p40) prevent or ameliorate disease in this model of spondyloarthritis.
Activation of the IL-23/IL-17 axis in human spondyloarthritis
Intestinal inflammation, although often asymptomatic, has been recognized for some time as a common component of spondyloarthritis in humans (68), with evolution to Crohn’s disease in up to 7% (69). Th17 T cells have emerged front and center in studies on the pathogenesis of many immune-mediated and/or autoimmune diseases, including inflammatory bowel disease (IBD) (70, 71) and now spondyloarthritis (72–75). Recent work has shown a greater frequency of CD4+ T cells expressing IL-17 in the peripheral blood of AS and other spondyloarthritis patients (74, 75), with many of the CD4+ Th17 T cells also expressing TNF-α and/or IFN-γ. The situation in the colon may be different. Ciccia and colleagues (76) found subclinical intestinal inflammation in 73% of subjects with AS, with IL-23p19 overexpression in the cecum and distal ileum similar to patients with Crohn’s disease (CD). While other Th1 pathway cytokines or signaling molecules (e.g. IFN-γ, IL-12p35, IL-27p28, STAT1) were increased in CD patients, this was not observed in AS. IL-23 protein was localized to cells that appeared to be macrophages or DCs in patients with AS and CD. Despite the strong upregulation of IL-23p19, there was no overexpression of IL-17 in AS patients, consistent with the lack of a fully developed Th17 response and the absence of clinical features of IBD. These novel observations strongly support a role for IL-23 in sub-clinical gut inflammation in human spondyloarthritis and raise interesting questions about why this does not result in Th17 activation. The answer to this question may be related to genetic polymorphisms in the IL-23 receptor (77).
Acute, adaptive, and non-adaptive changes in response to ER stress
Most of our discussion so far has focused on events occurring shortly after HLA-B27 upregulation, which represent the ‘acute’ response to protein misfolding. However, the UPR is a temporal phenomenon where its biological effects depend on when during the course of the response one looks as well as the nature and duration of the stimulus. Rutkowski and Kaufman (57) have provided a framework for the ER stress response where they define acute stressors as transient, occurring over minutes to hours, and only requiring that cells tolerate brief periods of stress with subsequent restoration of ER homeostasis. Chronic stressors are more prolonged occurring over days to years, which necessitate long-term adjustments to cellular function. Adaptation enables cells to maintain their function during periods of chronic stress, and avoid UPR-induced apoptosis.
The most rapid means of relieving acute stress on the ER is to reduce the load of proteins entering the organelle. Three aspects of the ER stress response are targeted at this level: (i) protein synthesis initiation is inhibited by PERK-induced eIF2α phosphorylation (78), (ii) protein translocation into the ER is inhibited (79), and (iii) IRE1α mediates the degradation of mRNAs encoding proteins that are destined for the ER (80). These are short-term fixes that are readily reversible. While they decrease stress on the ER, they do not facilitate adaptation. However, the ER stress sensors IRE1α, PERK, and ATF6α also contribute to a transcriptional response that enhances the folding and secretory capacity of the ER (58). Activated ATF6α is itself a transcription factor, as is the full length XBP1 protein translated from the spliced XBP1 transcript. The PERK-eIF2α-mediated inhibition of protein synthesis initiation results in preferential translation of certain mRNAs with upstream open reading frames, including ATF4. Through XBP1, ATF4, and ATF6, as well as additional downstream factors (e.g. CHOP is a transcription factor that is upregulated by ATF4), there is a robust transcriptional response that mediates the induction of genes encoding many ER chaperones (e.g. BiP, calreticulin), protein disulfide isomerases, ERAD components, and other gene products that enhance protein translocation, lipid and oligosaccharide synthesis, and mitochondrial function. Part of the adaptive response is also aimed at counteracting the acute response. For example, GADD34 is upregulated by the UPR and is involved in relieving the block on protein synthesis by promoting eIF2α dephosphorylation by protein phosphatase I (PPI) (81, 82). Even short periods of ER stress result in this transcriptional response, which becomes crucial for adaptation.
Failure to sufficiently resolve ER stress through UPR activation can trigger apoptosis. The threshold for determining this outcome may differ depending on the type and state of the cell. While the precise mechanism by which these divergent cell fates are decided is not clear, each arm of UPR signaling can be involved. One mechanism appears to be through activated IRE1α, which binds to the adapter molecule TRAF2. This can activate apoptosis signal-regulating kinase 1 (ASK1), resulting in phosphorylation of c-Jun N-terminal kinase (JNK) (83), which can convert a TNF-α response to pro-apoptotic (84). Caspase activation under conditions of ER stress may result from release of pro-caspase (12 in humans and 4 in mice) from TRAF2 upon IRE1α binding (56). However, this is an area of controversy, since conflicting results have been reported and caspase 12 in humans is non-functional. Other studies have implicated human caspases 4 and 11 in ER stress-induced cell death (85). Recent studies focused on distinguishing the kinase and endoribonuclease (RNase) functions of IRE1α support the idea that this molecule is a key component of the apoptotic switch mechanism (86). There are chemical kinase inhibitors that can be used to bypass autophosphorylation and partially activate the RNase function of IRE1α. While this causes XBP1 splicing to occur, the mRNA decay that is normally seen after IRE1α activation is subverted, as is the triggering of apoptosis. This associates mRNA decay with triggering of apoptosis, supporting the idea that the synthesis of key anti-apoptotic molecules may be prevented. The nature and specific roles of these molecules await further delineation. IRE1α has also been reported to bind to the pro-apoptotic proteins BAX (BCL2-associated X protein) and BAK (BCL2 antagonist of cell death), and in their absence, IRE1α signaling is affected (87). However, it remains unclear precisely how BAX and BAK influence UPR-induced apoptotic pathways.
