

Abstract
Glycolipids containing α-linked galactosyl and glucosyl moieties have been shown to possess unique immunostimulatory activity creating a need for access to diverse and anomerically pure sources of these compounds for immunological studies. To meet this demand, glycosyl iodides were enlisted in the synthesis of these biologically relevant glycoconjugates. In the first generation protocol per-O-benzyl galactosyl iodide was efficiently coupled with activated sphingosine acceptors, but fully functionalized ceramides were found to be unreactive. To overcome this obstacle, per-O-trimethylsilyl glycosyl iodides were investigated and shown to undergo highly efficient coupling with ceramide and glycerol ester acceptors. Contrary to what has been observed with other donors, we detected little difference between the reactivity of glucosyl and galactosyl iodides. The trimethylsilyl protecting groups play a dual role in activating the donor toward nucleophilic attack while at the same time providing transient protection: the silyl groups are readily removed upon methanolysis. All reactions proceeded with complete acceptor regioselectivity, eliminating the need for additional protecting group manipulations, and the desired α -anomers were formed exclusively. This three step one-pot synthetic platform provides rapid access to an important class of immunostimulatory molecules including the first reported synthesis of the glucosyl analog of the bacterial antigen BbGL-II.
Introduction
In 1993, researchers in Japan reported the isolation and bioactivity studies of the first known α-linked galactosylceramides (α-GalCer).1 These glycolipids, also known as agelasphins, were extracted from the marine sponge Agelas mauritianus and possessed unique anti-tumor activity. Subsequent structure activity relationship (SAR) studies led to the identification of the prototypical antigen, KRN7000 (Figure 1). The immunostimulatory effects of the agelasphins attracted considerable attention, as it was especially curious that natural products derived from a marine sponge could stimulate the immune system. Investigations aimed at identifying other possible sources of α-linked glycolipids commenced. One such study centered on Borrelia burgdorferi, which is the causative agent of Lyme disease. Lyme is the most common vector-borne disease in the United States and is a multisystemic disorder that affects the skin, nervous system, heart, muscles and joints.2 Its gram-negative spirochete is transmitted through the bite of infected ticks and contains the α-linked glycolipid BbGL-II (Figure 1), which exhibits similar antigenic activity to KRN7000. This discovery marked the first example of an NKT cell antigen expressed by a human pathogen and led to the hypothesis that the glycolipids isolated from sponge were of bacterial origin.3–7
Figure 1.

Structures of immunostimulatory glycolipids
Subsequent biological investigations revealed that these glycolipids interact with the lipid binding protein CD1d associated with antigen presenting cells (APC).8, 9 Once bound, the carbohydrate head group of the glycolipid-CD1d complex is presented to a T-cell receptor (TCR) on natural killer T-cells (NKT) which, upon binding, initiates an immunological cascade of events.10, 11 This immune response includes the release of cytokines mainly composed of the T helper 1 (Th1) cytokine IFN-γ and the T helper 2 (Th2) cytokine IL-4.12, 13 Th1 responses are important in controlling viral and bacterial infections, as well as tumor metastasis, whereas Th2 cytokine secretion is associated with certain autoimmune diseases such as type-1 diabetes and multiple sclerosis.14 However, simultaneous production of both Th1 and Th2 cytokines has an antagonizing effect resulting from reciprocal inhibition.15 Therefore, research into the identification of analogs that elicit biased cytokine production is of high interest for therapeutic use.16, 17
Since a Th1 response requires prolonged CD1d-glycolipid-TCR binding, the desired polarizing effects may be achieved through modulation of ternary complex longevity.18 Attempts to tune the stability of the CD1d-glycolipid-TCR construct have focused primarily on modifications to the lipid portion of the antigen. These alterations serve to attenuate complex stability by shifting the spatial orientation of the carbohydrate. Recently it has been shown that variation to the carbohydrate residue may also bias cytokine production. And while it is known that α-mannosyl ceramides (α-ManCer) exhibit no stimulatory activity, α-glucosyl ceramides (α-GlcCer) express similar activity profiles as their α-GalCer epimers.11, 19 As such, there is a need for access to diverse and anomerically pure sources of α-linked glycolipids to further probe the immunological effects of these antigens. To meet this demand, we recently communicated highly efficient syntheses of bioactive glycolipids with galactose head groups.20, 21 Herein we report a full account of those studies and extension of the methodology to include glucosyl analogs.
Results and Discussion
Within the last decade, glycosyl iodide chemistry has enjoyed a renaissance largely due to our ability to tame these reactive donors through careful choice of protecting groups. In the context of stereoselective sytheses of α-GalCer analogs, we first reported the coupling of per-O-benzyl protected galactosyl iodide (1) with azido phytosphingosine (2, Scheme 1).20 TBAI was added to the reaction mixture to promote in situ anomerizarion of the α-iodide to its more reactive β-anomer. Nucleophilic addition of 2 to the reactive intermediate provided α-galactosyl azido phytosphingosine (3) exclusively. In those investigations, we observed a correlation between yield and the protecting groups appended to the nucleophile. For example, when the secondary hydroxyls of 2 were protected with benzoates (Bz), only a 30% yield was observed. Benzyl (Bn) protecting groups, which are electron donating relative to the benzoates, afforded increased product yield (67%) and when p-methoxybenzyl (PMB) protecting groups were used, essentially quantitative yields were achieved. Subsequent reduction of the azide and amidation of the resulting amine followed by global deprotection afforded KRN7000 in arguably the most efficient series of reactions reported prior to the date of that publication.
Scheme 1.

1st generation synthesis of KRN7000
These results cemented our understanding of the electronic sensitivity of these reactions and the importance of matching donor and acceptor reactivity. It is well known that per-O-benzylated glycosyl iodides are orders of magnitude more reactive than their corresponding per-O-acetylated analogs. This is evidenced by the fact that per-O-acetylated glycosyl iodides can be easily purified by column chromatography and even crystallized,22, 23 whereas the benzyl analogs are far more reactive and can only be isolated with great care.24 Likewise, the more electron rich the acceptor alcohol, the better suited it is to couple with a donor. Conversely, more reactive donors are required for the glycosidation of unreactive acceptors. The concept of matching donor/acceptor reactivity changed our way of thinking and resulted in a new direction for our research.
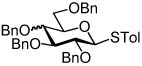
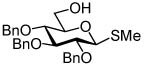
Our ultimate goal was to be able to react fully functionalized lipid acceptors with glycosyl iodide donors. We appreciated the fact that glycosidation with fully functionalized ceramides was notoriously difficult. For example, Sakai and co-workers attempted a reaction between per-O-benzyl galactosyl fluoride and a ceramide using stannous chloride/silver perchlorate activation in which only a 23% yield of the desired α-product was isolated.25 The main challenge with this strategy is the diminished nucleophilicity of the ceramide primary alcohol due to intramolecular hydrogen bonding with the amide proton.26 We alleviated this problem in our first generation syntheses by masking the amide functionality as an azide, however this process involves many additional steps and reduces the overall efficiency. As indicated in Scheme 2, starting from commercially available sphingosine (4), four steps are required to prepare the acceptor for glycosidation. First the amine is converted to an azide through the action of TfN3, the primary alcohol is then protected with a trityl group and the secondary alcohol is reacted with p-methoxybenzyl chloride before removal of the trityl protecting group. Reaction of azido sphingosine (5) with 1 is very efficient, however after glycosidation one must essentially reverse the reactions required to convert 4 to 5. First the azide is reduced, then it is amidated and finally the protecting groups are removed to afford the KRN7000 analog (7). In the case illustrated, the double bond on the lipid was reduced during hydrogenolysis although others have reported methods for selectively removing benzyl groups while leaving the double bond intact.25, 27
Scheme 2.

1st generation synthesis of desoxy-KRN7000 (7)
Wishing to avoid these protecting group manipulations, we turned our attention to the possibility of reacting glycosyl iodides with fully functionalized ceramide acceptors (Figure 2). Initial glycosylations between per-O-benzyl galactosyl iodide (1) and ceramide (8) met with limited success, as only low product yields were obtained. However, concurrent studies in our lab utilizing per-O-trimethylsilyl galactosyl iodide were more promising. We found this donor to be orders of magnitude more reactive than the per-O-benzyl analog. Per-O-TMS glycosides are readily prepared on large scale and the iodide can be generated quantitatively upon reaction with iodotrimethylsilane (TMSI).28 An additional benefit associated with the use of silyl ether protecting groups is that they can be readily removed by methanolysis upon completion of the reaction, leaving the glycosidic linkage and olefin unaffected. In this manner, the trimethylsilyl functionality serves as a transient protecting group.
Figure 2.

Structures of ceramide and glycerol containing products and acceptors
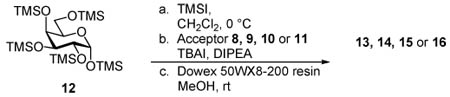
Our first attempts at developing this methodology focused on galactosyl glycolipids.21 The syntheses of 13 – 16 began with the addition of TMSI to a cooled solution of 12, generating per-O-TMS galactosyl iodide in situ. The donor was then added to a mixture containing the acceptor (8, 9, 10 or 11), TBAI and N,N-diisopropylethylamine (DIPEA). After the indicated time, the solvent was evaporated and the residue was subjected to hydrolysis using acidic resin in methanol affording the final products 13, 14, 15 and 16 (Table 1). Initially, the glycosidation of acceptor 9 proved problematic and at room temperature only low yields (30%, entry 2) were obtained. However, to our delight we found that when the reaction was conducted at 115 °C with 200W of microwave irradiation, the yield of 14 increased to 67% and the reaction time was reduced from 14 h to 90 min (Table 1, entry 3).
Table 1.
2nd generation synthesis of biologically relevant glycolipids
 | |||||
|---|---|---|---|---|---|
| Entry | Acceptor | Solvent | Conditions | Product | α:β ratio (Yield) |
| 1 | 8 | CH2Cl2 | TBAI (3 eq), rt, 48 h | 13 | only α (77%) |
| 2 | 9 | CH2Cl2 | TBAI (3 eq), rt, 48 h | 14 | only α (30%) |
| 3 | 9 | CH2Cl2 | TBAI (3 eq), ))), 1.5 h | 14 | only α (67%) |
| 4 | 10 | CH2Cl2 | TBAI (4 eq), rt, 24 h | 15 | only α (81%) |
| 5 | 11 | CH2Cl2 | TBAI (3 eq), rt, 36 h | 16 | only α (72%) |
We naturally thought that microwave irradiation would also promote reactions with 8, but that proved not to be the case. Instead we observed trans-silylation of the acceptor as evidenced by the disappearance of 8 by thin layer chromatography (TLC) and its reappearance after methanolysis. Indeed, we were able to confirm that trans-silylation had occurred by isolating and characterizing a silyl protected acceptor. We hypothesized that TBAI could be promoting desilylation of the sugar generating TMSI, which could in turn react with the acceptor. In an attempt to reduce this process, we explored the possibility of using fewer equiv of TBAI (Figure 3). These studies indicated that at least 1.5 equiv of TBAI are required for efficient conversion.29 We also attempted microwave irradiation with reduced TBAI and again observed primarily trans-silylation, indicating that room temperature conditions are best for this system.