Signaling through PERK has also been implicated in apoptosis. PERK activation leads to CHOP induction via ATF4 (81), which in turn causes GADD34 upregulation. By releasing the block on protein synthesis initiation, GADD34 promotes protein synthesis and the accumulation of reactive oxygen species (ROS) and oxidative stress (88, 89). CHOP may have other pro-apoptotic effects mediated in part by the induction of TRB3 (90). Although the third arm of UPR signaling, ATF6, has also been implicated in causing apoptosis in differentiating myoblasts and in a macrophage cell line, this activity may be cell type and/or condition specific (91, 92). ATF6α also contributes to CHOP and XBP1 upregulation and thus can influence apoptosis via crosstalk (93, 94). However, the generation of ATF6α-deficient mice has revealed that ATF6α also has a pro-survival role, as cells derived from these animals exhibit reduced survival during ER stress (95). Since UPR activation does not always lead to apoptosis, a key question that remains to be answered is how this decision is regulated.
HLA-B27 expression and adaptation to ER stress
We have not found evidence of differential cell death in HLA-B27-expressing rat macrophages exhibiting acute UPR activation after exposure to IFN-γ or other cytokines. However, only limited long-term experiments have been performed, and we have not carefully quantified cells that might be in early stages of apoptosis. It would not be surprising if the magnitude and duration of ER stress in HLA-B27-expressing macrophages is insufficient alone to trigger apoptosis, since the UPR in this situation is less robust response than that induced by pharmacologic means (42, unpublished observations). Nevertheless, further investigation of the response to cytokines and differential sensitivity to other inducers of apoptosis, such as what might occur during an inflammatory response, is warranted.
Several important questions about HLA-B27 and ER stress remain to be addressed. For example, it is unclear whether basal levels of HLA-B27 expression in transgenic rats cause low-level ER stress and adaptive changes, and if so whether adapted cells respond differently to further upregulation of HLA-B27 and/or to cytokines and other exogenous stimuli. Splenocytes from older transgenic rats exhibit higher levels of constitutive XBP1 splicing (~4% vs. ~1%) (43), and 1.5–2-fold higher BiP mRNA and protein levels (36, 43, 96). These cells accumulate disulfide-linked and BiP-bound HLA-B27 heavy chains, providing evidence for ongoing misfolding in the absence of acute upregulation (36, 43). Thus, these cells exhibit changes consistent with past UPR activation and/or a low-level ongoing UPR. Since they were isolated from rats with ongoing inflammatory disease, they have most likely been chronically exposed to IFN-γ and other cytokines (15). Although it seems likely that adaptation to ER stress can occur, we have no direct evidence to suggest that these cells are protected from additional ER stress. Breban and colleagues (97) have recently published a proteomic analysis of CD103+ DCs isolated from HLA-B27/hβ2m transgenic rats providing evidence for chronic adaptive changes. They show striking overexpression of MHC class I loading components and several ER chaperones at the protein level (e.g. BiP, GRP94, protein disulfide isomerase A3 (ERp57), and calreticulin) relative to cells from WT and HLA-B7/hβ2m-expressing rats (97). This confirms UPR activation specific for HLA-B27 expression in cells other than macrophages and evidence for in vivo IFN exposure with upregulation of the MHC class I-loading pathway. Interestingly, they also found reduced expression of several proteins involved in cytoskeleton reorganization, which associated with deficient motility and immunological synapse formation, and the DCs were more prone to apoptosis with antibody-mediated ligation of MHC class II. The basis for the latter changes was not identified. It is plausible that these are downstream effects of ER stress or some combination of soluble factors (e.g. cytokines) that cells from HLA-B27/hβ2m transgenic rats are exposed to as a consequence of the inflammatory disease. ER stress in cells can predispose them to apoptosis induced by infection or other means, even when they have adapted to the chronic stress (98). It is also conceivable that HLA-B27/hβ2m overexpression has additional specific immunobiological effects through an as yet unidentified pathway. Regardless of the mechanism, the DC abnormalities in HLA-B27/hβ2m transgenic rats have been hypothesized to play a role in the inflammatory phenotype, possibly through failure to induce (or loss of) immune tolerance (97, 99, 100). The effects of acute and chronic ER stress on cells from HLA-B27/hβ2m transgenic rats and possible links to DC dysfunction and the inflammatory disease phenotype remain an area of active investigation.
Overexpression of additional hβ2m: a new model of spondyloarthritis?
To address the question of whether HLA-B27 misfolding plays a role in rat spondyloarthritis, Tran et al. (96) introduced an additional 35 copies of the hβ2m transgene (from the 283-2 line) into different lines of rats already transgenic for HLA-B27/hβ2m (Note that rats of the 33-3 line with 55 copies of the HLA-B27 gene and 66 copies of hβ2m are the animals discussed so far in this review). The results were surprising. Additional hβ2m expression resulted in more severe and more frequent arthritis, including frequent spondylitis, with no change in colitis in 33-3 rats (33-3 × 283-2). The additional 35 copies of hβ2m were also able to induce arthritis and spondylitis in rats with 20 copies of HLA-B27 and 15 copies of hβ2m (21-3 × 283-2) that were previously healthy. The rationale for the experiment was that additional hβ2m would drive the folding of HLA-B27, perhaps preventing it from misfolding and activating the UPR. At first glance the results seemed to suggest this possibility, leading the authors to argue that misfolding was not critical to the development of HLA-B27-associated arthropathy. Indeed, the HLA-B27 heavy chain did fold faster, and there was a partial reduction in the formation of disulfide-linked complexes. BiP mRNA levels were reduced by ~25–30% in HLA-B27-expressing splenocytes with extra hβ2m. However, BiP binding to HLA-B27 heavy chains was not assessed, but more importantly, the response to HLA-B27 upregulation, which had been shown previously to be important for detecting UPR activation (42), was not examined (96). Thus, it is possible that the suggestion that the demise of HLA-B27 misfolding and UPR activation as a possible pathogenic mechanism was premature.