Figure 3.

TBAI optimization study
We next turned our attention to the syntheses of glucose containing bioactive glycolipids. While there are several published methods for the synthesis of α-GalCer derivatives, there are far fewer reports for the stereoselective synthesis of the corresponding glucosyl analogs. The most commonly employed donor for the synthesis of 1,2-cis glucosides has been 2,3,4,6-tetra-O-benzyl glucopyranosyl fluoride.30 Typical protocols require the use of toxic reagents (stannous chloride and silver perchlorate), result in α/β product mixtures, and proceed without regioselectivity with respect to the acceptor.31 When applied to the glucosidation of unprotected ceramides, mixtures of both mono and diglycosylated products are formed. Other donors show similar reactivities. For example, Fan and coworkers recently utilized 2,3,4,6-tetra-O-benzyl thiophenyl glucopyranoside in the NIS/TfOH promoted glycosidation of a serine-based ceramide. This protocol afforded GlcCer in 46% yield with 2:1 α:β selectivity.32 The reactivity, stereoselectivity and overall reaction efficiencies are typically reduced with glucosyl donors relative to galactosyl donors. As illustrated by competition studies comparing the reactivity of per-O-benzylated thioether and trichloroacetimidate donors (Table 2), the relative reactivity ratios were reported to be 6.4:1, 4:1 and 5:1 respectively.33–38 A number of theories have been proposed to account for these observations including stereoelectronic effects and torsional strain.34, 35, 38–42
Table 2.
Examples from competition studies


The key to extending our methodology to glucosyl systems hinged upon the unique reactivity of per-O-trimethylsilyl glycosyl iodides. We were hopeful that the synergistic effects of the TMS groups and the reactive nature of the glycosyl iodide would be sufficient to overcome the relative electron deficiency typically associated with glucosyl donors, resulting in a similar reactivity profile to their galactosyl counterparts. As a first step in our quest to extend this protocol, we initiated a competition study to quantify the relative reactivity of per-O-TMS Gal:Glc iodide donors (Scheme 3). Equal amounts of per-O-TMS-galactoside (12) and per-O-TMS-glucoside (17) were mixed and TMSI was added to the reaction to generate the respective glycosyl iodide donors in situ.28 Once iodide formation was complete, as evidenced by TLC, the solution was cannulated into a flask containing TBAI, DIPEA and (S)-(+)-1,2-isopropylidine glycerol (18), which was chosen as an acceptor because of its relevance to target glucoside 24. After 36 h the mixture was subjected to acidic resin mediated global deprotection followed by acetylation of the resulting free hydroxyls, which allowed for complete characterization of the products. In agreement with our previous reports and based upon 1H and 13C nuclear magnetic resonance (NMR) experiments, only the α-linked glycosides were formed.20, 21 The Gal:Glc product ratio was found to be 1.6:1, which is much closer than is usually observed with the aforementioned donors. In order to verify the results of the competition experiment, both reactions were conducted separately. In the individual experiments, 19 was obtained in 90% yield and 20 in 79% yield validating the competition experiment.
Scheme 3.

Competition experiment between per-O-TMS galactosyl and glucosyl iodides.
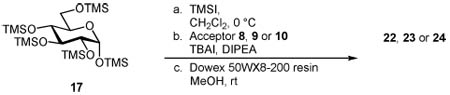
Next, we focused on the synthesis of the glucosyl analogs 22, 23 and 24. We began with the glucosidation of ceramide acceptor 8. As described earlier, per-O-TMS glucosyl iodide was transferred into a flask containing the acceptor, TBAI and DIPEA. After 48 h, the solvent was removed and the residue was reconstituted in methanol where it was subjected to acidic resin mediated deprotection, affording the desired final product after flash column chromatography. Once again, the reaction progressed in a completely stereoselective fashion, providing 22 in 52% yield from starting material to final product (Table 3, Entry 1). The reaction between 17 and glyceride acceptor 10 proceeded in a similar manner, resulting in the first reported synthesis of the glucosyl analog of the immunological antigen BbGL-II (24) in 58% overall yield (Table 3, Entry 3). The anomeric configuration of the products was determined using 1H and 13C NMR, and complete assignment was possible using a combination of 1D and 2D homonuclear and heteronuclear NMR experiments. The glucose H-H (3J1–2) coupling constants were all less than J = 4 Hz as expected for a 1,2-cis linkage. The presence of only one anomeric carbon, between 96–100 ppm, is in agreement with the literature and further indicated the desired α-linkage.
Table 3.
Results of the Glucosylation Study
 | |||||
|---|---|---|---|---|---|
| Entry | Acceptor | Solvent | Conditions | Product | α:β ratio (Yield) |
| 1 | 8 | CH2Cl2 | TBAI (2 eq), rt, 48 h | 22 | only α (52%) |
| 2 | 9 | CH2Cl2 | TBAI (3 eq), ))), 1.5 h | 23 | only α (32%) |
| 3 | 10 | CH2Cl2 | TBAI (2 eq), rt, 48 h | 24 | only α (58%) |
As was the case with our galactose study, initial attempts to incorporate acceptor 9 at room temperature were unsuccessful as none of the desired product (23) was recovered. The decreased reactivity is attributed to micelle formation.43–45 Various reaction conditions were explored including changes in the solvent, temperature and stoichiometry without success. The only thing we have found thus far to promote this reaction is microwave irradiation. Over the three steps, a 32% yield of 23 was obtained when the mixture was subjected to 200W and 115 °C for 1.5 h in a sealed microwave vessel (Table 3, Entry 2). Although we do not completely understand the mode of action, we have postulated that the microwave conditions increase acceptor accessibility by disrupting ordered lipid arrangement.
Conclusions
We have developed a robust platform for the synthesis of biologically relevant glycolipids based on reacting transiently protected glycosyl iodides with fully functionalized and unprotected acceptors. TBAI was utilized to promote in situ anomerization of α-glycosyl iodides to the corresponding β-linked reactive intermediates. Our first generation protocol involved the glycosidation of protected sphingosine or phytosphingosine acceptors with per-O-benzyl galactosyl iodide. While this process proceeded with complete α-selectivity, it was only efficient when the acceptor was outfitted with electron releasing protecting groups. These findings led to the hypothesis that electron rich glycosyl donors may undergo reaction with fully functionalized ceramide acceptors. Subsequent studies revealed that per-O-trimethylsilyl ethers are more reactive than the per-O-benzyl counterpart and are indeed sufficiently reactive to glycosidate ceramides. An extra added benefit of the trimethylsilyl protecting group is the ready removal upon methanolysis providing a one-pot method for the syntheses of bioactive α-linked galactosyl ceramides and glycerides. Encouraged by these results, we extended the methodology to encompass traditionally less reactive glucosyl donors but contrary to our expectation, per-O-TMS glucosyl iodide was nearly as reactive as the galactosyl iodide. This methodology provides selectivity for primary alcohols over allylic or secondary alcohols and no detectable amounts of either the β-glycosides or over glycosylated products were observed. Furthermore, this technology allows for the presence of other functional groups such as esters, amides and alkenes. This three step one-pot protocol is economical and provides rapid access to an important class of immunostimulatory molecules including the first reported synthesis of the glucosyl analog of the bacterial antigen BbGL-II. We are currently exploring the scope of this methodology for the synthesis of biologically relevant oligosaccharides and glycoconjugates.
Experimental Section
1,2-Di-O-acetyl-3-O-(2,3,4,6-tetra-O-α-d-galactopyranosyl)-(S)-glycerol (19)
A solution of per-O-trimethylsilyl-d-galactopyranoside (12, 1.31 g, 2.42 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C followed by the addition of TMSI (532 mg, 2.66 mmol). The reaction mixture was stirred for 20 min at 0 °C then quenched by adding anhydrous benzene (10 mL) followed by evaporation under reduced pressure to afford the glucosyl iodide as a viscous yellow oil. The iodide was next dissolved in CH2Cl2 (3.0 mL) and kept under an argon atmosphere. In a separate flask, TBAI (1.34 g, 3.63 mmol), (S)-(+)-1,2-isopropylidine glycerol (18, 107 mg, 0.81 mmol) and DIPEA (469 mg, 3.63 mmol) were dissolved in CH2Cl2 (5.0 mL) and the mixture was stirred at rt under argon. The glycosyl iodide solution was transferred dropwise, via cannula, to the acceptor flask. Once transferred, the reaction mixture was stirred at rt for 36 h. Next, the solvent was removed and the mixture was reconstituted in MeOH (15 mL) and stirred with Dowex® 50WX8-200 ion exchange resin (1 g) at rt for 4 h. The resin was then removed by filtration and the solvent was removed in vacuo to afford a brown oil. The crude mixture was acetylated using standard conditions followed by purification using flash column chromatography. (CH2Cl2:acetone = 97:3, Rf = 0.24) to afford 19 as a viscous oil (367 mg, 90%). 1H NMR (600 MHz, CDCl3) δ1.98 (s, 3H), 2.04 (s, 3H), 2.060 (s, 3H), 2.064 (s, 3H), 2.09 (s, 3H), 2.13 (s, 3H), 3.61–3.64 (dd, J = 11.4, 5.4 Hz, 1H, H-1a), 3.81–3.83 (dd, J = 11.4, 4.2 Hz, 1H, H-1b), 4.08 (d, J = 6.6 Hz, 2H, H-6’a, H-6’b), 4.13–4.16 (dd, J = 11.4, 5.4 Hz, 1H, H-3a), 4.20 (t, J = 6.6 Hz, 1H, H-5’), 4.30–4.33 (dd, J = 12.0, 4.2 Hz, 1H, H-3b), 5.08–5.10 (dd, J = 10.2, 3.6 Hz, 1H, H-2’), 5.12 (d, J = 3.6 Hz, 1H, H-1’), 5.17–5.20 (p, 1H, H-2), 5.30–5.32 (dd, J = 10.8, 3.0 Hz, 1H, H-3’), 5.45 (app d, J = 3.0 Hz, 1H, H-4’). 13C NMR (150 MHz, CDCl3) δ 20.7, 20.78, 20.81, 21.1, 61.8, 62.3, 66.6, 66.7, 67.6, 68.0, 68.1, 70.2, 96.7, 170.1, 170.30, 170.33, 170.5, 170.59, 170.61. ESI-MS calc for C21H30O14 [M+Na]+ = 529.15, found: 529.30.