We have now examined UPR activation in BM macrophages derived from HLA-B27/hβ2m transgenic rats expressing additional hβ2m (33-3 × 283-2 and 21-3 × 283-2) and stimulated with IFN-γ, LPS, or both. Indeed, additional hβ2m expression only attenuates UPR activation under these conditions. It does not prevent it from occurring (Fig. 7). BiP mRNA induction and XBP1 splicing are both quite robust in the presence of the extra hβ2m expressed from the additional 35 copies of the transgene. We also examined IFN-β and IL-23p19 induction under these conditions and found that even with additional hβ2m expression, the cytokines were still overexpressed compared to cells from WT rats. In fact there is some suggestion of a paradoxical effect, where additional hβ2m is associated with stronger induction of these cytokine mRNAs (Fig. 7). The question of how the additional hβ2m expression alters and exacerbates the phenotype and ultimately its relevance to human disease remain enigmatic. The possibility that β2m is exerting effects as a soluble mediator needs should be considered (101–103). Another interesting possibility is that the additional hβ2m is reducing basal levels of HLA-B27 misfolding, eliminating a chronic stress on the ER and preventing cells from adapting. This idea is consistent with the observed effects of additional hβ2m in lowering BiP mRNA expression in splenocytes (96). Removing the chronic stress could lead to very different biological effects in response to HLA-B27 upregulation, perhaps contributing to the phenotypic differences observed in gastrointestinal inflammation and arthritis. At this point, these ideas are speculative. However, what is clear is that a role for HLA-B27-induced UPR activation in spondyloarthritis-like disease in rats has not been ruled out and is likely to be complicated and require in-depth evaluation of effects of HLA-B27 on chronic as well as acute ER stress.
Fig. 7. Additional hβ2m does not prevent UPR activation in HLA-B27/hβ2m-expressing rat macrophages.

BM macrophages were derived from WT and 4 strains of HLA-B27/hβ2m transgenic rats that have different copy numbers of HLA-B27 and hβ2m transgenes. 33-3 rats have 55 copies of HLA-B27 and 66 copies of hβ2m; 33-3 × 283-2 (33-3 + hβ2m) have 35 additional copies of hβ2m from the 283-2 line. 21-3 rats have 20 copies of HLA-B27 and 15 copies of hβ2m; 21-3 × 283-2 (21-3 + hβ2m) have 35 additional copies of hβ2m. Macrophages remained untreated (NT) or were treated with 100 U/ml IFN-γ (IFN) for 24 h, 10 ng/ml LPS (LPS) for the last 3 h of culture, or IFN-γ + LPS (Both) with LPS present for the last 3 h of the 24 h IFN-γ treatment. RNA was isolated and mRNA for the various genes quantified with qPCR. Results are normalized to actin mRNA. XBP-1s is the percent of XBP1 mRNA transcripts that are spliced, and was measured as described previously (43, 65). Bars represent mean of triplicate cultures with standard deviation shown.
HLA-B27 subtypes
There is considerable heterogeneity within the HLA-B27 group of alleles referred to as subtypes. Up to this point we have been limited discussion to the most common subtype, B*2705, which is found in all populations around the world where there is HLA-B27. Subtypes are more closely related to one another than to other alleles, although precise criteria used to define different alleles on a molecular basis have not been established. Many subtypes have only been reported in a small number of individuals, and thus their prevalence and whether some represent mutations rather than polymorphisms are not certain. Since most subtypes of HLA-B27 occur infrequently, definitive disease association (or non-association) is lacking. The relatively common subtypes (e.g. B*2705, B*2702, and B*2704) that occur in multiple populations have been associated with AS, while two (B*2706 and B*2709) appear to be unassociated or perhaps weakly associated (104, 105). There may also be differences in the association of these subtypes with various forms of spondyloarthritis (106, 107). Reports of B*2706 and B*2709 being present in patients with AS have begun to emerge (108, 109).
When considering the question of which characteristics of the HLA-B27 protein are responsible for predisposing to disease, one approach has been to examine the subtypes that differ in disease association and narrow the possible mechanisms to only those characteristics that correlate with disease. In adopting this approach, one is assuming that structural or functional differences in the encoded protein are responsible and not other differences between the genes encoding the subtypes (or even closely linked genes). For example, there are entire haplotype differences between B*2705 and B*2709 in Sardinians that must be reconciled (110). Assessment of genetic variation between the genes (or regions) encoding the subtypes in question has not typically included regulatory regions or introns. For the most part, analyses have focused on non-synonymous SNPs that result in amino acid differences, yet SNPs that affect expression differences could be important (111). Recent studies suggest that copy number variation can be involved in disease susceptibility and/or phenotype variation (112), and regulatory RNAs (miRNAs) that are encoded within introns have been implicated as well (113). Another important consideration is the assumption that if the subtype is present in the population, it will be capable of predisposing to disease. This assumes that other susceptibility genes and/or necessary environmental factors are present. Given the dominant role of HLA-B27 in spondyloarthritis, this assumption seems a reasonable. However, even B*2705, which has been associated with disease virtually around the world, does not appear to confer risk in West Africa (114).