1,2-Di-O-acetyl-3-O-(2,3,4,6-tetra-O-α-d-glucopyranosyl)-(S)-glycerol (20)
A solution of per-O-trimethylsilyl-d-glucopyranoside (17, 1.31 g, 2.42 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C followed by the addition of TMSI (532 mg, 2.66 mmol). The reaction mixture was stirred for 20 min at 0 °C then quenched by adding anhydrous benzene (10 mL) followed by evaporation under reduced pressure to afford the glucosyl iodide as a viscous yellow oil. The iodide was next dissolved in CH2Cl2 (3.0 mL) and kept under an argon atmosphere. In a separate flask, TBAI (1.34 g, 3.63 mmol), (S)-(+)-1,2-isopropylidine glycerol (18, 107 mg, 0.81 mmol) and DIPEA (469 mg, 3.63 mmol) were dissolved in CH2Cl2 (5.0 mL) and stirred at rt under argon. The glycosyl iodide solution was transferred dropwise, via cannula, to the acceptor flask. Once transferred, the reaction mixture was stirred at rt for 36 h. Next, the solvent was removed and the mixture was reconstituted in MeOH (15 mL) and stirred with Dowex® 50WX8-200 ion exchange resin (1 g) at rt for 4 h. The resin was then removed by filtration and the solvent was removed in vacuo to afford a brown oil. The crude mixture was acetylated using standard conditions followed by purification using flash column chromatography. (CH2Cl2:Acetone = 97:3, Rf = 0.24) to afford 20 as a viscous oil (323 mg, 79%). 1H NMR (600 MHz, CDCl3) δ 1.99 (s, 3H), 2.02 (s, 3H), 2.05 (s, 3H), 2.06 (s, 3H), 2.079 (s, 3H), 2.083 (s, 3H), 3.62–3.65 (dd, J = 11.4, 5.2 Hz, 1H, H-1a), 3.80–3.83 (dd, J = 10.8, 4.2 Hz, 1H, H-1b), 3.97–4.00 (dq, J = 10.2, 2.4 Hz, 1H, H-5’), 4.07–4.09 (dd, J = 12.6, 2.4 Hz, 1H, H-6’a), 4.14–4.17 (dd, J = 12.0, 6.0 Hz, 1H, H-3a), 4.23–4.26 (dd, J = 12.6, 4.8 Hz, 1H, H-6’b), 4.30–4.33 (dd, J = 12.0, 4.8 Hz, 1H, H-3b), 4.82–4.85 (dd, J = 10.2, 4.2 Hz, 1H, H-2’), 5.04 (app t, J = 10.2, 9.6 Hz, 1H, H-4’), 5.09 (d, J = 4.2 Hz, 1H, H-1’), 5.17–5.20 (p, 1H, H-2), 5.44 (app t, J = 10.2, 9.6 Hz, 1H, H-3’). 13C NMR (150 MHz, CDCl3) δ 20.70, 20.73, 20.8, 21.0, 61.9, 62.3, 66.7, 67.7, 68.6, 70.1, 70.2, 70.8, 96.3, 169.7, 170.2, 170.3, 170.4, 170.6, 170.7. ESI-MS calc for C21H30O14 [M+Na]+ = 529.15, found: 529.30.
1-O-Palmitoyl-2-O-oleoyl-3-O-α-d-glucopyranosyl-sn-glycerol (Glc-BbGL-II, 24)
A solution of per-O-trimethylsilyl-d-glucopyranoside (17, 136 mg, 0.25 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C followed by the addition of TMSI (56 mg, 0.28 mmol). The reaction was stirred for 45 min at 0 °C then quenched by adding anhydrous benzene (10 mL) followed by evaporation under reduced pressure to afford the glucosyl iodide as a viscous yellow oil. The iodide was next dissolved in CH2Cl2 (2.0 mL) and kept under under an argon atmosphere. In a separate flask, TBAI (186 mg, 0.50 mmol), 10 (50 mg, 0.08 mmol) and DIPEA (65 mg, 0.50 mmol) were dissolved in CH2Cl2 (3.0 mL) and stirred at rt under argon. The glycosyl iodide solution was transferred dropwise, via cannula, to the acceptor flask. Once transferred, the reaction mixture was stirred at rt for 48 h. Next, the solvent was removed and the mixture was reconstituted in MeOH (10 mL) and stirred with Dowex® 50WX8-200 ion exchange resin (0.5 g) at rt for 4 h. The resin was then removed by filtration and the solvent was removed in vacuo to afford a brown oil which was purified using flash column chromatography. (CHCl3:MeOH = 95:5, Rf = 0.36) to afford 24 as a white foam (36 mg, 58%). [α]D25+34.4° (C = 1.0, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 0.87 (t, J = 6.6 Hz, 7H), 1.25–1.29 (m, 54H), 1.60 (bs, 4H), 2.01 (q, J = 12.0, 6.0 Hz, 4H), 2.31 (dt, J = 7.2, 4.2 Hz, 4H), 3.49 (app d, J = 13.2 Hz, 1H, H-2’), 3.56–3.60 (m, 3H, H-3a, H-4’, H-5’), 3.72 (bt, 1H, H-3’), 3.82 (bs, 3H, H-3b, H-6’a, H-6’b), 4.15 (dd, J = 11.4, 6.0 Hz, 1H, H-1a), 4.38 (d, J = 9.6 Hz, 1H, H-1b), 4.86 (app s, 1H, H-1’), 5.25 (bt, 1H, H-2), 5.34 (dt, J = 10.8, 6.0 Hz, 2H, HC=CH). 13C NMR (150 MHz, CDCl3) δ 14.2, 22.8, 22.9, 25.0, 27.35, 27.38, 29.27, 29.34, 29.41, 29.48, 29.49, 29.53, 29.69, 29.71, 29.83, 29.84, 29.88, 29.91, 29.92, 32.06, 32.08, 34.3, 34.4, 61.7, 62.7, 66.3, 69.9, 72.0, 72.2, 74.3, 99.3, 129.8, 130.2, 173.4, 173.9. ESI-HRMS calc for C43H80O10 [M-H]− = 755.5668, found: 755.5665.
(2S, 3R, 4E)-1-O-(α-d-Glucopyranosyl)-2-(N-octadecanosylamino)-4-1,3-octadecenediol (22)
A solution of per-O-trimethylsilyl-d-glucopyranoside (17, 162 mg, 0.3 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C followed by the addition of TMSI (56 mg, 0.3 mmol). The reaction mixture was stirred for 45 min at 0 °C then quenched by adding anhydrous benzene (10 mL) followed by evaporation under reduced pressure to afford the glucosyl iodide as a viscous yellow oil. The iodide was next dissolved in CH2Cl2 (2.0 mL) and kept under an argon atmosphere. In a separate flask, TBAI (222 mg, 0.6 mmol), 8 (56 mg, 0.1 mmol) and DIPEA (77 mg, 0.6 mmol) were dissolved in CH2Cl2 (3.0 mL) and stirred at rt under argon. The glycosyl iodide solution was transferred dropwise, via cannula, to the acceptor flask. Once transferred, the reaction mixture was stirred at rt for 48 h. Next, the solvent was removed and the mixture was reconstituted in MeOH (10 mL) and stirred with Dowex® 50WX8-200 ion exchange resin (0.5 g) at rt for 4 h. The resin was then removed by filtration and the solvent was removed in vacuo to afford a brown oil which was purified using flash column chromatography (CH2Cl2:MeOH = 90:10, Rf = 0.36). The product (22) was obtained as a white foam (37 mg, 52%). [α]D25 -31.6° (C = 1.0, CH2Cl2). 1H NMR (600 MHz, CDCl3:CD3OD) δ 0.69 (t, J = 7.2 Hz, 8H), 1.07–1.11, m, 66H), 1.16–1.18 (m, 4H), 1.39–1.41 (m, 2H), 1.84 (q, J = 7.2 Hz, 2H), 2.01 (t, J = 7.2 Hz, 2H), 3.17 (p, J = 1.4 Hz, 2H), 3.19 (t, J = 9.6 Hz, 1H, H-4’), 3.26 (dd, J = 9.6, 4.2 Hz, 1H, H-2’), 3.36 (dq, J = 4.2, 2.4 Hz, 1H, H-5’), 3.47 (t, J = 9.0 Hz, 1H, H-3’), 3.53 (dd, J = 4.2, 1.2 Hz, 1H, H-1a), 3.54 (app t, J = 4.8, 3.0 Hz, 1H, H-6’a), 3.58 (app t, J = 4.2, 3.0 Hz, 1H, H-6’b), 3.61 (t, J = 3.0 Hz, 1H, H-1b), 3.77 (p, J = 3.6 Hz, 1H, H-2), 3.91 (app t, J = 6.6 Hz, 1H, H-3), 4.63 (d, J = 3.6 Hz, 1H, H-1’), 5.26 (dd, J = 15.0, 7.2 Hz, 1H, HC=CH), 5.54 (dt, J = 15.0, 7.2 Hz, 1H, HC=CH). 13C NMR (150 MHz, CDCl3:CD3OD) δ 13.8, 22.5, 25.8, 29.10, 29.17, 29.23, 29.28, 29.39, 29.46, 29.47, 29.51, 31.7, 32.2, 36.3, 53.4, 61.3, 67.2, 70.0, 71.86, 71.88, 73.6, 99.3, 128.9, 134.1, 174.5. ESI-HRMS calc for C42H81NO8 [M+H]+ = 728.6035, found: 728.6031.