There is no clear consensus on the immunobiological behavior of disease-associated and non-associated (or weakly associated) subtypes, except that they do exhibit differences, including different but overlapping peptide repertoires (115). Two studies examined the folding efficiency (half-life of endoglycosidase-H sensitive β2m-free heavy chains) of HLA-B27 subtypes and other class I molecules differentially associated with AS and claimed that ‘misfolding’ could not account for their role in disease (116, 117). However, characteristics of HLA-B27 misfolding, such as the formation of aberrant disulfide-linked complexes and BiP binding in the ER that are well documented in the literature (7, 32, 36, 41–43), were not assessed. The ability of additional β2m expression in rat cells to dramatically improve the folding efficiency of the HLA-B27 heavy chain (96), yet not prevent it from misfolding and activating the UPR (Fig. 7), underscores the importance of properly measuring misfolding before drawing conclusions about its role in disease. At this point, correlations between the behavior of HLA-B27 subtypes and disease association do not provide a clear answer as to which mechanism of disease is most likely to be relevant. At this point, a more fruitful approach may be to establish how B*2705 causes spondyloarthritis-like disease in transgenic rats with corroborative studies in humans. In-depth genetic comparisons of the MHC region between affected and unaffected with different subtypes including deep sequencing approaches may also be worth considering.
Complex genetics of spondyloarthritis: beyond HLA-B27
The dominance of a single gene in a complex genetic disease places HLA-B27 and spondyloarthritis in an unusual place among immune-mediated and autoimmune inflammatory disorders. In AS, where >90% of affected individuals have HLA-B27, one can say that it is almost necessary for disease, yet it is clearly not sufficient, since only about 5% of HLA-B27 carriers develop a form of spondyloarthritis. This appreciation fueled the belief for some time that AS was essentially monogenic (118). Yet, it is now well established that susceptibility is complex, and HLA-B27 is only one of many genes involved in determining predisposition and phenotype (12). In this context, it is remarkable that expression of a single human risk allele in rats results in a phenotype that so closely mimics key features of human spondyloarthritis. Perhaps the fact that spondyloarthritis-like disease in rats is not a phenocopy of human AS should not be surprising, and we will need to understand the role of other polymorphic genes before other unique aspects of the phenotype can be understood. Ongoing genome-wide association studies will greatly expand the number of known risk alleles for AS and related spondyloarthritides over the next few years, which promise to be an exciting time.
What do other genes tell us about pathogenesis and the role of HLA-B27 in disease?
Alleles of IL23R and ERAP1 (also known as ARTS-1) were found recently to be associated with AS, and together may contribute as much as a third of overall susceptibility (77). Both of these genetic discoveries are noteworthy for several reasons. IL23R variants have been associated with both IBD and psoriasis previously (119, 120), and the data so far suggest that similar polymorphisms are involved, with the minor allele serving a protective role. Importantly, the effect of IL23R polymorphisms in AS appears to be independent of co-existing IBD or psoriasis, indicating that it is a separate risk factor for this disease.
The IL23R finding, together with a previous report of elevated serum IL-17 levels (72) and now Th17 activation (73–76, 121) in AS and spondyloarthritis patients, and combined with evidence from the transgenic rat model (65), strongly support the idea that the Th17 axis is important in spondyloarthritis pathogenesis (122, 123). In addition to increased numbers of Th17 T cells, there appears to be a subset making TNF-α and IFN-γ (74), and in another study a significant proportion of IL-17-producing T cells from AS patients were producing IL-22 and IFN-γ (75). While these data support activation of the Th17 axis in spondyloarthritis, Th17 activation is by no means unique to this group of diseases, and it alone cannot account for the unique phenotype.
A major goal of unraveling the complex genetics of AS is to begin to better understand pathogenesis through the identification of involved pathways and perhaps how variants of gene products interact to cause dysregulated immunity. In Fig. 8, we present one view of how some of the findings discussed in this review on HLA-B27 may inform current concepts of pathogenesis. Most notable is the potential connection between HLA-B27 misfolding and activation of the IL-23/IL-17 axis through effects of ER stress and the UPR IL-23 production. If IL23R polymorphisms influence responsiveness of Th17 T cells to IL-23, it could provide a plausible link between the risk conferred by these two gene products.
Fig. 8. Hypothetical mechanism linking HLA-B27 misfolding and other disease associated genes to the IL-23/IL-17 axis.

Cytokines that upregulate the MHC class I expression and assembly pathway, such as IFNs and/or TNF-α, may trigger an UPR in antigen-presenting cells of HLA-B27-expressing individuals when a critical threshold of misfolding is reached. ERAP1 polymorphisms could influence the efficiency of HLA-B27 folding, its propensity to misfold, and/or its peptide repertoire, through its effect on peptide trimming in the ER. The UPR will polarize cells responding to PRR agonists toward the production of greater amounts of IL-23 over IL-12, which in turn provides a stimulus for Th17 survival and activation in individuals with permissive IL23R polymorphisms. Th17 cells are shown producing IFN-γ, which has been documented in several situations, but would also be expected to come from Th1 cells. IL-1 locus polymorphisms (possibly IL1A) could play a role in Th17 development as well as in other inflammatory processes. This schema is meant as a framework to provide testable hypotheses regarding the role of various polymorphic susceptibility genes in the pathogenesis of spondyloarthritis. Additional susceptibility genes that fit within this schema are being identified (unpublished data). This figure is reproduced in a modified form from “Molecular Mechanisms of Spondyloarthropathies”, edited by Carlos López-Larrea and Roberto Diaz-Peña (c2009 Landes Bioscience and Springer Science+Business Media) with permission.