(2S, 3S, 4R)-1-O-(α-d-Glucopyranosyl)-2-(N-octadecanosylamino)-1,3,4-octadecanetriol (23)
The microwave-assisted one-pot glycosylation began with the cooling of a solution of per-O-trimethylsilyl-d-glucopyranoside (17, 82 mg, 0.15 mmol) in CH2Cl2 (3 mL) to 0 °C followed by the addition of TMSI (30 mg, 0.15 mmol). The reaction mixture was stirred for 45 min at 0 °C then quenched by adding anhydrous benzene (10 mL) followed by evaporation under reduced pressure to afford the glucosyl iodide as a viscous yellow oil. The iodide was next dissolved in CH2Cl2 (2.0 mL) and kept under an argon atmosphere. Next, TBAI (167 mg, 0.45 mmol), 9 (30 mg, 0.05 mmol) and DIPEA (60 mg, 0.45 mmol) were added to a 10 mL microwave vessel containing CH2Cl2 (3.0 mL) and stirred at rt under argon. The glycosyl iodide solution was transferred, via cannula, to the sealed microwave reaction vessel and it was placed into a microwave reactor. The reaction was conducted at 115 °C for 90 min at 200 watts. The reaction mixture was transferred to a round bottomed flask and the solvent was removed affording a brown viscous oil which was reconstituted in MeOH (10 mL) and stirred with Dowex® 50WX8-200 ion exchange resin (0.5 g) at rt for 4 h. The resin was then removed by filtration and the solvent was removed in vacuo to afford a brown oil, which was purified using flash column chromatography as described above to afford 23 as a white foam (12 mg, 32%). [α]D25 −36.8° (C = 1.0, CH2Cl2:MeOH, 93:7). 1H NMR (600 MHz, C5D5N) δ 0.98 (t, J = 6.6 Hz, 6H), 1.33–1.39 (m, 50H), 1.42–1.53 (m, 6H), 1.75 (m, 1H), 1.91 (m, 2H), 2.37 (m, 1H), 2.52 (t, J = 7.2 Hz, 2H), 4.24 (dd, J = 9.6, 3.6 Hz, 1H, H-2’), 4.32 (t, J = 9.6 Hz, 1H, H-4’), 4.40–4.50 (m, 4H, H-1a, H-6’a, H-4, H-3), 4.52–4.54 (m, 1H, H-5’), 4.58 (dd, J = 11.4, 1.8 Hz, 1H, H-6’b), 4.67 (t, J = 9.0 Hz, 1H, H-3’), 4.83 (dd, J = 10.8, 6.0 Hz, 1H, H-1b), 5.37 (m, 1H, H-2), 5.69 (d, J = 4.2 Hz, 1H, H-1’), 8.47 (d, J = 8.4 Hz, 1H, N-H). 13C NMR (150 MHz, C5D5N) δ14.8, 23.4, 26.85, 26.94, 30.1, 30.2, 30.3, 30.35, 30.41, 30.47, 30.49, 30.50, 30.51, 30.6, 30.9, 32.6, 34.9, 37.3, 51.8, 63.2, 68.6, 72.4, 72.8, 74.0, 75.1, 75.9, 77.2, 101.5, 173.7. ESI-HRMS calc for C42H83NO9 [M+H]+ = 746.6141, found: 746.6143.
Supplementary Material
Acknowledgements
This work is supported by the National Institutes of Health, NIH Grant R01GM090262. NSF CRIF program (CHE-9808183), NSF Grant OSTI 97-24412, and NIH Grant RR11973 provided funding for the NMR spectrometers used on this project. HRMS samples were analyzed by Dr. William Jewell at the UC Davis, Campus Mass Spectrometry Facilities.
Footnotes
Supporting Information
1H, 13C, 2D COSY and HETCOR NMR spectral data is available free of charge via the internet at http://pubs.acs.org.
References and Notes
- 1.Natori T, Koezuka Y, Higa T. Tetrahedron Lett. 1993;34(35):5591–5592. [Google Scholar]
- 2.Orloski KA, Hayes EB, Campbell GL, Dennis DT. MMWR CDC Surveill. Summ. 2000;49:1–11. [PubMed] [Google Scholar]
- 3.Hossain H, Wellensiek H-J, Geyer R, Lochnit G. Biochimie. 2001;83(7):683–692. doi: 10.1016/s0300-9084(01)01296-2. [DOI] [PubMed] [Google Scholar]
- 4.Kulkarni SS, Gervay-Hague J. Org. Lett. 2006;8(25):5765–5768. doi: 10.1021/ol062354m. [DOI] [PubMed] [Google Scholar]
- 5.Cerundolo V, Silk JD, Masri SH, Salio M. Nat Rev Immun. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 6.Godfrey DI, Berzins SP. Nat Immunol. 2006;7(9):904–906. doi: 10.1038/ni0906-904. [DOI] [PubMed] [Google Scholar]
- 7.Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR-E-I, Zajonc DM, Ben-Menachem G, Ainge GD, Painter GF, Khurana A, Hoebe K, Behar SM, Beutler B, Wilson IA, Tsuji M, Sellati TJ, Wong C-H, Kronenberg M. Nat. Immunol. 2006;7(9):978–986. doi: 10.1038/ni1380. [DOI] [PubMed] [Google Scholar]
- 8.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. Science. 1997;278(5343):1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 9.Zajonc DM, III, C C, Mattner J, Zhou D, Savage PB, Bendelac A, Wilson IA, Teyton L. Nat. Immunol. 2005;6(8):810–818. doi: 10.1038/ni1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulkarni SS, Gervay-Hague J. In Chemical Glycobiology I: Glycoconjugates and Carbohydrate-Protein Interactions; ACS Symposium Series; American Chemical Society; 2008. pp. 153–166. [Google Scholar]