Potential roles of ERAP1 in this schema are also worth considering. ERAP1 encodes an ER aminopeptidase involved in the processing of peptides presented by MHC class I molecules (124–129) (Fig. 8). Polymorphisms in ERAP1 would be anticipated to influence the quality and quantity of MHC class I complexes assembled in the cell and presented on the cell surface. While this argues for a functional interaction between HLA-B27 and ERAP1, it does not tell us which feature of HLA-B27 is critically affected and how this is relevant to disease pathogenesis. If the transgenic rat model is wrong and spondylogenic peptides are important in disease, then the effect of ERAP1 might be mediated through effects on the HLA-B27 peptide repertoire. Alternatively, by influencing peptide supply, ERAP1 could alter the folding characteristics of HLA-B27 and its tendency to misfold and/or its propensity to dimerize after reaching the cell surface. Interestingly, ERAP1 was discovered independently as ARTS1, and named for its role in promoting TNF receptor shedding (aminopeptidase regulator of TNF receptor shedding) (130). ARTS1 also promotes shedding of the IL-1 and IL-6 receptors (131, 132). Its role in receptor shedding does not require its aminopeptidase activity. Determining how ERAP1 influences the development of spondyloarthritis may be as complicated as deciphering the role of HLA-B27.
Conclusions
The role of HLA-B27 in susceptibility to spondyloarthritis remains an enigma. This review has focused on the discovery of unusual features of HLA-B27, how these observations came about, and what we have learned about their potential consequences. Our focus has been on the propensity of newly made HLA-B27 to form aberrant disulfides and bind BiP, and hence to misfold in the ER, and to dimerize after reaching the cell surface. While these processes share a common feature, i.e. abnormal disulfide bond formation, they differ temporally, spatially, and mechanistically, and have different consequences, and thus need to be understood in this context. The journey through the ER has resulted in novel observations about protein misfolding and the convergence of UPR and PRR signaling pathways in cytokine production that have implications for this and other diseases. There is evidence for UPR activation in cells from humans with spondyloarthritis (133, 134), although more needs to be done to understand when and where this occurs, and to validate findings from animal models. For spondyloarthritis, HLA-B27 misfolding may provide a mechanistic link between this gene and new genetic and translational findings implicating the IL-23/IL-17 axis in spondyloarthritis pathogenesis. The rapid pace at which new susceptibility genes are being discovered is both exciting and daunting, when one considers the challenges involved in establishing the functional significance of these variants and then how they interact with other risk genes. Nevertheless, this is the task that we now face if we are to translate these findings into new therapeutic approaches.
Acknowledgments
This work was supported by a Pfizer Scholar Award, and grants from the National Institutes of Health (AR46177 and AR48372) and the Arthritis Foundation (Biomedical Science Grant) to RAC. The authors thank members of the Colbert lab who, over the years, have contributed to our understanding of the immunobiology of HLA-B27, including Shuzhen Bai, Nandita Dangoria, Thomas Griffin, Daniel Kingsbury, Gerlinde Layh-Schmitt, John Mear, Rajashree Mohapatra, Kathy Schreiber, Dawn Sowders, Judith Smith, and Matthew Turner. We also thank Joel Taurog for contributing the HLA-B27 transgenic rats used for the experiment shown in Fig. 7.
References
- 1.Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369:1379–1390. doi: 10.1016/S0140-6736(07)60635-7. [DOI] [PubMed] [Google Scholar]
- 3.Caffrey MF, James DC. Human lymphocyte antigen association in ankylosing spondylitis. Nature. 1973;242:121. doi: 10.1038/242121a0. [DOI] [PubMed] [Google Scholar]
- 4.Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DCO, Sturrock RD. Ankylosing spondylitis and HL-A 27. Lancet. 1973;1:904–907. doi: 10.1016/s0140-6736(73)91360-3. [DOI] [PubMed] [Google Scholar]
- 5.Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288:704–706. doi: 10.1056/NEJM197304052881403. [DOI] [PubMed] [Google Scholar]
- 6.Mear JP, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163:6665–6670. [PubMed] [Google Scholar]
- 7.Colbert RA. The immunobiology of HLA-B27: variations on a theme. Curr Mol Med. 2004;4:21–30. doi: 10.2174/1566524043479293. [DOI] [PubMed] [Google Scholar]
- 8.Allen RL, O’Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J Immunol. 1999;162:5045–5048. [PubMed] [Google Scholar]
- 9.Bowness P. HLA B27 in health and disease: a double-edged sword? Rheumatology. 2002;41:857–868. doi: 10.1093/rheumatology/41.8.857. [DOI] [PubMed] [Google Scholar]
- 10.Kollnberger S, et al. HLA-B27 heavy chain homodimers are expressed in HLA-B27 transgenic rodent models of spondyloarthritis and are ligands for paired Ig-like receptors. J Immunol. 2004;173:1699–1710. doi: 10.4049/jimmunol.173.3.1699. [DOI] [PubMed] [Google Scholar]
- 11.Chan AT, Kollnberger SD, Wedderburn LR, Bowness P. Expansion and enhanced survival of natural killer cells expressing the killer immunoglobulin-like receptor KIR3DL2 in spondylarthritis. Arthritis Rheum. 2005;52:3586–3595. doi: 10.1002/art.21395. [DOI] [PubMed] [Google Scholar]
- 12.Brown MA. Genetics and the pathogenesis of ankylosing spondylitis. Curr Opin Rheumatol. 2009;21:318–323. doi: 10.1097/bor.0b013e32832b3795. [DOI] [PubMed] [Google Scholar]