- 11.Wu D, Fujio M, Wong C-H. Bioorg. Med. Chem. 2008;16(3):1073–1083. doi: 10.1016/j.bmc.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinjo Y, Wu D, Kim G, Xing G-W, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong C-H, Kronenberg M. Nature. 2005;434(7032):520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 13.Wu D, Xing G-W, Poles MA, Horowitz A, Kinjo Y, Sullivan B, Bodmer-Narkevitch V, Plettenburg O, Kronenberg M, Tsuji M, Ho DD, Wong C-H. Proc. Natl. Acad. Sci. U. S. A. 2005;102(5):1351–1356. doi: 10.1073/pnas.0408696102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage PB, Teyton L, Bendelac A. Chem. Soc. Rev. 2006;35:771–779. doi: 10.1039/b510638a. [DOI] [PubMed] [Google Scholar]
- 15.Miyamoto K, Miyake S, Yamamura T. Nature. 2001;413(6855):531–534. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]
- 16.Fujio M, Wu D, Garcia-Navarro R, Ho DD, Tsuji M, Wong CH. J. Am. Chem. Soc. 2006;128(28):9022–9023. doi: 10.1021/ja062740z. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M, Nakayama T, Taniguchi M, Koezuka Y, Tsuji M. J. Exp. Med. 2002;195(5):617–624. doi: 10.1084/jem.20011889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berkers CR, Ovaa H. Trends Pharmacol Sci. 2005;26(5):252–257. doi: 10.1016/j.tips.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Trappeniers M, Van Beneden K, Decruy T, Hillaert U, Linclau B, Elewaut D, Van Calenbergh S. J. Am. Chem. Soc. 2008;130(49):16468–16469. doi: 10.1021/ja8064182. [DOI] [PubMed] [Google Scholar]
- 20.Du W, Gervay-Hague J. Org. Lett. 2005;7(10):2063–2065. doi: 10.1021/ol050659f. [DOI] [PubMed] [Google Scholar]
- 21.Du W, Kulkarni SS, Gervay-Hague J. Chem. Commun. 2007;(23):2336–2338. doi: 10.1039/b702551c. [DOI] [PubMed] [Google Scholar]
- 22.Fisher E, Fischer H. Ber. 1910;43:2535. [Google Scholar]
- 23.Mukhopadhyay B, Kartha KPR, Russell DA, Field RA. J. Org. Chem. 2004;69(22):7758–7760. doi: 10.1021/jo048890e. [DOI] [PubMed] [Google Scholar]
- 24.Caputo R, Kunz H, Mastroianni D, Palumbo G, Pedatella S, Solla F. Eur. J. Org. Chem. 1999;1999(11):3147–3150. [Google Scholar]
- 25.Sakai T, Ueno H, Natori T, Uchimura A, Motoki K, Koezuka Y. J. Med. Chem. 1998;41:650–652. doi: 10.1021/jm970613v. [DOI] [PubMed] [Google Scholar]
- 26.Polt R, Szabo L, Treiberg J, Li Y, Hruby VJ. J. Am. Chem. Soc. 1992;114(26):10249–10258. [Google Scholar]
- 27.Kinjo Y, Pei B, Bufali S, Raju R, Richardson SK, Imamura M, Fujio M, Wu D, Khurana A, Kawahara K, Wong C-H, Howell AR, Seeberger PH, Kronenberg M. Chemistry & Biology. 2008;15(7):654–664. doi: 10.1016/j.chembiol.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhat A, Gervay-Hague J. Org. Lett. 2001;3(13):2081–2084. doi: 10.1021/ol0160405. [DOI] [PubMed] [Google Scholar]
- 29.We note that the authors of ref. 27 experienced difficulty in removal of TBAI during the synthesis of a galactosyl ceramide in which 3 eq of TBAI was employed and a yield was not reported. Reduction to 1.5 eq may help alleviate purification complications.
- 30.Mukaiyama T, Murai Y, Shoda S-i. Chem. Lett. 1981:431–432. [Google Scholar]
- 31.Morita M, Natori T, Akimoto K, Osawa T, Fukushima H, Koezuka Y. Bioorg. Med. Chem. Lett. 1995;5(7):699–704. [Google Scholar]
- 32.Fan G-T, Pan Y-s, Lu K-C, Cheng Y-P, Lin W-C, Lin S, Lin C-H, Wong C-H, Fang J-M, Lin C-C. Tetrahedron. 2005;61(7):1855–1862. [Google Scholar]
- 33.Bulow A, Meyer T, Olszewski TK, Bols M. Eur. J. Org. Chem. 2004;(2):323–329. [Google Scholar]
- 34.Namchuk MN, McCarter JD, Becalski A, Andrews T, Withers SG. J. Am. Chem. Soc. 2000;122:1270–1277. [Google Scholar]
- 35.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J. Am. Chem. Soc. 1999;121(4):734–753. [Google Scholar]
- 36.Lahmann M, Oscarson S. Can. J. Chem. 2002;80:889–893. [Google Scholar]
- 37.Hadd MJ, Gervay-Hague J. J. Org. Chem. 1997;62:6961–6967. [Google Scholar]
- 38.Jensen HH, Bols M. Acc. Chem. Res. 2006;39(4):259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 39.Crich D, de la Mora M, Vinod AU. J. Org. Chem. 2003;68(21):8142–8148. doi: 10.1021/jo0349882. [DOI] [PubMed] [Google Scholar]
- 40.Edward JT. Chem. Ind. (London) 1955:1102–1104. [Google Scholar]
- 41.Khan SH, O'Neill RA. Modern Methods in Carbohydrate Synthesis. Harwood Academic Publishers; 1996. p. 558. [Google Scholar]
- 42.Pedersen CM, Nordstrom LU, Bols M. J. Am. Chem. Soc. 2007;129(29):9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- 43.Nakano M, Inoue R, Koda M, Baba T, Matsunaga H, Natori T, Handa T. Langmuir. 2000;16(18):7156–7161. [Google Scholar]
- 44.Karttunen M, Haataja MP, Saily M, Vattulainen I, Holopainen JM. Langmuir. 2009;25(8):4595–4600. doi: 10.1021/la803377s. [DOI] [PubMed] [Google Scholar]
- 45.Van Veldhoven PP, Bishop WR, Yurivich DA, Bell RM. Biochem Mol Biol Int. 1995;36(1):21–30. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.