- 13.Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer RE, Maika SD, Richardson JA, Tang J-P, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human β2-m: an animal model of HLA- B27-associated human disorders. Cell. 1990;63:1099–1112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- 15.Taurog JD, et al. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–223. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 16.May E, et al. CD8αβ T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol. 2003;170:1099–1105. doi: 10.4049/jimmunol.170.2.1099. [DOI] [PubMed] [Google Scholar]
- 17.Taurog JD, et al. Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheum. 2009;60:1977–1984. doi: 10.1002/art.24599. [DOI] [PubMed] [Google Scholar]
- 18.Hoentjen F, Tonkonogy SL, Qian BF, Liu B, Dieleman LA, Sartor RB. CD4(+) T lymphocytes mediate colitis in HLA-B27 transgenic rats monoassociated with nonpathogenic Bacteroides vulgatus. Inflamm Bowel Dis. 2007;13:317–324. doi: 10.1002/ibd.20040. [DOI] [PubMed] [Google Scholar]
- 19.Cook JL, Ikle DN, Routes BA. Natural killer cell ontogeny in the athymic rat. Relationship between functional maturation and acquired resistance to E1A oncogene-expressing sarcoma cells. J Immunol. 1995;155:5512–5518. [PubMed] [Google Scholar]
- 20.Kievits F, Ivanyi P, Krimpenfort P, Berns A, Ploegh HL. HLA-restricted recognition of viral antigens in HLA transgenic mice. Nature. 1987;329:447–449. doi: 10.1038/329447a0. [DOI] [PubMed] [Google Scholar]
- 21.Krimpenfort P, Rudenko G, Hochstenbach F, Gussow D, Berns A, Ploegh H. Crosses of two independently derived transgenic mice demonstrate functional complementation of the genes encoding heavy (HLA-B27) and light (β2-microglobulin) chains of HLA class I antigens. EMBO J. 1987;6:1673–1676. doi: 10.1002/j.1460-2075.1987.tb02416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taurog JD, Lowen L, Forman J, Hammer RE. HLA-B27 in inbred and non-inbred transgenic mice: cell surface expression and recognition as an alloantigen in the absence of human β2-microglobulin. J Immunol. 1988;141:4020–4023. [PubMed] [Google Scholar]
- 23.Khare SD, Luthra HS, David CS. Spontaneous inflammatory arthritis in HLA-B27 transgenic mice lacking β2-microglobulin: a model of human spondyloarthropathies. J Exp Med. 1995;182:1153–1158. doi: 10.1084/jem.182.4.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khare SD, Hansen J, Luthra HS, David CS. HLA-B27 heavy chains contribute to spontaneous inflammatory disease in B27/human β2-microglobulin (β2m) double transgenic mice with disrupted mouse β2m. J Clin Invest. 1997;98:2746–2455. doi: 10.1172/JCI119100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khare SD, Hansen J, Luthra HS, David CS. Spontaneous inflammatory disease in HLA-B27 transgenic mice is independent of MHC class II molecules: a direct role for B27 heavy chains and not B27-derived peptides. J Immunol. 1998;160:101–106. [PubMed] [Google Scholar]
- 26.Kingsbury DJ, Mear JP, Witte DP, Taurog JD, Roopenian DC, Colbert RA. Development of spontaneous arthritis in b2-microglobulin-deficient mice without expression of HLA-B27: association with deficiency of endogenous major histocompatibility complex class I expression. Arthritis Rheum. 2000;43:2290–2296. doi: 10.1002/1529-0131(200010)43:10<2290::AID-ANR17>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 27.Benjamin RJ, Parham P. Guilt by association: HLA-B27 and ankylosing spondylitis. Immunol Today. 1990;11:137–142. doi: 10.1016/0167-5699(90)90051-a. [DOI] [PubMed] [Google Scholar]
- 28.Colbert RA, Rowland-Jones SL, McMichael AJ, Frelinger JA. Allele-specific B pocket transplant in class I major histocompatibility complex protein changes requirement for anchor residue at P2 of peptide. Proc Natl Acad Sci USA. 1993;90:6879–6883. doi: 10.1073/pnas.90.14.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes EA, Hammond C, Cresswell P. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc Natl Acad Sci USA. 1997;94:1896–1901. doi: 10.1073/pnas.94.5.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 31.Whelan MA, Archer JR. Chemical reactivity of an HLA-B27 thiol group. Eur J Immunol. 1993;23:3278–3285. doi: 10.1002/eji.1830231233. [DOI] [PubMed] [Google Scholar]
- 32.Dangoria NS, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem. 2002;277:23459–23468. doi: 10.1074/jbc.M110336200. [DOI] [PubMed] [Google Scholar]
- 33.Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol. 2003;33:748–759. doi: 10.1002/eji.200323678. [DOI] [PubMed] [Google Scholar]
- 34.Urban RG, et al. A subset of HLA-B27 molecules contains peptides much longer than nonamers. Proc Natl Acad Sci USA. 1994;91:1534–1538. doi: 10.1073/pnas.91.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malik P, Klimovitsky P, Deng LW, Boyson JE, Strominger JL. Uniquely conformed peptide-containing beta 2-microglobulin-free heavy chains of HLA-B2705 on the cell surface. J Immunol. 2002;169:4379–4387. doi: 10.4049/jimmunol.169.8.4379. [DOI] [PubMed] [Google Scholar]
- 36.Tran TM, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172:5110–5119. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 37.Fussell H, et al. Novel detection of in vivo HLA-B27 conformations correlates with ankylosing spondylitis association. Arthritis Rheum. 2008;58:3419–3424. doi: 10.1002/art.23990. [DOI] [PubMed] [Google Scholar]
- 38.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noessner E, Parham P. Species-specific differences in chaperone interaction of human and mouse major histocompatibility complex class I molecules. J Exp Med. 1995;181:327–337. doi: 10.1084/jem.181.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158:247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem. 2004;279:8895–8902. doi: 10.1074/jbc.M311757200. [DOI] [PubMed] [Google Scholar]
- 42.Turner MJ, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 43.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56:215–223. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 44.Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional structure of HLA-B27 at 2.1 Å resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992;70:1035–1048. doi: 10.1016/0092-8674(92)90252-8. [DOI] [PubMed] [Google Scholar]
- 45.Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 46.Peace-Brewer AL, Tussey LG, Matsui M, Li G, Quinn DG, Frelinger JA. A point mutation in HLA-A*0201 results in failure to bind the TAP complex and to present virus-derived peptides to CTL. Immunity. 1996;4:505–514. doi: 10.1016/s1074-7613(00)80416-1. [DOI] [PubMed] [Google Scholar]
- 47.Hosokawa N, et al. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 49.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 50.Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J Biol Chem. 2003;278:26287–26294. doi: 10.1074/jbc.M303395200. [DOI] [PubMed] [Google Scholar]
- 51.Peh CA, et al. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity. 1998;8:531–542. doi: 10.1016/s1074-7613(00)80558-0. [DOI] [PubMed] [Google Scholar]
- 52.Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity. 2009;30:21–32. doi: 10.1016/j.immuni.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purcell AW, et al. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J Immunol. 2001;166:1016–1027. doi: 10.4049/jimmunol.166.2.1016. [DOI] [PubMed] [Google Scholar]
- 54.Sesma L, et al. Qualitative and quantitative differences in peptides bound to HLA-B27 in the presence of mouse versus human tapasin define a role for tapasin as a size-dependent peptide editor. J Immunol. 2005;174:7833–7844. doi: 10.4049/jimmunol.174.12.7833. [DOI] [PubMed] [Google Scholar]
- 55.Gregersen N, Bross P, Vang S, Christensen JH. Protein Misfolding and Human Disease. Annu Rev Genomics Hum Genet. 2006;7:103–124. doi: 10.1146/annurev.genom.7.080505.115737. [DOI] [PubMed] [Google Scholar]
- 56.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 59.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9:378–387. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 61.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 63.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 64.Mintz M, et al. Time series proteome profiling to study endoplasmic reticulum stress response. J Proteome Res. 2008;7:2435–2444. doi: 10.1021/pr700842m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 68.Mielants H, Veys EM, Cuvelier C, de Vos M. Ileocolonoscopic findings in seronegative spondylarthropathies. Br J Rheumatol. 1988;27 (Suppl):95–105. doi: 10.1093/rheumatology/xxvii.suppl_2.95. [DOI] [PubMed] [Google Scholar]
- 69.De Vos M, Mielants H, Cuvelier C, Elewaut A, Veys E. Long-term evolution of gut inflammation in patients with spondyloarthropathy. Gastroenterology. 1996;110:1696–1703. doi: 10.1053/gast.1996.v110.pm8964393. [DOI] [PubMed] [Google Scholar]
- 70.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 71.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74:304–305. doi: 10.1016/j.jbspin.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Singh R, Aggarwal A, Misra R. Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J Rheumatol. 2007;34:2285–2290. [PubMed] [Google Scholar]
- 74.Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008;58:2307–2317. doi: 10.1002/art.23655. [DOI] [PubMed] [Google Scholar]
- 75.Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009;60:1647–1656. doi: 10.1002/art.24568. [DOI] [PubMed] [Google Scholar]
- 76.Ciccia F, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009;60:955–965. doi: 10.1002/art.24389. [DOI] [PubMed] [Google Scholar]
- 77.Burton PR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 79.Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 81.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1021. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 84.Nishitoh H, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hitomi J, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han D, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. [Google Scholar]
- 88.Zinszner H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gotoh T, Oyadomari S, Mori K, Mori M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J Biol Chem. 2002;277:12343–12350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- 92.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoshida H, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 95.Wu J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 96.Tran TM, et al. Additional human beta(2)-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 2006;54:1317–1327. doi: 10.1002/art.21740. [DOI] [PubMed] [Google Scholar]
- 97.Dhaenens M, et al. Dendritic cells from spondylarthritis-prone HLA-B27-transgenic rats display altered cytoskeletal dynamics, class II major histocompatibility complex expression, and viability. Arthritis Rheum. 2009;60:2622–2632. doi: 10.1002/art.24780. [DOI] [PubMed] [Google Scholar]
- 98.Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol. 2006;172:395–407. doi: 10.1083/jcb.200508016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hacquard-Bouder C, et al. Alteration of antigen-independent immunologic synapse formation between dendritic cells from HLA-B27-transgenic rats and CD4+ T cells: selective impairment of costimulatory molecule engagement by mature HLA-B27. Arthritis Rheum. 2007;56:1478–1489. doi: 10.1002/art.22572. [DOI] [PubMed] [Google Scholar]
- 100.Hacquard-Bouder C, et al. Defective costimulatory function is a striking feature of antigen-presenting cells in an HLA-B27-transgenic rat model of spondylarthropathy. Arthritis Rheum. 2004;50:1624–1635. doi: 10.1002/art.20211. [DOI] [PubMed] [Google Scholar]
- 101.Miyata T, et al. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J Clin Invest. 1996;98:1088–1094. doi: 10.1172/JCI118889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hou FF, et al. beta(2)-Microglobulin modified with advanced glycation end products delays monocyte apoptosis. Kidney Int. 2001;59:990–1002. doi: 10.1046/j.1523-1755.2001.059003990.x. [DOI] [PubMed] [Google Scholar]
- 104.Lopez-Larrea C, et al. HLA-B27 subtypes in Asian patients with ankylosing spondylitis: Evidence for new associations. Tissue Antigens. 1995;45:169–176. doi: 10.1111/j.1399-0039.1995.tb02436.x. [DOI] [PubMed] [Google Scholar]
- 105.Fiorillo MT, et al. Susceptibility to ankylosing spondylitis correlates with the C-terminal residue of peptides presented by various HLA-B27 subtypes. Eur J Immunol. 1997;27:368–373. doi: 10.1002/eji.1830270205. [DOI] [PubMed] [Google Scholar]
- 106.Olivieri I, et al. The HLA-B*2709 subtype in a patient with undifferentiated spondarthritis. Ann Rheum Dis. 2000;59:654–655. doi: 10.1136/ard.59.8.654a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Olivieri I, et al. The HLA-B*2709 subtype confers susceptibility to spondylarthropathy. Arthritis Rheum. 2002;46:553–554. doi: 10.1002/art.10127. [DOI] [PubMed] [Google Scholar]
- 108.Olivieri I, D’Angelo S, Scarano E, Santospirito V, Padula A. The HLA-B*2709 subtype in a woman with early ankylosing spondylitis. Arthritis Rheum. 2007;56:2805–2807. doi: 10.1002/art.22821. [DOI] [PubMed] [Google Scholar]
- 109.Hou TY, Chen HC, Chen CH, Chang DM, Liu FC, Lai JH. Usefulness of human leucocyte antigen-B27 subtypes in predicting ankylosing spondylitis: Taiwan experience. Intern Med J. 2007;37:749–752. doi: 10.1111/j.1445-5994.2007.01450.x. [DOI] [PubMed] [Google Scholar]
- 110.Cascino I, et al. Identification of previously unrecognized predisposing factors for ankylosing spondylitis from analysis of HLA-B27 extended haplotypes in Sardinia. Arthritis Rheum. 2007;56:2640–2651. doi: 10.1002/art.22820. [DOI] [PubMed] [Google Scholar]
- 111.Cauli A, et al. Increased level of HLA-B27 expression in ankylosing spondylitis patients compared with healthy HLA-B27-positive subjects: a possible further susceptibility factor for the development of disease. Rheumatology. 2002;41:1375–1379. doi: 10.1093/rheumatology/41.12.1375. [DOI] [PubMed] [Google Scholar]
- 112.Fanciulli M, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ying SY, Lin SL. Intron-derived microRNAs--fine tuning of gene functions. Gene. 2004;342:25–28. doi: 10.1016/j.gene.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 114.Hill AVS, Allsop CEM, Kwiatkowski D, Antsey NM, Greenwood BM, McMichael AJ. HLA class I typing by PCR: HLA-B27 and an African B27 subtype. Lancet. 1991;337:640– 642. doi: 10.1016/0140-6736(91)92452-8. [DOI] [PubMed] [Google Scholar]
- 115.Marcilla M, Lopez de Castro JA. Peptides: the cornerstone of HLA-B27 biology and pathogenetic role in spondyloarthritis. Tissue Antigens. 2008;71:495–506. doi: 10.1111/j.1399-0039.2008.01051.x. [DOI] [PubMed] [Google Scholar]
- 116.Galocha B, de Castro JA. Folding of HLA-B27 subtypes is determined by the global effect of polymorphic residues and shows incomplete correspondence to ankylosing spondylitis. Arthritis Rheum. 2008;58:401–412. doi: 10.1002/art.23164. [DOI] [PubMed] [Google Scholar]
- 117.Merino E, Galocha B, Vazquez MN, Lopez de Castro JA. Disparate folding and stability of the ankylosing spondylitis-associated HLA-B*1403 and B*2705 proteins. Arthritis Rheum. 2008;58:3693–3704. doi: 10.1002/art.24045. [DOI] [PubMed] [Google Scholar]
- 118.Brown MA. Breakthroughs in genetic studies of ankylosing spondylitis. Rheumatology. 2008;47:132–137. doi: 10.1093/rheumatology/kem269. [DOI] [PubMed] [Google Scholar]
- 119.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cargill M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang X, Lin Z, Wei Q, Jiang Y, Gu J. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatol Int. 2009;29:1343–1347. doi: 10.1007/s00296-009-0883-x. [DOI] [PubMed] [Google Scholar]
- 122.Colbert RA, et al. 2007 annual research and education meeting of the Spondyloarthritis Research and Therapy Network (SPARTAN) J Rheumatol. 2008;35:1398–1402. [PubMed] [Google Scholar]
- 123.Layh-Schmitt G, Colbert RA. The interleukin-23/interleukin-17 axis in spondyloarthritis. Curr Opin Rheumatol. 2008;20:392–397. doi: 10.1097/BOR.0b013e328303204b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 125.Saric T, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–1176. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 126.York IA, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 127.Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol. 2006;7:103–112. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- 128.Kanaseki T, Blanchard N, Hammer GE, Gonzalez F, Shastri N. ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity. 2006;25:795–806. doi: 10.1016/j.immuni.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8:101–108. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 130.Cui X, et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest. 2002;110:515–526. doi: 10.1172/JCI13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cui X, Rouhani FN, Hawari F, Levine SJ. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol. 2003;171:6814–6819. doi: 10.4049/jimmunol.171.12.6814. [DOI] [PubMed] [Google Scholar]
- 132.Cui X, Rouhani FN, Hawari F, Levine SJ. An aminopeptidase, ARTS-1, is required for interleukin-6 receptor shedding. J Biol Chem. 2003;278:28677–28685. doi: 10.1074/jbc.M300456200. [DOI] [PubMed] [Google Scholar]
- 133.Gu J, et al. Clues to the pathogenesis of spondyloarthropathy derived from synovial fluid mononuclear cell gene expression profiles. J Rheumatol. 2002;29:2159–2164. [PubMed] [Google Scholar]
- 134.Dong W, Zhang Y, Yan M, Liu H, Chen Z, Zhu P. Upregulation of 78-kDa glucose-regulated protein in macrophages in peripheral joints of active ankylosing spondylitis. Scand J Rheumatol. 2008:1–8. doi: 10.1080/03009740802213310. [DOI] [PubMed] [Google Scholar]